
Abstract
This chapter gives an overview on the cellular expression and function of ABCG2/BCRP/MXR and ABCB6. Inborn errors of ABCG2 are implicated in cancer multidrug resistance, hematological diseases and gout, while those of ABCB6 cause rare and poorly defined conditions, affecting eye development or pigmentation. We discuss the basic biochemical, physiological, and pathophysiological properties of these transporters, focusing on polymorphisms and mutations that lead to pathological conditions. Since in several cases the related diseases are caused by aberrant protein folding, trafficking or degradation, we describe potential correction strategies for prevention or treatment. In this chapter we also provide an improved database for the analysis of disease-causing mutations in ABC transporters, with the hope of promoting further basic research and clinical studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Human ABC transporters may form channels, active transporters and may also act as regulators, thus are involved in a wide range of physiological functions. A large group of human ABC transporters are active, ATP-dependent exporters of endo- and xenobiotics, and protect our body against the accumulation of harmful chemicals or various drugs. In this chapter we focus on two so-called “ABC half-transporters” both of which form homodimers in order to become functional. Their exact function may not be fully understood, but problems of their cellular localization and processing were shown to result in various human diseases. Interestingly, reduced expression and function of both ABCG2 and ABCB6 is relatively frequent and was thought to be asymptomatic. Still, during stress conditions, metabolic alterations, food or drug exposures the function of these proteins becomes clearly important. Under such conditions even relatively frequent polymorphisms may play a decisive role in disease development. An interesting, recently realized connection between these two ABC transporters is their relatively high level expression in the red cell membrane, and their contribution to the human blood group antigen repertoire. We document the potential utilization of this feature in analyzing transporter regulation and promoting diagnostics. Since a better understanding of the biochemical and physiological consequences of mutations in clinically relevant human ABC transporters may lead to potential cures or disease prevention, here we also provide a significantly extended and improved database for the analysis of disease-causing ABC protein mutations.
Diseases Connected to the Expression of Too Much or Too Little of ABCG2 (BCRP/MXR)
Structure, Transport Properties, Substrates, and Localization of ABCG2
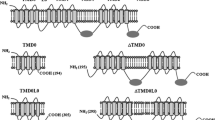
The ABCG2 protein is a “half ABC transporter”, containing one TMD with six transmembrane helices and one NBD, while the functional form of ABCG2 is a homodimer or homo-oligomer (Ozvegy et al. 2001; Gottesman et al. 2002; Dean et al. 2005; Sarkadi et al. 2006). ABCG2 resides in the plasma membrane and in polarized cells in the apical (luminal) membranes. As in the case of all eukaryotic ABC transporters working as “pumps”, this protein is an ATP-dependent exporter. ABCG2 is a multidrug-type transporter, and thus has a promiscuous capacity of recognizing and extruding a large number of transported substrates—including many hydrophobic toxic compounds, amphiphilic positively or negatively charged agents, as well as practically water-soluble molecules. This protein is a physiologically important member of the xenobiotic defense systems in our body, and a major player in cancer drug resistance (Sarkadi et al. 2006). Interestingly, human ABCG2 is also an efficient uric acid transporter, and mutations or polymorphic variants of this transporter are causative in the development of gout (Woodward et al. 2009). ABCG2 may also be transporting heme and its metabolites (Krishnamurthy et al. 2004).
In the human body ABCG2 is expressed in many cell types and tissues. High level expression can be observed in the major metabolic and toxin elimination centers, the liver (bile canalicular membranes) and the kidney (proximal tubular luminal membranes). Other key sites for ABCG2 expression are the barrier forming tissues, including the lumenal side of intestinal epithelial cells, the amnion epithelial cells and the chorion trophoblasts of the placenta, and the endothelial cells of the blood–brain barrier. ABCG2 is preferentially expressed in both pluripotent and some multipotent stem cells, e.g., in the hematopoietic progenitors, the proliferating keratinocytes of the epidermis, or the stem cells of limbal epithelia (Dean et al. 2005; Watanabe et al. 2004). In fact, ABCG2, causing a side-population phenotype of a Hoechst dye uptake, is considered as a marker for both normal and cancer stem cells (Ding et al. 2010). In the hematopoietic stem cells, ABCG2 may be involved in protection against hypoxic challenges (Krishnamurthy et al. 2004) and may permit enhanced stem cell survival in oxygen-poor environments by reducing the accumulation of toxic heme metabolites. These are the reasons why ABCG2 has been named as a “transporter for all seasons” (Sarkadi et al. 2004).
The Role of ABCG2 in Cancer Multidrug Resistance and ADME-Tox Parameters
In cancer cells the expression of ABCG2 results in chemotherapy resistance against a wide variety of anticancer agents that have to cross the cell membrane to reach their intracellular targets. In addition, it has been suggested that similarly to normal stem cells, the putative cancer stem cells (efficient cancer-initiating cells) preferentially express ABCG2, and this expression pattern may be an important factor in the inherent chemotherapy resistance of cancer stem cells. Similarly to that found in the case of the other key cancer multidrug transporter, ABCB1/MDR1/Pgp (see Chapter ‘Genetic Polymorphisms of P-glycoprotein: Echoes of Silence’), a long list of chemotherapeutic agents testifies the huge variety of experimentally verified ABCG2 export substrates (e.g., mitoxantrone, camptothecin, methotrexate, daunorubicin, and doxorubicin—for details see (Brózik et al. 2011; Cusatis et al. 2006; Hegedus et al. 2009a).
A long sought-after solution to counteract cancer multidrug resistance has been the application of specific inhibitors of key ABC multidrug transporters. Several generations of high affinity, specific, or more general inhibitors have been developed and investigated in large clinical studies. However, these studies resulted in unexpected failures and drug developers became uninterested in this particular therapeutic intervention. The explanation of these failures is discussed in detail in (Szakacs et al. 2006), but in addition to several technical problems (e.g., improper diagnosis of transporter expression) the key limitation was an interference with the inherent physiological role of the ABC multidrug transporters—that is the protection of our body against chemical invaders.
ABCG2 is one of the key players in the chemical defense network of the human body. While its function is dispensable under the conditions of a protected environment (see below), full inhibition of this protein may lead to increased exposure of pharmacological sanctuaries like the CNS during cancer chemotherapy. Inhibition of intestinal ABCG2 results in increased absorption of normally extruded toxins, and may strongly disturb the metabolic protection against untoward side-effects of xenobiotics. Thus, general inhibition of the ABCG2 protein may not be the optimal solution to improve the efficacy of cancer therapy even if high ABCG2-expressing cells are the targets.
An especially interesting area in this regard is the recently developed large number of targeted small molecule anticancer agents, including signal transduction or proteasome inhibitors. It has been documented for numerous such molecules that they are transported substrates of ABCG2 (or in some cases of ABCB1). These targeted agents have to reach their targets in the cell interior, and are usually hydrophobic and rapidly cross the plasma membrane. If they are ABCG2 substrates, in spite of their specifically targeted nature for affecting the mutated signaling or regulatory proteins and/or pathways, the cancer cells, and especially the cancer stem cells, are protected by the promiscuous transporter. Accordingly, overexpression (too much) of ABCG2 in cancer is an adverse prognostic factor for therapeutic response in numerous cancer types (see Dean et al. 2005; Robey et al. 2007; Chen et al. 2011). However, a potential hope in this regard is provided by the observations that several targeted anticancer agents, including, e.g., imatinib or nilotinib, are strong inhibitors of the ABCG2 protein at therapeutically relevant concentrations. Thus, the cancer-related cellular action of these molecules is not prevented by active extrusion and, in addition, they provide chemotherapy sensitivity for co-administered agents, otherwise exported by this ABC protein. Detailed in vitro and clinical studies may further explore this therapeutically important phenomenon.
ABCG2 Mutations, Polymorphisms, and Human Diseases Related to “Less” ABCG2
For a long time, ABCG2 expression has been implicated only in multidrug resistance, and, on the positive side, a protective mechanism against toxic xenobiotics. Polymorphic variants or nonsense mutations of ABCG2 were found to be associated with higher sensitivity and interindividual variability to various drug treatments (see Basseville et al. 2012). Numerous ABCG2 genetic variants have been studied in model cell lines. The most frequent/relevant polymorphic variant, Q141K ABCG2, has been shown to be associated with lower plasma membrane expression and/or reduced transport function (see Morisaki et al. 2005). Still, until recently, “too little” of ABCG2 has not been linked to specific human disease conditions.
In ABCG2 knock-out mice studies, when these animals were kept under controlled conditions, practically no phenotype was observed. Detailed experiments thereafter indicated that a diet containing the chlorophyll metabolite pheophorbide resulted in a UV-dependent skin damage as a result of the altered absorption and metabolism (Vlaming et al. 2009). In the ABCG2 knock-out animals, the fetuses accumulate higher levels of cytotoxic drugs, and the concentration of cytotoxic agents and xenobiotics was increased in milk. Despite the increased sensitivity of Abcg2 KO mice to xenobiotics, the human disease-causing effect of less ABCG2 was thought to be not significant.
This paradigm was significantly changed by two key observations. The first was the recognition of a significant role of an ABCG2 polymorphism, the Q141K alteration in the development of gout by GWAS studies, and the demonstration of the role of the ABCG2 protein in uric acid metabolism (Woodward et al. 2009). Gout is a relatively human-specific condition, as in primates and humans, the end-product of the purine metabolism is uric acid, which can accumulate in the joints. ABCG2 is involved both in the renal and extrarenal transport of uric acid, thus counteracting systemic and local hyperuricemia (Matsuo et al. 2009, 2014; Ichida et al. 2012). The Q141K polymorphic variant of ABCG2, present with 5–30 % allele frequency in various human populations (Cervenak et al. 2006), has been shown already in 2005 to be less efficiently expressed in the plasma membrane of model cells (Morisaki et al. 2005; Mizuarai et al. 2004). Later, it was shown to undergo rapid degradation, and to possess less efficient transport properties (see Woodward et al. 2009). Together with both in vitro and in vivo data, by now it has been firmly established that human ABCG2 is an efficient uric acid transporter, and mutations or polymorphic variants, causing a lower level functional expression of this transporter, are causative in the development of gout (Dehghan et al. 2008).
The other unexpected discovery was the recognition of the ABCG2 protein as a blood group antigen. Two papers published within the same issue of Nature Genetics in 2012 (Saison et al. 2012; Zelinski et al. 2012) linked the rare blood group Jun- to the ABCG2 protein, showing that Jun- individuals have no ABCG2 expression in their red cell membranes. These individuals have mutations in their ABCG2 genes on both alleles, resulting in early termination of transcription. Although Jun- individuals have no apparent disease conditions, they may have anti-Jr(a) antibodies in their serum, which can cause transfusion reactions or hemolytic disease of the fetus or newborn (Saison et al. 2012). Corresponding to the higher incidence of ABCG2 nonsense mutations, the Jr- phenotype has a higher frequency in Asian populations.
Detection and Potential Correction of ABCG2 Protein Expression
As discussed above, polymorphic variants and mutations in ABCG2 may have important consequences regarding drug treatment, xenobiotic metabolism, the development of gout, or rare blood group related diseases. The related genetic diagnostics may help to devise chemotherapy protocols, combined medical treatments, or dietary precautions in a large number of patients (Basseville et al. 2012). However, it has to be considered that ABCG2 expression is regulated by several signal transduction pathways, especially by nuclear receptors, and may be directly influenced by posttranslational modifications (Tóth et al. 2015). It has been documented for numerous membrane proteins, and clearly for ABCG2, that mRNA expression is not directly related to the level of protein expression, and especially not necessarily to proper plasma membrane localization (see Robey et al. 2007). Thus, in many cases the determination of ABCG2 protein expression levels in the relevant membrane of a specific tissue is warranted.
Diagnosis of “too much” ABCG2 in drug-resistant cancer relies on functional assays measuring fluorescent dye accumulation (see Hegedus et al. 2009b). “Too little” ABCG2, that is, reduction of ABCG2 expression and function cannot be directly deciphered from genetic analysis, as numerous minor polymorphisms, mutations, not easily detectable splicing problems, or other cellular processing alterations may variably alter membrane protein levels.
In order to approach this problem, we have recently developed a rapid, simple, and reliable, antibody-based flow cytometry assay for the quantitative determination of the ABCG2 protein in the human red blood cell (RBC) membranes (Kasza et al. 2012). We hypothesized that RBC expression may closely correlate with general tissue expression levels, as the potential genetic defects or processing problems should also affect ABCG2 expression during erythropoiesis, and the red cell membrane provides a long-term, relatively stable indicator for measuring this expression. Indeed, in healthy volunteers we have detected significant differences between the expression levels of the wild-type ABCG2 protein and the heterozygous Q141K polymorphic variant. In addition, we found several individuals with about 50 % reduction in RBC ABCG2 expression, and by sequencing the ABCG2 gene we found the related monoallelic nonsense mutations in these individuals. By now we have extended this observation to several other membrane proteins (see Várady et al. 2013; Koszarska et al. 2014) to suggest that the RBC membrane protein levels may be applied to detect genotype-dependent tissue expression patterns. Such a diagnostic approach may significantly facilitate appropriate therapeutic interventions.
As to the potential correction of reduced ABCG2 expression, studies related to the polymorphism causing both processing, functional or trafficking problems, as in the case of the Q141K variant, may help to devise rescue strategies. There have been numerous studies examining these cellular mechanisms (see Sarkadi et al. 2006). In our recent work (Sarankó et al. 2013) we have studied the stability and cellular processing of this variant and a related mutation (ΔF142 ABCG2), corresponding to the processing deficient ΔF508 mutation in the ABCC7 (CFTR) protein. Similarly to that seen in ABCC7, the Q141K variant had a mild processing defect, which could be rescued by low temperature and partially by a chemical chaperone (phenylbutyrate, PBA). Mutations and chemical interventions, resulting in a corrected folding, trafficking, and functional properties of ABCG2 may provide new hopes for rescue strategies.
The Enigmatic Localization and Function of ABCB6
Structure, Function, and Localization of ABCB6
ABCB6 is widely expressed in many tissues, especially in the heart, skeletal muscles (Mitsuhashi et al. 2000), and skin (Zhang et al. 2013). ABCB6 is a half-transporter of 842 amino acids, containing a unique N-terminal region followed by the ABC core consisting of a transmembrane domain and a cytoplasmic nucleotide-binding domain. ABCB6 forms homodimers (Krishnamurthy et al. 2006) and was shown to possess ATPase and transport activities after purification and functional reconstitution into liposomes (Chavan et al. 2013).
ABCB6 was first identified as the human ortholog of the yeast mitochondrial ABC transporter Atm1p (Mitsuhashi et al. 2000). In 2006, Krishnamurthy and colleagues published a paper showing that ABCB6 catalyzes the mitochondrial uptake of a heme synthesis intermediate, coproporphyrinogen III, thereby serving as an important regulator of cellular porphyrin biosynthesis (Krishnamurthy et al. 2006). Based on these findings ABCB6 is usually discussed in the context of mitochondrial ABC transporters, despite mounting evidence supporting its extramitochondrial localization and function. ABCB6 differs in several aspects from the three canonical (ABCB7, ABCB8, and ABCB10) inner membrane mitochondrial ABC transporters: it lacks a mitochondrial targeting sequence; humans and mice seem to tolerate its absence without any obvious phenotype; and its glycosylated form has been detected along the classical secretory pathway including the ER, the Golgi apparatus and the plasma membrane. ABCB7, ABCB8, and ABCB10 reside in the inner mitochondrial membrane, where they promote the export of various solutes from the mitochondrial matrix. Although the transported substrates remain to be identified, it is generally accepted that the function of the mitochondrial ABC transporters is linked to erythropoiesis and heme metabolism. ABCB7 and ABCB8 are believed to be involved in mitochondrial iron export (Allikmets et al. 1999; Ichikawa et al. 2012, 2014), and recent evidence suggests that ABCB10 plays an essential role in the protection against oxidative stress during erythropoiesis (Liesa et al. 2011, 2012).
As mentioned above, Krishnamurthy and colleagues assigned ABCB6 to the outer mitochondrial membrane with the nucleotide-binding domain facing the cytoplasm. This orientation implies an inward transport (i.e., mitochondrial import), which was consistent with experiments showing the uptake of 55Fe-labeled hemin by mitochondria purified from ABCB6-overexpressing cells (Krishnamurthy et al. 2006). Although initial findings suggested that loss of one Abcb6 allele in embryonic stem (ES) cells impairs porphyrin synthesis (Krishnamurthy et al. 2006), mice derived from these stem cells were phenotypically normal (Ulrich et al. 2012). In 2012, the group of Arnaud Lionel at the National Institute of Blood Transfusion in Paris identified ABCB6 as the molecular basis of a rare blood group antigen called Langereis (Lan), and showed that Lan(−) individuals do not show any phenotype, suggesting that porphyrin import into the mitochondrial intermembrane space may not be dependent on ABCB6 (Helias et al. 2012). Several other groups have identified ABCB6 in other extramitochondrial compartments, challenging the paradigm linking the expression and function of ABCB6 to mitochondria (Kiss et al. 2012). Based on these results it appears that ABCB6 is localized in the endolysosomal continuum including the plasma membrane (Helias et al. 2012; Paterson et al. 2007), the Golgi apparatus (Tsuchida et al. 2008), and organelles of the vesicular system (Bagshaw et al. 2005; Schroder et al. 2007; Della Valle et al. 2011; Jalil et al. 2008) such as secreted exosomes (Kiss et al. 2012). Regulation of the intracellular trafficking and the molecular details of targeting of the ABCB6 protein are not known. More work is needed to identify cellular and experimental conditions that result in the targeting of ABCB6 to mitochondria or the secretory pathway.
Expression and Function of ABCB6 in Pathological Conditions: Genotype–Phenotype Correlations
Elucidation of the intracellular targeting and trafficking of ABCB6 should provide hints to the physiological functions of ABCB6. Whereas lack of ABCB6 in Lan-negative individuals does not result in an overt phenotype, ABCB6 mutations have been associated with various pathological conditions such as ocular coloboma (Wang et al. 2012), dominant familial pseudohyperkalemia (Andolfo et al. 2013), and dyschromatosis universalis hereditaria (DUH) (Zhang et al. 2013). The genotype–phenotype correlations or the pathophysiological role of ABCB6 in these conditions are not known, as it has been difficult to correlate sequence variations to ABCB6 function or expression. This is partly due to the lack of our understanding of the physiological function and the structure–activity relationships within ABCB6, and to the complex regulation of synthesis, maturation, trafficking, and posttranslational modifications of membrane proteins.
Genotyping of cryopreserved blood showing weak or no reactivity with anti-Lan antibodies has identified 34 ABCB6 sequence variants in association with reduced ABCB6 expression. All the Lan-negative individuals genotyped so far have inherited two recessive null mutations. Heterozygous mutations result in reduced erythrocytic ABCB6 protein levels, indicating bi-allelic expression of ABCB6. It is not known how Lan mutations that do not result in gross sequence alterations result in lower or absent erythrocytic ABCB6 expression. Whereas the R192W mutation causes ER retention in model cells (Koszarska et al. 2014; Saison et al. 2013), other point mutations associated with reduced red blood cell expression did not influence the distinct endolysosomal expression pattern of ABCB6 (Koszarska et al. 2014). These results highlight the relevance of erythrocyte-specific trafficking events that may not be readily studied in cell lines that are routinely used to model protein expression and function. Erythroid cells undergo significant membrane remodeling during the late stages of differentiation. During this process proteins residing in intracellular membrane compartments are either lost through secreted exosomes or are redistributed to the plasma membrane of mature erythrocytes.
Currently, there is no information available on the distribution of ABCB6 expression levels across the general population, healthy subpopulations, and disease-diagnosed individuals. Lan negativity is extremely rare, estimated to appear in approximately 0.005 % of the Caucasian population (Reid et al. 2014). Interestingly, ABCB6 expression levels in red blood cells display a large variation even in healthy individuals with an unexpectedly high frequency of low expressors, suggesting that genetic variations underlying low red blood cells ABCB6 expression are more common than inferred based on the frequency of Lan-negative blood type (Koszarska et al. 2014).
Ocular coloboma is a developmental defect of the eye linked to the abnormal or incomplete closure of the optic fissure. Mutations in two conserved residues of ABCB6 were identified by positional cloning and the targeted sequencing of the Abcb6 gene in sporadic cases of microphthalmia with coloboma (MAC) (Wang et al. 2012). ABCB6 is highly expressed in human retina and retinal pigment epithelial (RPE) cells. Confocal microscopy experiments found that the wild-type and mutant ABCB6 variants carrying the A57T and L811V mutations associated with coloboma are localized to the endoplasmic reticulum and Golgi apparatus of RPE cells. It remains to be shown how a 50 % reduction of the protein levels and/or function explains the developmental defects identified in heterozygous coloboma patients. Neither the wild-type nor the mutant proteins showed colocalization with mitochondria, suggesting that the coloboma mutations do not affect the protein’s subcellular localization (Wang et al. 2012). The pathological relevance of coloboma mutations was demonstrated in zebra fish, where knockdown of ABCB6 produced a phenotype characteristic of coloboma. Importantly, the knockdown phenotype could be corrected with coinjection of wild-type ABCB6 suggesting that the phenotype observed in zebra fish is due to insufficient ABCB6 function (Wang et al. 2012). Despite the compelling results obtained in the zebrafish model, it is not clear how ABCB6 function is related to the formation and closure of the optic fissure. It is worth noting that a similar experiment examining the effect of DUH mutations (see below) did not reproduce any coloboma-related eye defects (Liu et al. 2014).
DUH is a pigmentary disorder characterized by hyperpigmented and hypopigmented macules distributed randomly over the body. The first DUH mutations were identified in families (L356P) and sporadic cases (S170G, G579E). Subsequently, the mutational spectrum of the ABCB6 gene was expanded through the analysis of further autosomal dominant (A453V, G555K) and sporadic DUH patients (459delC, 776delC, S322K) (Zhang et al. 2013; Liu et al. 2014; Cui et al. 2013). No difference was found in the expression of ABCB6 between patient and control samples. Electron microscopy revealed an abnormal pattern of mature melanosomes and immature melanosomes in the basal layer of hypo- and hyperpigmented skin obtained from a DUH patient (Cui et al. 2013). Expression of the tagged forms of the mutant and wild-type proteins in B16 melanoma cells revealed endosomal localization for the wild-type protein, and a distinct perinuclear localization corresponding to the Golgi apparatus in the case of the mutant ABCB6 variants (Zhang et al. 2013). DUH is essentially a deficiency of melanin synthesis and/or melanosome sorting. Melanin synthesis takes place within melanosomes that are derived from the endoplasmic reticulum. Several melanogenic proteins are sorted in exosomes along melanosome maturing—the presence of ABCB6 in exosomes (Kiss et al. 2012) may be related to this process.
Functional gene mapping identified the segregation of missense ABCB6 mutations with familial pseudohyperkalemia (FP), which is a dominant red cell trait characterized by increased serum [K+] in whole blood stored at or below room temperature (Andolfo et al. 2013). The reported FP mutations (R375Q and R375W) do not alter protein levels or localization. At present, it is unclear how ABCB6 mutations contribute to increased cation leak from red blood cells.
The absence of significant phenotypic alterations in ABCB6 loss-of-function models and in Lan-negative individuals demonstrates that hypomorphic and null mutations of ABCB6 expression are of only modest pathological importance under unstressed conditions (Andolfo et al. 2013). Yet heterozygous mutations at different positions in the ABCB6 gene result in seemingly independent phenotypes: DUH mutations do not result in any phenotype associated with coloboma (and vice versa). Clearly, further studies are needed to explain how specific mutations associate with distinct phenotypes.
A Comprehensive Mutational Database, ABCMdb, for ABC Transporters
In order to design new drugs, strategies for rescuing the function or the proper localization of a specific mutant protein, knowledge on the effect of mutation, or the mutation at the same position in a homologous gene is an important prerequisite. During the last decade, numerous databases have been created to connect information on diseases, mutations, and their mechanisms. However, most of these databases exhibit serious shortcomings. First, most of the data collected focus on genetic alterations found in patients, while a plethora of mutations has also been experimentally generated. Since these mutations are well characterized in many aspects (effect on function, trafficking, etc.), they could serve as a comparative basis for targeting the disease-causing mutations observed in patients. Second, in many cases a specific mutation at a given position in the target protein has not been studied yet, but data on mutations at similar positions in homologous proteins from the same or different species are available. This information can also help investigations for rescuing mutant variants.
Recently we have generated a web application and a connected database (ABCMdb; http://abcmutations.hegelab.org) to facilitate structure/function studies of ABC proteins (Gyimesi et al. 2012). This database contains any mention of amino acid substitutions occurring in searchable full text files, as compared to the reference sequence, including natural and experimental mutations or polymorphisms, found in genotypic or protein in vitro studies. The key features of this database include the searchable presentation of missense mutations at the protein level and the corresponding nucleotide numbers in the coding region, the sequences of homologous proteins with potential mutations in the related areas, and in many cases the available 3D structure information.
As a manual collection of the published mutations is highly resource expensive, we decided to automatically mine mutational data from the literature, in spite of the somewhat increased level of errors produced by such automatic approaches. In this regard it is important to note that a manual collection of data from publications and even the publications themselves are also tainted by human errors. During generation of the database containing ABCC6 variations (http://www.ncbi.nlm.nih.gov/lovd/home.php?select_db=ABCC6) approximately 40 % of the mutations mentioned in published papers turned out to be erroneous (e.g., typos either in the allele or locus—A. Váradi, personal communication). In an automatic collection, the pattern describing a protein mutation is relatively simple (a.a., position, another a.a.), while its correct identification is complex. It is generally assumed that the name of the protein harboring the mentioned mutation is close to the mutations described in the text (e.g., in the same sentence). Moreover, the a.a. at the given position can be confirmed by the reference sequence of the protein. Still we could experience the worst-case scenario, namely the protein mentioned in the same sentence with the mutation is not the target (mutated) protein but exhibits the same amino acid at the same position as the wild-type target protein. While the MutationFinder software (Caporaso et al. 2007) was used in the last years in different pipelines (Gyimesi et al. 2012; Vohra and Biggin 2013) to identify mutation patterns, the recent approach of tmVar (Wei et al. 2013) seems to exhibit a better performance. In addition, the analysis from any type of database must be performed very carefully because of human errors in the original publications.
Since a simple list of mutations identified for the target proteins is not user-friendly, implementation of a useful presentation layer is an important objective. To facilitate discoveries we provide three specific layers for researchers: (1) The sentences, in which the queried mutation was matched, are also listed together with the publication’s PubMed ID that makes manual verification of the hit easier, if needed. (2) Deposited alignments of homologous sequences help both visualizing the a.a. position of interest in the target ABC protein in the context of mutations described in other ABC proteins, and searching for mutations in homologous and nearby positions. (3) Structural models for certain proteins and domains allow the users to investigate mutations in 3D, to decipher their possible effect at the atomic level.
In ABCMdb we elected to present only missense or nonsense point mutations in the coding region, as we have been focusing on studies at the protein level. There is an increasing demand to make additional information accessible, including insertions, deletions, and mutations in the non-coding regions, etc. Additional information on the effect of the mutation (e.g., disease associated, function disrupting, trafficking–effecting) would also effectively guide further research. However, automatic detection of nucleotide mutations is not really effective and decoding the functional effects of mutations by employing language processing tools is currently not potent enough (Peterson et al. 2013).
References
Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM (1999) Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 8:743–749
Andolfo I, Alper SL, Delaunay J, Auriemma C, Russo R, Asci R, Esposito MR, Sharma AK, Shmukler BE, Brugnara C et al (2013) Missense mutations in the ABCB6 transporter cause dominant familialpseudohyperkalemia. Am J Hematol 88:66–72
Bagshaw RD, Mahuran DJ, Callahan JW (2005) A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol Cell Proteomics 4:133–143
Basseville A, Tamaki A, Ierano C, Trostel S, Ward Y, Robey RW, Hegde RS, Bates SE (2012) Histone deacetylase inhibitors influence chemotherapy transport by modulating expression and trafficking of a common polymorphic variant of the ABCG2 efflux transporter. Cancer Res 72:3642–3651
Brózik A, Hegedüs C, Erdei Z, Hegedűs T, Özvegy-Laczka C, Szakács G, Sarkadi B (2011) Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol 7:623–642
Caporaso JG, Baumgartner WA, Randolph DA, Cohen KB, Hunter L (2007) MutationFinder: a high-performance system for extracting point mutation mentions from text. Bioinforma Oxf Engl 23:1862–1865
Cervenak J, Andrikovics H, Ozvegy-Laczka C, Tordai A, Nemet K, Varadi A, Sarkadi B (2006) The role of the human ABCG2 multidrug transporter and its variants in cancer therapy and toxicology. Cancer Lett 234:62–72
Chavan H, Khan MMT, Tegos G, Krishnamurthy P (2013) Efficient purification and reconstitution of ATP binding cassette transporter B6 (ABCB6) for functional and structural studies. J Biol Chem 288:22658–22669
Chen Y-J, Huang W-C, Wei Y-L, Hsu S-C, Yuan P, Lin HY, Wistuba II, Lee JJ, Yen C-J, Su W-C et al (2011) Elevated BCRP/ABCG2 expression confers acquired resistance to gefitinib in wild-type EGFR-expressing cells. PLoS ONE 6:e21428
Cui Y-X, Xia X-Y, Zhou Y, Gao L, Shang X-J, Ni T, Wang W-P, Fan X-B, Yin H-L, Jiang S-J et al (2013) Novel mutations of ABCB6 associated with autosomal dominant dyschromatosis universalis hereditaria. PLoS ONE 8:e79808
Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, Ludovini V, Villa E, Hidalgo M, Sparreboom A et al (2006) Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 98:1739–1742
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284
Dehghan A, Köttgen A, Yang Q, Hwang S-J, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC et al (2008) Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372:1953–1961
Della Valle MC, Sleat DE, Zheng H, Moore DF, Jadot M, Lobel P (2011) Classification of subcellular location by comparative proteomic analysis of native and density-shifted lysosomes. Mol Cell Proteomics 10:M110 006403
Ding X, Wu J, Jiang C (2010) ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci 86:631–637
Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58
Gyimesi G, Borsodi D, Sarankó H, Tordai H, Sarkadi B, Hegedűs T (2012) ABCMdb: a database for the comparative analysis of protein mutations in ABC transporters, and a potential framework for a general application. Hum Mutat 33:1547–1556
Hegedus C, Ozvegy-Laczka C, Szakács G, Sarkadi B (2009a) Interaction of ABC multidrug transporters with anticancer protein kinase inhibitors: substrates and/or inhibitors? Curr Cancer Drug Targets 9:252–272
Hegedus C, Szakács G, Homolya L, Orbán TI, Telbisz A, Jani M, Sarkadi B (2009b) Ins and outs of the ABCG2 multidrug transporter: an update on in vitro functional assays. Adv Drug Deliv Rev 61:47–56
Helias V, Saison C, Ballif BA, Peyrard T, Takahashi J, Takahashi H, Tanaka M, Deybach J-C, Puy H, Le Gall M et al (2012) ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet 44:170–173
Ichida K, Matsuo H, Takada T, Nakayama A, Murakami K, Shimizu T, Yamanashi Y, Kasuga H, Nakashima H, Nakamura T et al (2012) Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun 3:764
Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Jairaj Naik T, Ardehali H (2012) Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci USA 109:4152–4157
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H (2014) Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 124:617–630
Jalil YA, Ritz V, Jakimenko A, Schmitz-Salue C, Siebert H, Awuah D, Kotthaus A, Kietzmann T, Ziemann C, Hirsch-Ernst KI (2008) Vesicular localization of the rat ATP-binding cassette half-transporter rAbcb6. Am J Physiol Cell Physiol 294:C579–C590
Kasza I, Várady G, Andrikovics H, Koszarska M, Tordai A, Scheffer GL, Németh A, Szakács G, Sarkadi B (2012) Expression levels of the ABCG2 multidrug transporter in human erythrocytes correspond to pharmacologically relevant genetic variations. PLoS ONE 7:e48423
Kiss K, Brozik A, Kucsma N, Toth A, Gera M, Berry L, Vallentin A, Vial H, Vidal M, Szakacs G (2012) Shifting the paradigm: the putative mitochondrial protein ABCB6 resides in the lysosomes of cells and in the plasma membrane of erythrocytes. PLoS ONE 7:e37378
Koszarska M, Kucsma N, Kiss K, Varady G, Gera M, Antalffy G, Andrikovics H, Tordai A, Studzian M, Strapagiel D et al (2014) Screening the expression of ABCB6 in erythrocytes reveals an unexpectedly high frequency of lan mutations in healthy individuals. PLoS ONE 9:e111590
Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD (2004) The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem 279:24218–24225
Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, Schuetz JD (2006) Identification of a mammalian mitochondrial porphyrin transporter. Nature 443:586–589
Liesa M, Luptak I, Qin F, Hyde BB, Sahin E, Siwik DA, Zhu Z, Pimentel DR, Xu XJ, Ruderman NB et al (2011) Mitochondrial transporter ATP binding cassette mitochondrial erythroid is a novel gene required for cardiac recovery after ischemia/reperfusion. Circulation 124:806–813
Liesa M, Qiu W, Shirihai OS (2012) Mitochondrial ABC transporters function: the role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim Biophys Acta 1823:1945–1957
Liu H, Li Y, Hung KKH, Wang N, Wang C, Chen X, Sheng D, Fu X, See K, Foo JN et al (2014) Genome-wide linkage, exome sequencing and functional analyses identify ABCB6 as the pathogenic gene of dyschromatosis universalis hereditaria. PLoS ONE 9:e87250
Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Ikebuchi Y, Ito K, Kusanagi Y, Chiba T, Tadokoro S et al (2009) Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med 1:5ra11
Matsuo H, Nakayama A, Sakiyama M, Chiba T, Shimizu S, Kawamura Y, Nakashima H, Nakamura T, Takada Y, Oikawa Y et al (2014) ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci Rep 4:3755
Mitsuhashi N, Miki T, Senbongi H, Yokoi N, Yano H, Miyazaki M, Nakajima N, Iwanaga T, Yokoyama Y, Shibata T et al (2000) MTABC3, a novel mitochondrial ATP-binding cassette protein involved in iron homeostasis. J Biol Chem 275:17536–17540
Mizuarai S, Aozasa N, Kotani H (2004) Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer J Int Cancer 109:238–246
Morisaki K, Robey RW, Ozvegy-Laczka C, Honjo Y, Polgar O, Steadman K, Sarkadi B, Bates SE (2005) Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol 56:161–172
Ozvegy C, Litman T, Szakács G, Nagy Z, Bates S, Váradi A, Sarkadi B (2001) Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem Biophys Res Commun 285:111–117
Paterson JK, Shukla S, Black CM, Tachiwada T, Garfield S, Wincovitch S, Ernst DN, Agadir A, Li X, Ambudkar SV et al (2007) Human ABCB6 localizes to both the outer mitochondrial membrane and the plasma membrane. Biochemistry (Mosc.) 46:9443–9452
Peterson TA, Doughty E, Kann MG (2013) Towards precision medicine: advances in computational approaches for the analysis of human variants. J Mol Biol 425:4047–4063
Reid ME, Hue-Roye K, Huang A, Velliquette RW, Tani Y, Westhoff CM, Lomas-Francis C, Zelinski T (2014) Alleles of the LAN blood group system: molecular and serologic investigations. Transfusion (Paris) 54:398–404
Robey RW, Polgar O, Deeken J, To KW, Bates SE (2007) ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev 26:39–57
Saison C, Helias V, Ballif BA, Peyrard T, Puy H, Miyazaki T, Perrot S, Vayssier-Taussat M, Waldner M, Le Pennec P-Y et al (2012) Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat Genet 44:174–177
Saison C, Helias V, Peyrard T, Merad L, Cartron J-P, Arnaud L (2013) The ABCB6 mutation p.Arg192Trp is a recessive mutation causing the Lan−blood type. Vox Sang 104:159–165
Sarankó H, Tordai H, Telbisz Á, Özvegy-Laczka C, Erdős G, Sarkadi B, Hegedűs T (2013) Effects of the gout-causing Q141K polymorphism and a CFTR ΔF508 mimicking mutation on the processing and stability of the ABCG2 protein. Biochem Biophys Res Commun 437:140–145
Sarkadi B, Ozvegy-Laczka C, Nemet K, Varadi A (2004) ABCG2—a transporter for all seasons. FEBS Lett 567:116–120
Sarkadi B, Homolya L, Szakács G, Váradi A (2006) Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol Rev 86:1179–1236
Schroder B, Wrocklage C, Pan C, Jager R, Kosters B, Schafer H, Elsasser HP, Mann M, Hasilik A (2007) Integral and associated lysosomal membrane proteins. Traffic 8:1676–1686
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234
Tóth A, Brózik A, Szakács G, Sarkadi B, Hegedüs T (2015) A novel mathematical model describing adaptive cellular drug metabolism and toxicity in the chemoimmune system. PLoS ONE 10:e0115533
Tsuchida M, Emi Y, Kida Y, Sakaguchi M (2008) Human ABC transporter isoform B6 (ABCB6) localizes primarily in the golgi apparatus. Biochem Biophys Res Commun 369:369–375
Ulrich DL, Lynch J, Wang Y, Fukuda Y, Nachagari D, Du G, Sun D, Fan Y, Tsurkan L, Potter PM et al (2012) ATP-dependent mitochondrial porphyrin importer ABCB6 protects against phenylhydrazine toxicity. J Biol Chem 287:12679–12690
Várady G, Cserepes J, Németh A, Szabó E, Sarkadi B (2013) Cell surface membrane proteins as personalized biomarkers: where we stand and where we are headed. Biomark Med 7:803–819
Vlaming MLH, Lagas JS, Schinkel AH (2009) Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev 61:14–25
Vohra S, Biggin PC (2013) Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS ONE 8:e71711
Wang L, He F, Bu J, Zhen Y, Liu X, Du W, Dong J, Cooney JD et al (2012) ABCB6 mutations cause ocular coloboma. Am J Hum Genet 90:40–48
Watanabe K, Nishida K, Yamato M, Umemoto T, Sumide T, Yamamoto K, Maeda N, Watanabe H, Okano T, Tano Y (2004) Human limbal epithelium contains side population cells expressing the ATP-binding cassette transporter ABCG2. FEBS Lett 565:6–10
Wei C-H, Harris BR, Kao H-Y, Lu Z (2013) tmVar: a text mining approach for extracting sequence variants in biomedical literature. Bioinforma Oxf Engl 29:1433–1439
Woodward OM, Köttgen A, Coresh J, Boerwinkle E, Guggino WB, Köttgen M (2009) Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci 106:10338–10342
Zelinski T, Coghlan G, Liu X-Q, Reid ME (2012) ABCG2 null alleles define the Jr(a−) blood group phenotype. Nat Genet 44:131–132
Zhang C, Li D, Zhang J, Chen X, Huang M, Archacki S, Tian Y, Ren W, Mei A, Zhang Q et al (2013) Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J Invest Dermatol 133:2221–2228
Acknowledgement
GS was supported by a Momentum Grant of the Hungarian Academy of Sciences and the Austrian Science Fund SFB35 (F3525). TH was supported by a Bolyai Research Fellowship of the Hungarian Academy of Sciences and OTKA 111678. Funding from TET_13_DST-1-2013-0012 is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Szakács, G., Hegedűs, T., Sarkadi, B. (2016). Inborn Errors of the Cellular Expression and Localization of ABCG2 and ABCB6. A Database for ABC Transporter Mutations. In: George, A. (eds) ABC Transporters - 40 Years on. Springer, Cham. https://doi.org/10.1007/978-3-319-23476-2_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-23476-2_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23475-5
Online ISBN: 978-3-319-23476-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)