
Abstract
Guanidinoacetic acid (GAA) is a natural precursor of creatine, and a possible substrate for the creatine kinase (CK) enzyme system, serving as a creatine mimetic. Its direct role in cellular bioenergetics has been confirmed in several studies, however GAA utilization by CK seems to be a second-rate as compared to creatine, and compartment-dependent. Here we discuss various factors that might affect GAA use in high-energy phosphoryl transfer in the cytosol and mitochondria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Creatine / creatine kinase (CK) system is well recognized as a major natural regulator of energy utilization in the cell. Its controlling capacity to replenish cellular levels of adenosine triphosphate (ATP) or accumulate immediate available cellular energy seems to be of particular importance for tissues with high-energy turnover, such as skeletal muscle, heart and brain (Wallimann et al. 2011). Both sufficient availability of CK substrates (e.g. creatine, phosphocreatine, ATP) and functional catalysis are traditionally considered prerequisites for normal cellular bioenergetics (Wyss and Kaddurah-Daouk 2000). Many studies have reported disturbed energy metabolism, and structural and physiological abnormalities in different disorders of creatine biosynthesis and/or transport (Nasrallah et al. 2010). However, several studies questioned the paradigm that creatine is essential in temporal and spatial energy buffering. Mice deficient in creatine due to knockout of the biosynthetic enzyme (S-adenosylmethionine:guanidinoacetate methyltransferase, GAMT) were viable and showed only minor overt abnormalities (Schmidt et al. 2004). Lygate et al. (2013) reported unaltered exercise capacity and response to chronic myocardial infarction in GAMT-deficient knockout mice. Creatine-deficient mice voluntarily ran just as fast and as far as controls (>10 km/night) and performed the same level of work when tested to exhaustion on a treadmill, suggesting consistent bioenergetics without creatine through unknown compensatory adaptation. Kan et al. (2004) indicated that guanidinoacetic acid (GAA), a natural precursor of creatine, might serve as an alternative energy donor, and compensate for the lack of creatine. However, the functional role of GAA in high-energy phosphoryl transfer in health and disease is open to debate. In addition, its possible bioenergetic utilization might be compartment-dependent, and different in the cytosol and within mitochondria.
Guanidinoacetic acid: an alternative or substitute for creatine?
GAA (also known as glycocyamine or guanidinoacetate) is a glycine derivative, and a direct precursor of creatine (Ostojic et al. 2013). Besides its role in creatine synthesis (Fig. 1), GAA has no known physiological role in the normal mammalian metabolism.

Creatine synthesis pathway. GAA guanidinoacetic acid, AGAT L-arginine:glycine amidinotranferase, GAMT S-adenosylmethionine:guanidinoacetate methyltransferase
This intermediary product is synthesized from glycine and arginine by the enzyme L-arginine:glycine amidinotranferase (AGAT), with synthesis taking place mainly in the kidney and pancreas. After transported to the liver (and pancreas), GAA is methylated to yield creatine by the action of GAMT. Creatine itself can be phosphorylated by CK to form phosphocreatine, which is used as an energy reserve in skeletal muscles and the brain. However, central nervous system insures its own creatine synthesis through expression of AGAT, GAMT and the creatine transporter, due to low permeability of blood brain barrier for creatine (Braissant et al. 2010). Creatine deficiency syndromes (CDS), a group of inborn errors of creatine synthesis and transport, induce a notable deficiency of creatine/phosphocreatine in the brain and an accumulation of GAA (Schulze 2013), with creatine levels slightly reduced in skeletal muscle of patients with CDS (Schulze et al. 2003). In this case, GAA can interact directly with CK and form phospho-GAA that might act as a compensatory phosphagen, and an energy reservoir for cellular bioenergetics (Boehm et al. 1996). However, the utilization of guanidinoacetate by CK seems to depend on many physiological and pathophysiological factors (Table 1).
During normal conditions, when availability of creatine is unimpaired, the potential of GAA to act as a substrate for CK is probably negligible. GAA competes with creatine, with the flux through the CK reaction is ~ 100 times lower for GAA as compared to creatine in vitro (Rowley et al. 1971). The low affinity of CK for GAA is probably due to the lack of the N-methyl group, which is considered an important structural feature for the CK reaction (James and Morrison 1966). On the other hand, in creatine-deficient conditions GAA might completely saturate CK and act as a substitutional phosphagen. In GAMT-knockout mice, the resting metabolite level of phospho-GAA (which is not normally present) did not differ from the phosphocreatine peak in wild-type mice (Lygate et al. 2013), implying a notable utilization of GAA by CK. Creatine-deficient mice are able to cope with energetically compromised conditions by using phospho-GAA (Kan et al. 2004). However, animals showed a slower recovery after ischemia and reduced enzyme kinetics. Dephosporylation of phospho-GAA by CK seems to be a rate-limiting step in GAA utilization (Lygate et al. 2013). This suggests that GAA might act as a possible proxy for creatine in CDS yet its ability for replenishment of cellular energy seems inferior as compared to creatine. In addition, it appears that GAA can be effective as an energy donor under stress conditions (e.g. ischemia, acidosis) in creatine deficiency (Lim et al. 2010). This is probably related to the chemical stability of unsubstituted internal nitrogen atom and the absence of the methyl group in GAA (Ellington 2001). Elevated GAA levels were also found by Amayreh et al. (2014) in the blood and cerebrospinal fluid of patients with arginase deficiency, an autosomal recessive disease caused by deficiency of arginase-1, yet the degree of GAA utilization as an energy donor in this disorder is currently unknown.
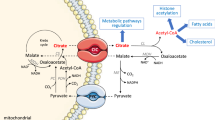
Mitochondrial utilization of guanidinoacetic acid
High-energy phosphoryl transfer through the CK system occurs in both the cytosol and mitochondria. Separate CK isoenzymes and the highly diffusible CK reaction product (such as phosphocreatine) provide a temporal and spatial energy buffer to maintain cellular energy homeostasis (Schlattner et al. 2006). Thus, the compartmental utilization by the CK system depends on, at least, two factors: (1) the affinity of a particular CK isoenzyme for a candidate phosphagen, and (2) the favorable diffusion of a compound through mitochondrial biomembranes. Boehm et al. (1996) reported that GAA is utilized by cytosolic CK in vivo. The accumulation of phosphorylated GAA and creatine in the cytosol was not significantly different (as determined by measuring peak areas via 31P-NMR) although GAA and creatine has different reactivities in vitro. It seems that GAA can be readily utilized in a living tissue containing high concentration of cytosolic CK, and the rate of utilization might be determined by the ATPase rate and not CK (Boehm et al. 1996). On the other hand, GAA had no significant effect on the mitochondrial response to ATP, suggesting low affinity of mitochondrial isoenzymes of CK for GAA. Mitochondrial CK was unable to phosphorylate GAA in isolated mitochondria and saponin-permeabilised myocardial fibers (Boehm et al. 1996). This is probably due to the octameric structure of mitochondrial CK that has a lower structural affinity for GAA due to substrate size and geometry. Another aspect that might limit mitochondrial utilization of GAA is related to the saturation of the membrane transporter required to deliver this compound into mitochondria. Since GAA is transported through biomembranes via creatine transporter (Tachikawa et al. 2009), when the creatine concentration is high a membrane transport protein might become saturated and inhibit GAA transport. However, creatine/GAA transporter (SLC6A8; also known as CrT, CT1 and CreaT) is located on the plasma membrane, and nothing is known so far on the way creatine and/or GAA may enter or exit the mitochondria through SLC6A8. The data published on the potential localization of SLC6A8 in the mitochondrial membranes were due to an antibody artifact (Speer et al. 2004). In addition, pH gradient inside the organelle (basic inside) might limit the transfer of non-polar molecules (such as GAA) into the mitochondria. Even if delivered to mitochondria in creatine-deficient conditions, the utilization of GAA by the organelle is rather poor or absent. Insufficient mitochondrial availability of GAA probably affects its net high-energy phosphoryl transfer in the cell.
Conclusion
Guanidinoacetic acid is a possible energy donor in creatine deficiency, yet its bioenergetics seems to be subordinate to creatine due to gradual high-energy phosphoryl transfer, poor delivery to mitochondria, and low affinity of mitochondrial CK for GAA. Further studies are needed to evaluate GAA compartment-dependent utilization in mitochondrial dysfunction and after exogenous administration of GAA. However, caution should be taken before using GAA as an exogenous energy-donor agent since enough GAA accumulation might be deleterious in CDS (Hanna-El-Daher et al. 2015) and negatively affect brain cell development.
References
Amayreh W, Meyer U, Das AM (2014) Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev Med Child Neurol 56:1021–1024
Boehm EA, Radda GK, Tomlin H, Clark JF (1996) The utilisation of creatine and its analogues by cytosolic and mitochondrial creatine kinase. Biochim Biophys Acta 1274:119–128
Braissant O, Béard E, Torrent C, Henry H (2010) Dissociation of AGAT, GAMT and SLC6A8 in CNS: relevance to creatine deficiency syndromes. Neurobiol Dis 37:423–433
Ellington WR (2001) Evolution and physiological roles of phosphagen systems. Annu Rev Physiol 63:289–325
Hanna-El-Daher L, Béard E, Henry H, Tenenbaum L, Braissant O (2015) Mild guanidinoacetate increase under partial guanidinoacetate methyltransferase deficiency strongly affects brain cell development. Neurobiol Dis 79:14–27
James E, Morrison JF (1966) The reaction of phosphagens with ATP:creatine phosphotransferase. Biochim Biophys Acta 128:327–336
Kan HE, Renema WK, Isbrandt D, Heerschap A (2004) Phosphorylated guanidinoacetate partly compensates for the lack of phosphocreatine in skeletal muscle of mice lacking guanidinoacetate methyltransferase. Physiol 560:219–229
Lim K, Pullalarevu S, Surabian KT, Howard A, Suzuki T, Moult J, Herzberg O (2010) Structural basis for the mechanism and substrate specificity of glycocyamine kinase, a phosphagen kinase family member. Biochemistry 49:2031–2041
Lygate CA, Aksentijevic D, Dawson D, ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag-Montefiore L, Hunyor I, Channon KM, Clarke K, Zervou S, Watkins H, Balaban RS, Neubauer S (2013) Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ Res 112:945–955
Nasrallah F, Feki M, Kaabachi N (2010) Creatine and creatine deficiency syndromes: biochemical and clinical aspects. Pediatr Neurol 42:163–171
Ostojic SM, Niess B, Stojanovic M, Obrenovic M (2013) Creatine metabolism and safety profiles after six-week oral guanidinoacetic acid administration in healthy humans. Int J Med Sci 10:141–147
Rowley GL, Greenleaf AL, Kenyon GL (1971) On the specificity of creatine kinase. New glycocyamines and glycocyamine analogs related to creatine. J Am Chem Soc 93:5542–5551
Schlattner U, Tokarska-Schlattner M, Wallimann T (2006) Mitochondrial creatine kinase in human health and disease. Biochim Biophys Acta 1762:164–180
Schmidt A, Marescau B, Boehm EA, Renema WK, Peco R, Das A, Steinfeld R, Chan S, Wallis J, Davidoff M, Ullrich K, Waldschütz R, Heerschap A, De Deyn PP, Neubauer S, Isbrandt D (2004) Severely altered guanidino compound levels, disturbed body weight homeostasis and impaired fertility in a mouse model of guanidinoacetate N-methyltransferase (GAMT) deficiency. Hum Mol Genet 13:905–921
Schulze A (2013) Creatine deficiency syndromes. Handb Clin Neurol 113:1837–1843
Schulze A, Bachert P, Schlemmer H, Harting I, Polster T, Salomons GS, Verhoeven NM, Jakobs C, Fowler B, Hoffmann GF, Mayatepek E (2003) Lack of creatine in muscle and brain in an adult with GAMT deficiency. Ann Neurol 53:248–251
Speer O, Neukomm LJ, Murphy RM, Zanolla E, Schlattner U, Henry H, Snow RJ, Wallimann T (2004) Creatine transporters: a reappraisal. Mol Cell Biochem 256–257:407–424
Tachikawa M, Kasai Y, Yokoyama R, Fujinawa J, Ganapathy V, Terasaki T, Hosoya K (2009) The blood-brain barrier transport and cerebral distribution of guanidinoacetate in rats: involvement of creatine and taurine transporters. J Neurochem 111:499–509
Wallimann T, Tokarska-Schlattner M, Schlattner U (2011) The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40:1271–1296
Wyss M, Kaddurah-Daouk R (2000) Creatine and creatinine metabolism. Physiol Rev 80:1107–1213
Acknowledgments
This work was supported by the Serbian Ministry of Science (Grant No. 175037).
Conflict of interest
The author declares no conflicts of interest related to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ostojic, S.M. Cellular bioenergetics of guanidinoacetic acid: the role of mitochondria. J Bioenerg Biomembr 47, 369–372 (2015). https://doi.org/10.1007/s10863-015-9619-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-015-9619-7