
Abstract
15N R2 relaxation measurements are key for the elucidation of the dynamics of both folded and intrinsically disordered proteins (IDPs). Here we show, on the example of the intrinsically disordered protein α-synuclein and the folded domain PDZ2, that at physiological pH and near physiological temperatures amide—water exchange can severely skew Hahn-echo based 15N R2 relaxation measurements as well as low frequency data points in CPMG relaxation dispersion experiments. The nature thereof is the solvent exchange with deuterium in the sample buffer, which modulates the 15N chemical shift tensor via the deuterium isotope effect, adding to the apparent relaxation decay which leads to systematic errors in the relaxation data. This results in an artificial increase of the measured apparent 15N R2 rate constants—which should not be mistaken with protein inherent chemical exchange contributions, Rex, to 15N R2. For measurements of 15N R2 rate constants of IDPs and folded proteins at physiological temperatures and pH, we recommend therefore the use of a very low D2O molar fraction in the sample buffer, as low as 1%, or the use of an external D2O reference along with a modified 15N R2 Hahn-echo based experiment. This combination allows for the measurement of Rex contributions to 15N R2 originating from conformational exchange in a time window from µs to ms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteins are inherently dynamic systems with motions that cover a several orders of magnitude wide time scale from femtosecond to more than seconds (Mittermaier and Kay 2009; Palmer 2015). Such dynamics may be local, concerted, correlated or of anti-correlated nature (Salvi et al. 2012; Pelupessy et al. 2003; Fenwick et al. 2011; Vogeli et al. 2014; Vogeli and Yao 2009). Nuclear magnetic resonance spectroscopy (NMR) is one of the major methods to study protein dynamics. A plethora of NMR experiments have been and are further being developed to elucidate protein motions (Palmer 2015, 2004; Vogeli et al. 2014; Mittermaier and Kay 2006; Vallurupalli et al. 2012; Lange et al. 2008; Fawzi et al. 2011; Charlier et al. 2016, 2013). One of the standard experiments are 15N R1, R2 relaxation measurements and the 15N NOE experiment for the detection of the rotational correlation time of the molecule under study as well as local fast dynamics at a residue-specific resolution (i.e. for each 15N–1H moiety along the amino acid sequence) (Kay et al. 1989; Farrow et al. 1994). These measurements have been complemented with more sophisticated experiments and analyses to obtain also intermediate and slow time scale information from µs up to ms. This includes the 15N CPMG- or 15N R1ρ based relaxation dispersion experiments (Loria et al. 1999; Mulder et al. 2001), CEST or DEST measurements (Vallurupalli et al. 2012; Fawzi et al. 2011) and alternatively 13C methyl relaxation measurements covering protein side-chain dynamics (Kiteyski-LeBlanc et al. 2018; Tugarinov and Kay 2005). Towards a more comprehensive picture of dynamics, residual-dipolar couplings (Lange et al. 2008; Tolman et al. 2001; Peti et al. 2002), cross-correlated relaxation (Pelupessy et al. 2003; Vogeli and Yao 2009; Vogeli 2017), paramagnetic relaxation enhancement (PRE) (Pintacuda and Otting 2002; Iwahara and Clore 2006; Xu et al. 2008) and eNOE-based (Vogeli et al. 2014, 2012) data have been acquired and can be used in combination with molecular dynamics simulation (Showalter and Bruschweiler 2007; Markwick et al. 2009) or ensemble averaging (Fenwick et al. 2011; Lange et al. 2008; Vogeli et al. 2012; Bouvignies et al. 2005; Lindorff-Larsen et al. 2005) and chemical-shift based structural ensemble prediction (Camilloni and Vendruscolo 2012; Kannan et al. 2014; Case 2013).
For the investigation of µs–ms dynamics, 15N R2 measurements are among the most frequently used experiments. The 15N R2 rate constant, which describes the decay of 15N transverse magnetization as measured, e.g. in a Hahn-echo experiment, has an exchange contribution, Rex, due to conformational and chemical exchange that modulates the 15N chemical shift tensor (Luginbuhl and Wuthrich 2002; Cavanagh et al. 2007) that adds to the R2,0 auto-relaxation rate constant: R2 = R2,0 + Rex.
It is probably surprising that the presented work identifies a systematic error in several 15N R2 relaxation measurements for the characterization of µs-ms dynamics that deteriorates the dynamics analysis of proteins and in particular intrinsically disordered proteins (IDP) and protein loops when measured under physiological conditions (i.e. pH ~ 7.4 and at a temperature of ~ 37 °C). The identified culprit is the fast exchange of the amide protons with water and simultaneously with the internal reference substance, D2O, resulting in an exchange contribution induced by the deuterium-induced isotope shift of 15N, that becomes particularly acute at physiological pH and temperatures. We exemplify this effect using Hahn-echo based 15N R2 measurements that do not suppress exchange contributions and CPMG relaxation dispersion measurements on α-synuclein, which is an IDP associated to Parkinson’s disease, as well as the PDZ2 domain of human phosphatase and provide a straightforward solution (i.e. the use of a very low D2O molar fraction, as low as 1%, or, alternatively, the use of an external D2O lock and the appropriate pulse sequence).
Materials and methods
Protein expression and purification
Acetylated α-synuclein was expressed using co-expression of the N-terminal acetyltransferase B (NatB) complex and the α-synuclein plasmid (pRK172), as described earlier (Johnson et al. 2010). Expression and purification were performed as described earlier (Huang et al. 2005), with some modification. Briefly, after transformation, colonies containing both plasmids (NatB and pRK172) were grown at 37 °C in 10 ml Lysogeny Broth (LB) medium overnight and were then transferred into 1 L of LB media. After reaching an OD600 of around 0.5, cells were harvested by centrifugation and resuspended into 1 L M9 minimal media containing 15NH4Cl and grown till an OD600 of 1.0 was reached. Protein expression was carried out overnight at 37 °C, after induction with 1 mM IPTG. Cells were harvested by centrifugation and α-synuclein, present in the periplasm, were purified using ion exchange chromatography and hydrophobic interaction chromatography as described earlier (Campioni et al. 2014).
The PDZ2 domain from human phosphatase (hPTP1E) was encoded into a pET21 expression system with a T7 promoter and Histidine tag. Expression and purification were performed as described earlier (Gianni et al. 2005), with some modifications. After transformation, a single colony was inoculated overnight in 10 ml LB medium at 37 °C and then transferred into 1 L M9 minimal media containing 15NH4Cl and grown till an OD600 of 0.5 was reached. Protein expression was induced by adding 1 mM IPTG and cells were harvested by centrifugation after 5 h. A Ni-affinity column (HisTrap FF) was used for purification of protein and the histidine-tag was cleaved with Human Rhinovirus 3C (HRV 3C) followed by another Ni-affinity column purification step.
NMR Measurements
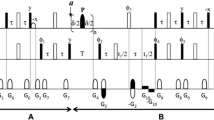
NMR spectra were recorded with 500 µM of 15N-labeled acetylated α-synuclein dissolved in 20 mM Tris (pH 7.4) and 100 mM NaCl, unless indicated differently. Spectra of 15N-labeled PDZ domain, dissolved in 50 mM sodium phosphate buffer (pH 8.0) and 150 mM NaCl, were recorded at an experimental temperature of 303 K. 15N TROSY- \({R}_{2}^{\beta }\) rate constants were measured by applying the NMR experiment described earlier (Lakomek et al. 2013). 15N R2 experiments, applying proton decoupling during the relaxation delay period (Cavanagh et al. 2007), were recorded using the pulse sequence described in Fig. 1. For proton decoupling, waltz64 with an RF amplitude of 2.5 kHz was applied. 15N R1ρ rate constants were recorded using the NMR experiment described in (Lakomek et al. 2012). CPMG-based 15N R2 rate constants were determined using a proton-decoupled CPMG experiment, similar to the one described by Yuwen et al. (2014), however using waltz64 with an RF amplitude of 2.5 kHz for proton decoupling rather than DIPSI2. 15N R2 rate constants were measured for the two CPMG frequencies, 20 Hz and 100 Hz. All NMR experiments were performed on a Bruker 600 MHz Avance III HD spectrometer equipped with cryogenic probe. Spectral dimensions were Ω(1H) x Ω(15N) = 14.014 ppm × 35 ppm. 512 complex points were recorded in the direct dimension (1H) and 80 complex points in the indirect dimension (15N), resulting in an acquisition time of 60.08 ms in the direct and 37.5 ms in the indirect dimension, respectively. The 1H carrier was set to 4.7 ppm and the 15N carrier to 118 ppm, respectively. The magnetization decay was recorded using four different relaxation decay periods, in an inter-leaved manner (Lakomek et al. 2012). For α-synuclein, R2 relaxation delays were set to 0 ms, 200 ms, 100 ms, and 50 ms and for the 15N R1ρ experiments, delays were set to 1 ms, 120 ms, 60 ms, and 20 ms, respectively. The spin-lock RF field strength in the 5N R1ρ measurement was set to 2 kHz. For experiments using the 15N TROSY-\({R}_{2}^{\beta }\) sequence decay periods differed slightly; at pH 7.4 and at a temperature of 303 K or 283 K delays were 0, 100, 50, and 20 ms. Spectral intensities for the different decay periods were recorded in an inter-leaved manner, 16 scans were recorded for each decay period. The total experimental time was 4.75 h. For the PDZ domain, relaxation delays were set to 0 ms, 50 ms, 30 ms, and 10 ms. For the CPMG experiments, a fixed relaxation delay of 200 ms was used. Relaxation data where recorded for two different CPMG frequencies, 100 Hz and 20 Hz by adjusting the number of 180° (N) pulses and the inter-pulse delay accordingly.

Pulse scheme for the 15N R2 relaxation (Hahn-echo) experiment. 1H magnetization is transferred to 15N in-phase magnetization via a refocused INEPT transfer. After a z-filter, a Hahn echo (Δ − 180–(N) − Δ) with the variable relaxation delay Δ is performed. 1H decoupling during the Hahn echo minimizes the evolution of the anti-phase term during the relaxation period. Echo/ anti-echo encoding for quadrature detection is performed prior to the t1 evolution period. Narrow rectangles indicate hard 90° pulses and broader rectangles hard 180° pulses. The rectangular 1H pulses marked—x are low power 90° pulses (1.2 ms at 600 MHz); shaped low power 1H pulses (1.9 ms) correspond to the center lobe of a (sinx)/x function, all serving to return the water magnetization to z prior to detection (Pervushin et al. 1998). For application to samples that also are enriched in 13C: durations of 13C pulses (all 180°) are equal to \(\frac{\sqrt{3}}{2\varOmega }\) (47.4 µs at 600 MHz), where Ω is the frequency difference between 13Cα and 13C′. Delay durations are δ = 2.65 ms and ε corresponds to the duration of the decoding gradient G4 (60.8 µs; the slight offset (ε/2) relative to the 15N 180° pulse enables insertion of the decoding gradient G4, without introducing a linear phase error in the 1H dimension. Gradients: G0 (1000 µs; 21 G/cm), G1 (2650 µs; 0.7 G/cm), G2 (2550 µs; 1.4 G/cm), G3 (500 µs; 42 G/cm), G4 (1000 µs; 7 G/cm), G5 (300 µs; -23 G/cm), G6 (300 µs; 7 G/cm), G7 (1000 µs; 35 G/cm) and G8 (60.8 µs; 23 G/cm) are sine-bell shaped. Phase cycling: φ1 = 8(y), 8(-y); φ2 = y; and φ3 = y, x, − y, − x, − y, − x ,y ,x; φ4 = y; φ5 = y and φrec = y, − x, − y, x, − y, x, y, − x, − y, x, y, − x, y, − x, − y, x. Quadrature detection is implemented using the Rance-Kay echo/anti-echo scheme (Kay et al. 1992), with the polarity of gradients G5 and—G5 inverted, and φ3 = y, − x, − y, x, − y, − x, y, x, φ4 = − y and φ5 = − y for the second FID generated for each quadrature pair. The relaxation decay of 15N (in-phase) coherence is sampled at different delay durations Δ in an inter-leaved manner
The software Sparky 3.115 (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, USA) and Bruker Topspin 3.5pl7 (Bruker, Inc., Billerica, MA, USA) were used for analyzing the spectra and extracting the rate constants.
NMR spectra recorded on samples containing D2O in the sample buffer were measured using a regular Shigemi tube (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). For samples without D2O in the sample buffer, D2O was added as an external reference using a Wilmad coaxial insert (stem length 50 mm, 2 mm diameter) and the sample was kept in a thin wall 5 mm NMR tube (Wilmad NMR tubes, 5 mm diam., precision, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). The coaxial insert containing D2O was inserted into the 5 mm thin-wall NMR tube containing the sample.
Simulation of CPMG relaxation dispersion curves
CPMG relaxation dispersion curves were calculated using the formula:
as described in (Palmer et al. 2001; Ishima and Torchia 1999), with the basic CPMG element τCP/2 − 180° − τCP/2. The inter-pulse delay τCP relates to the CPMG frequency νCPMG via νCPMG = 1/(2τCP). Τhe chemical shift difference induced by the deuterium isotope effect is \(\Delta \delta ({\text{N}})=687 \pm 35\) ppb (Tugarinov 2014) which amounts to ca. Δω = 250 rad s−1 and Δω/2π = 40 Hz at a magnetic field strength of 14.1 T (corresponding to a proton Larmor frequency of 600 MHz) and kex is equal to the assumed amide solvent exchange rate constant (see Results section for derivation); pD is the population of deuterium in the sample buffer (e.g. 0.1 for 10% D2O) and pH the population of H2O in the sample buffer.
Results
Pulse sequence for the measurement of 15N R2 relaxation
The average backbone amide exchange rate constant shows a strong pH dependence, with a minimum around pH 3 (10−1/min at 298 K) and a tenfold increase for each pH unit (Wagner and Wuthrich 1979; Dempsey 2001), resulting in ca. 102 / min at pH 6 (298 K) and roughly 103 / min at pH 7.4 (298 K). Indeed, for α-synuclein at 288 K, measured amide solvent exchange rates varied between 2 and 20 s−1 for different residues at low salt concentration (20 mM) and between 10 s−1 and 80 s−1 for high salt concentrations (300 mM) (Croke et al. 2008). With the emphasis to measure 15N relaxation of α-synuclein at physiological conditions including physiological temperature (i.e. 303 K) we have therefore selected an NMR pulse sequence that measures the transverse relaxation of 15N in-phase coherence, with proton decoupling applied during the relaxation period to alleviate the impact of exchange of the 15N–1H moiety with water. By that, evolution into anti-phase Nx/yHz coherence is minimized (Fig. 1). This is different from e.g. 15N TROSY- \({R}_{2}^{\beta }\) experiments using a Hahn-echo based pulse sequence element (Fig. S1) (Lakomek et al. 2013; Wang et al. 2003). Because if anti-phase Nx/yHz coherence is present or evolves during the Hahn-echo relaxation delay in presence of amide exchange, amide exchange will lead to decorrelation of two spin-order (Skrynnikov and Ernst 1999). This loss of the Nx/yHz coherence will lead to an artificial extra relaxation contribution to the measured 15N R2, rate constant as illustrated in Fig. S2 (this artificial extra relaxation contribution is denoted Rex,amide in Fig. S2).
In Fig. 1, a TROSY-based and Hahn-echo based 15N R2 experiment is shown that avoids this bias introduced by amide exchange. In details, anti-phase 15N magnetization generated after the first INEPT transfer is transferred further to in-phase 15N magnetization in the second step of the re-focused INEPT transfer (b). Therefore, at the beginning of the relaxation period, Nx in-phase magnetization is present. After a z-filter (c), in-phase Nx magnetization is subject to transverse 15N R2 relaxation during the Hahn-echo element. Importantly, the generation of anti-phase magnetization is minimized by 1H decoupling (d). After a second z-filter (e), gradient as well as phase-cycling based echo/anti-echo encoding is achieved prior to t1 evolution. After t1 evolution (f), Nx/yHβ coherence is transferred to Hx/yNβ coherence during a TROSY-read out scheme, opening this pulse sequence also for large systems (Pervushin et al. 1997) (g), which then evolves during acquisition. Further, 15N magnetization, transferred from 1H during the TROSY read-out scheme is destroyed by a 90° pulse on 15N (h) (Lakomek et al. 2012; Favier and Brutscher 2011). Note that in this experiment, 15N R2,0 auto-relaxation (plus Rex contribution) and therefore the average of fast and slowly relaxing NH doublet components is measured rather than the decay of the slowly relaxing Nx/yHβ line, which is measured in the 15N TROSY-\({R}_{2}^{\beta }\)experiment (Lakomek et al. 2013). The relevance of selecting a 15N-inphase-based pulse sequence becomes apparent when comparing the 15N relaxation rate constants measured for the intrinsically disordered protein α-synuclein using the pulse sequence of Fig. 1 compared to those measured using the 15N TROSY-\({R}_{2}^{\beta }\)experiment (Lakomek et al. 2013) (see Fig. S3).
15N R2 relaxation contribution by the deuterium isotope effect
Measuring 15N relaxation of α-synuclein at physiological conditions (i.e. pH 7.4 and 303 K) using the pulse sequence shown in Fig. 1, we noticed a variation of extracted rate constants, depending on the D2O molar fraction in the sample buffer. This is demonstrated in Fig. 2, for which 15N R2 rate constants of α-synuclein were measured in 4%, 10%, and 50% D2O, respectively. The apparent rate constants measured are significantly elevated with increased D2O, apart from C-terminal residues 110–140 that do not show any significant increase, due to exchange protection through hydrogen bond formation of acidic-side chains with amide groups (Khare et al. 1999).

D2O-dependent 15N relaxation rate constants. 15N R2 rate constants of 15N-labeled α-synuclein, measured using the pulse sequence shown in Fig. 1 and in the presence of (A) 4% (black), 10% D2O (red), or 50% D2O (blue). The experimental temperature was 303 K and the pH was 7.4. The increase of the relaxation rates with increase of D2O identifies D2O as a culprit for 15N transverse relaxation measurements
This finding points to a Rex contribution because of the deuterium isotope effect that modulates the 15N chemical shift tensor due to exchange between protons and deuterons in the amide group at an exchange rate constant, kex, which is equal to the solvent exchange rate constant (Henry et al. 1987). This can be seen as follows: The residue-specific kex is the sum of the forward and backward pseudo first order reaction rate constant, kHD and kDH, respectively, where kHD is the product of the amide solvent exchange rate constant, kNH, describing the exchange of amide protons with water, and the likelihood that an exchange to a deuteron takes place instead of a proton, which is equal to the population of D2O in the sample buffer, pD: \({k}_{HD}= {k}_{NH}\times {p}_{D}\). Vice versa, the rate constant for the backward reaction is \({k}_{DH}= {k}_{ND}\times {p}_{H}\) where pH is the population of H2O in the sample buffer and kND the exchange of the amide deuterium with water. It is assumed that kND = kNH (Connelly et al. 1993). This yields:
Figure 3 illustrates the described process.

Chemical exchange between and amide N–H and N–D moiety changes the resonance frequency of the 15N nucleus by changing the chemical shift tensor via the deuterium isotope effect. In equilibrium, the rate constant that describes the conversion from N–H to N–D is described by the solvent amide exchange rate multiplied with the population of D2O in the sample buffer. The backward reaction from N–D to N–H is described by the solvent amide exchange rate times the population of H2O in the sample buffer. It is thereby assumed that the exchange of the amide deuterium with water is equal to the exchange of an amide proton with water (Connelly et al. 1993)
The exchange rate constant describing the modulation of the chemical shift tensor can also be obtained from a kinetic derivation as we shall see:
Equations (3a) and (3b) are of pseudo first order, since both the H2O and D2O molar fractions are much higher than the protein molar fraction in water. Therefore, we obtain:
with the pseudo first order rate constants:
and
where \({\left[{H}_{2}O\right]}_{0}\) is the molar fraction of H2O in the sample buffer in the absence of D2O; pD is the population of D2O and pH is the population of H2O in the sample buffer, resulting in an actual molar fraction \(\left[{H}_{2}O\right]= {\left[{H}_{2}O\right]}_{0} \times {p}_{H}\) of H2O and \(\left[{D}_{2}O\right]= {\left[{H}_{2}O\right]}_{0} \times {p}_{D}\) of D2O in the sample buffer. (Note that for low molar fractions of D2O, actually HDO is present in the sample buffer which has twice the molar fraction as D2O. However, the likelihood of an exchange from NH to ND is only half in the presence of HDO compared to D2O. Both pre-factors cancel out such that the final pseudo first order rate constant is the same. We therefore use the simplified description involving D2O).
For the rate equation describing the time evolution of the molar fraction of the amide group \(\left[NH\right]\) we obtain:
Solving the linear differential equation and using the initial condition [NH] (t = 0) = [NH]0 and [ND] (t = 0) = 0, as only 15N bound to protons is present after the refocused INEPT and start of the relaxation period, yields:
Using the relations described in Eq. (5a, 5b) this yields:
which further simplifies with \({p}_{D}\) to
an thus \({k}_{ex}= {k}_{NH}\). That means that the chemical shift tensor gets modulated at the amide solvent exchange rate.
Τhe chemical shift difference induced by the deuterium isotope effect is \(\varDelta {\updelta }\left(\text{N}\right)=687\pm 35\) ppb (Tugarinov 2014) which amounts to ca. Δω = 250 rad s−1 and Δω/2π = 40 Hz at a magnetic field strength of 14.1 T (corresponding to a proton Larmor frequency of 600 MHz). With an amide exchange rate constant, kex, in the range between 10 and 100 s−1 at pH 7.4 and 303 K, the exchange process is neither in the fast exchange limit, Δω \(\ll\) k, nor in the slow exchange limit, Δω \(\gg k\), but rather on an intermediate timescale. To estimate the exchange contribution on 15N R2 as a result of solvent exchange in the sample buffer, the following formula was used:
as described in (Millet et al. 2000). For 4% D2O, described by pa = 0.96 and pb = 0.04, Eq. (8) yields Rex = 3.31 s−1 for kex = 100 s− 1, Rex = 0.38 s− 1 for kex = 10 s− 1 and Rex = 0.04 s− 1 for kex = 1 s− 1. While for 50% D2O Rex = 21.55 s− 1 for kex= 100 s− 1, Rex = 2.5 s− 1 for kex= 10 s− 1 and Rex = 0.25 s− 1 for kex= 1 s− 1 are estimated. Please note Eq. (8) is strictly speaking no longer fulfilled in the latter case because pa =pb = 0.5 but can be used to get approximate values. Furthermore, effects like a different dipolar coupling interaction for 15N–D versus 15N–H or the quadrupole moment of the deuteron have not been taken into consideration.
Nonetheless, with this rough estimate, an idea on the order of magnitude of the exchange contribution caused by the deuterium isotope effect modulating the 15N chemical shift tensor as a result of chemical exchange between amide protons and deuterons is obtained. It is in good agreement with the experimentally observed D2O dependency of the 15N R2. There is an additional loss mechanism by solvent exchange from a 15N–1H moiety to 15N–D moiety during the relaxation delay making the latter moiety impossible to detect by 1H acquisition (Kim et al. 2013). This effect sales linear with the D2O concentration and can explain partly the observed increase in 15N R2 rate constants for the sample containing 50% D2O. As deuterium is not decoupled during the relaxation period, also 15N-D anti-phase magnetization will evolve during the relaxation period and contribute by scalar relaxation of the second kind. This effect also scales linear with the D2O concentration in the sample buffer; further an15N–H spin pair will show a higher 15N R2 rate constant than a 15N-D spin pair (Vasos et al. 2006; Xu et al. 2005). Scalar relaxation of the second kind induced by the exchange of amide protons can also be an additional loss mechanism in Hahn-echo based 15N R2 measurements (Kateb et al. 2007), however we did not observe any significant differences when changing the RF amplitude of the waltz64 1H decoupling scheme from 2.5 kHz to 6 kHz (Fig. S4).
The use of an external deuterium lock for 15N R2 relaxation measurements
The findings discussed above request 15N relaxation R2 measurements in absence of D2O in the sample buffer. This is achieved by using a coaxial insert by Wilmad comprising D2O inserted into a 5 mm thin-wall NMR tube containing the 15N-labeled α-synuclein in its D2O-free buffer. The external D2O reference is added by inserting a 2 mm capillary which leads to a loss of 16% effective sample volume for a 5 mm NMR tube. No line broadening as result of potential B0 inhomogeneity was observed, however the quality of water suppression was slightly worse and the spectral noise increased slightly.
This approach allows for 15N relaxation measurements using external D2O as a lock substance. Figure 4 shows a comparison of the Hahn-echo based 15N R2, relaxation rate constants of α-synuclein in presence of 4% D2O and in the absence of any D2O in the sample buffer, at two temperatures 283 K and 303 K. Interestingly, while rate constants at pH 7.4 and 283 K vary little (Fig. 4a), at 303 K rate constants measured in the absence of D2O are systematically lower than in the presence of only 4% of D2O, with the exception of the last ~ 30 residues (Fig. 4b). Similar observations have been made with a D2O-free sample that lacked an external locking substance and was thus measured without locking the magnetic field (data not shown).

The importance of using a D2O molar fraction as low as 1% or an external deuterium lock for the measurement of Hahn-echo based 15N relaxation rates of 15N-labeled α-synuclein: 15N relaxation rates in the absence of D2O versus a D2O content of 4% and 1% in the sample buffer. Hahn-echo based 15N R2 rate constants of α-synuclein measured with the pulse sequence shown in Fig. 1. Rate constants measured in the presence of 4% D2O (black) are compared to those measured without D2O (light blue) in the sample buffer at pH 7.4 and temperatures of a 283 K and b 303 K (using an external deuterium lock). When using a D2O molar fraction of 1% (red), even at 303 K the effect is small
The effect of sample internal D2O on the 15N R2 relaxation measurements on the folded protein domain PDZ2
To illustrate that the documented deuterium exchange effects are visible not only for IDPs as illustrated above for α-synuclein, relaxation measurements on the 15N-labeled PDZ2 domain of human phosphatase (Gianni et al. 2005) were performed at pH 8.0 and a temperature of 303 K. The impact of the presence of D2O in the sample buffer on the measured Hahn-echo based 15N R2 rate constants for the PDZ2 domain is illustrated in Fig. 5. Some residues in loop regions (i.e. Asn16, Gly19, Gly24, Gly25, Gly34, Gly50, and Gly63) show a systematic increase in the 15N R2 rate constants when measured in the presence of only 4% D2O in the sample buffer compared to the sample without any D2O in the sample buffer, using an external D2O reference. Glycine residues appear thereby to be overrepresented which is attributed to their overall fast intrinsic amide-water exchange (Bai et al. 1993).

domain (PDB ID: 3PDZ) shown in a ribbon representation
Impact of D2O on the Hahn-echo based 15N R2 rate constants of the globular domain PDZ2. a 15N R2 relaxation rates using the pulse sequence of Fig. 1 were measured on the PDZ2 domain of human phosphatase (Gianni et al. 2005) in the presence of 4% D2O (black) and absence of D2O (blue) in the buffer. In the latter case, a sample-external D2O inside an insert was used for locking the magnetic field. The influence of D2O is pronounced for a few residues in loops (i.e. Asn16, Gly19, Gly24, Gly25, Gly34, Gly50, and Gly63) with a strong overrepresentation of glycine residues attributed to their overall fast intrinsic amide-water exchange. b The residues for which the relaxation was altered by the absence of D2O are highlighted in blue on the structure of PDZ2
The impact of internal D2O on CPMG-based relaxation dispersion experiments
Because of the significant Rex contribution on the measured Hahn-echo based 15N R2 rate constant caused by D2O in the sample buffer, we simulated the anticipated Rex contributions in a CPMG relaxation dispersion experiment. At pH 7.4 and 25 °C (298 K), for solvent-exposed residues the amide exchange rate will assume values in the order of kNH = 10 s−1 to kNH = 100 s−1, (Croke et al. 2008) depending on the extend of solvent exposure of the respective residue. Calculations for different amounts of D2O in the sample buffer (1%, 4% and 10%) are shown in Fig. 6. As illustrated in Fig. 6a, in the presence of 10% D2O for a residue showing fast amide exchange with = 100 s−1 the Rex contribution due to D2O in the sample buffer is present at CPMG frequencies less than 100 s−1, but is significantly reduced for CPMG frequencies νcpmg > 100 s−1, and fully averaged out for a CPMG frequency νcpmg = 500 s−1. The observed effects scales approximately linearly with the amount of D2O in the sample buffer (Fig. 6a). However, even for a D2O molar fraction as low as 1%, the maximum Rex contribution goes up to 1 s−1 (at low CPMG frequencies with νcpmg < 100 s−1). While this may be negligible for the structured part of a large globular protein with an R2 rate constants of e.g. 50 s−1, it amounts to an error of 50% for an IDP with a rate constant of e.g. 2 s−1. For an amide exchange rate constant of kNH < 10 s− 1, the effect is reduced by approximately ten-fold and therefore less critical for only low amounts of D2O in the sample buffer (Fig. 6b). Overall, the Rex contribution roughly scales linearly with the percentage of D2O in the sample buffer and the given amide exchange rate constant kNH. Therefore, at lower pH < 6.5 and temperatures around or below room temperature, where the amide exchange rate will usually be less than kNH < 10 s− 1, at 1% of D2O in the sample buffer the Rex contribution by D2O can be safely ignored. When approaching physiological pH and temperature however, the amide exchange rate constants for many residues can approach values of 100 s− 1 (Croke et al. 2008). Then, for νcpmg < 100 s− 1 the Rex contribution by D2O in the sample buffer can add a significant systematic error on measured R2 relaxation dispersion profiles of IDPs that have low R2,0 auto-relaxation constants.

Simulated CPMG relaxation dispersion curves in presence of different amounts of D2O in the sample buffer. The Rex contribution to 15N R2 is shown as a function of the applied CPMG frequency, with ν_cpmg = 1/(2 τ_cp) and τ_cpmg/2 − 180˚ (N) − τ_cpmg/2 constituting the basic CPMG block. Data are shown for an amide exchange rate constant of a kNH = 100 s− 1 and 10% (blue), 4% (red) and 1% (yellow) D2O in the sample buffer as well as for amide exchange rate constant of b kNH = 10 s− 1 and 10% (blue), 4% (red) and 1% (yellow) D2O in the sample buffer. See “Materials and Methods” for further details.
We have tested the impact of D2O in sample buffer on the extracted CPMG-based 15N R2 rate constants experimentally with α-synuclein. Indeed, at a CPMG frequency of 100 Hz, the exchange contribution induced by D2O appears to be reduced substantially. However, at a low CPMG frequency of 20 Hz we observe substantial Rex contributions, leading to increased 15N R2 rate constants in the presence of 10% D2O (Fig. 7).

Impact of D2O on CPMG-based 15N R2 rate constants of α-synuclein with weak CPMG frequencies (i.e. 20 Hz and 100 Hz). CPMG-based 15N R2 rate constants of α-synuclein were measured at pH 7.4 and at 303 K in the presence of 10% D2O (black), 1% D2O (red) and in the absence of D2O in the sample buffer (light blue). The CPMG frequency was a 20 Hz and b 100 Hz
Finally, we measured also standard 15N R1ρ relaxation measurements (Lakomek et al. 2012) with a spin-lock RF amplitude of 2 kHz on α-synuclein (pH 7.4, 303 K). As expected, when comparing 15N R1ρ rate constants in the presence of 10% D2O and absence of D2O in the sample buffer, we observe only little differences, which is attributed to the loss of measurable magnetization from the exchange to a N-D moiety during the relaxation delays (Fig. 8).

Little impact of D2O on 15N R1ρ rate constants of α-synuclein under a spin-lock frequency (i.e. 2 kHz). 15N R1ρ rate constants for α-synuclein (pH 7.4, 303 K) were measured using a spin-lock RF amplitude of 2 kHz in absence (blue) and presence (black) of 10% D2O
Therefore, the Rex contribution induced by D2O in the sample buffer appears to be suppressed for a standard 15N R1ρ experiment employing a spin-lock RF amplitude of 2 kHz.
Discussion
The presented data shows that at near physiological pH (i.e. pH 7.4) and physiological temperatures of 30–37 °C, solvent exchange of the amide protons with deuterium in the sample buffer can impact Hahn-echo based 15N R2 measurements significantly due to the deuterium isotope effect even at low molar fraction of D2O in the sample buffer (as low as 4%). This effect is pronounced for several loop residues in the folded protein domain PDZ2 but is most prominent in the intrinsically disordered protein α-synuclein. As many IDPs show very low 15N R2 rate constants (< 5 s−1) due to their high intrinsic flexibility, even a small systematic artifactual Rex contribution of e.g. 1 s− 1 can lead to a large error in the data. Therefore, for Hahn-echo based 15N R2 measurements the use of only a very low D2O molar fraction in the sample buffer, as low as 1%, is necessary or, alternatively, the use of an external D2O lock using a coaxial capillary insert.
Since IDPs form a large part of the human proteome (30–40%) and play an essential role in cellular signaling and regulation of many biomolecular interactions (Tompa 2002; Wright and Dyson 1999, 2015), over the last two decades solution-state NMR provided important insights to characterize secondary structure propensity, conformational space (Abyzov et al. 2016; Salvi et al. 2017) and non-local and local dynamics of IDPs using mainly 15N CPMG based relaxation dispersion experiments (Kay et al. 1989; Farrow et al. 1994; Loria et al. 1999; Mulder et al. 2001; Wright and Dyson 2015; Salvi et al. 2017; Sugase et al. 2007; Rezaei-Ghaleh et al. 2012; Maltsev et al. 2013; Bah et al. 2015; Zweckstetter 2016; Delaforge et al. 2018; Kurzbach et al. 2015; Lakomek et al. 2016; Schneider et al. 2015; Arai et al. 2015; Charlier et al. 2017). Several experimental strategies have been designed to allow the recording of 1H-15N correlation spectra (Lopez et al. 2016; Yuwen and Skrynnikov 2014) and CPMG relaxation experiments of IDPs under physiological conditions and obviate the influence of amide exchange (Yuwen et al. 2014; Kim et al. 2013a, b), but the adverse impact of D2O through the isotope effect has to our knowledge escaped attention. At physiological pH and near physiological temperatures, we observed a substantial Rex contribution induced by D2O in the sample buffer that is not suppressed for a low CPMG frequency of 20 kHz. That finding is in agreement with simulated data that predict a substantial contribution for CPMG frequencies < 100 Hz. At a CPMG frequency of 100 Hz the Rex contribution induced by D2O in the sample buffer appears however suppressed, in agreement between experimental and simulated data.
For standard 15N R2 measurements (aiming at the investigation of ps-ns dynamics), employing a CPMG frequency of at least 100 Hz as well as proton decoupling (Kim et al. 2013a, b; Yuwen et al. 2014) (to counteract the adverse effect of amide exchange), the effect is however reduced substantially and will not lead to an artificial increase of the 15N R2 rate constants. Also, in standard 15N R1ρ experiments that spin-lock 15N transverse magnetization, deuterium isotope effects will be suppressed, as long as the spin-lock RF amplitude, given in frequency units, is significantly faster than the amide exchange rate constants—which is usually the case, e.g. for an RF amplitude of 2 kHz and an amide exchange rate in the order of 100 s− 1.
Therefore, the discussed effect is uncritical for standard 15N R2 experiments that aim at characterizing ns-ps dynamics and therefore suppress Rex contributions by spin-lock fields with high RF amplitude in the case of R1ρ measurements or high CPMG frequencies in the case of 15N CPMG-based 15N R2 experiments. But it is important for standard 15N R2 experiments that aim at the quantification of Rex contributions due to conformational dynamics on a µs-ms timescale, such as Hahn-echo based 15N R2 experiments or the more popular CPMG-based 15N R2 relaxation dispersion experiments. (For both experiments it is important to employ proton decoupling during the relaxation period such as suggested by (Kim et al. 2013a, b) and (Yuwen et al. 2014) to counteract adverse effects of amide exchange.) The deuterium induced Rex contribution is less critical for globular proteins with higher 15N R2 rate constants and in the presence of large Rex effects due to conformational dynamics, in the order of, e.g. 10 s− 1. But it can become very critical for the interpretation of CPMG-based relaxation dispersion curves or Hahn-echo based 15N R2 experiments of intrinsically disordered proteins (IDPs) that are characterized by low 15N R2 rate constants (in the order of a few s− 1) and where even a small Rex contribution in the order of e.g. 0.5 s− 1 or more can add to the 15N R2 rate constant substantially.
Indeed, for CPMG-based relaxation dispersion experiments the CPMG frequency for the first, low frequency data points, can be lower than the solvent-exchange rate, depending on the settings for the minimal frequency of the CPMG block in relaxation dispersion experiments, and thus exchange with deuterons in the sample buffer may adversely affect the accuracy of the extracted results and may lead to artificial Rex effects, which originate from deuterium exchange and a modulation of the 15N chemical shift tensor through the deuterium isotope effect rather than conformational dynamics. For Hahn-echo based 15N R2 measurements, that do not suppress but detect all Rex contributions, Rex contributions caused by the deuterium isotope effect are never suppressed and therefore most severe. For high precision CPMG-based relaxation measurements as well as Hahn-echo based 15N R2 experiments that aim at the quantification of µs-ms dynamics, we therefore recommend also the use of a very low D2O content, as low as 1%, or, alternatively, the use of an external deuterium reference, which is easily possible using commercially available NMR tube inserts. This is most critical for IDPs or very flexible loop region in globular proteins that are characterized by low 15N R2 rate constants.
Conclusion
The determination of the 15N R2 relaxation rate constants is a standard NMR experiment in the evaluation of the dynamics of proteins, including both folded and intrinsically disordered protein entities. While measurements at low pH (< 6.5) or low temperatures (< 10 °C) are usually uncritical because of low solvent amide exchange rates, at physiological pH and temperatures, effects related to solvent amide exchange can lead to artifactual Rex contributions.
The presented results show that the presence of D2O > 1% in the sample buffer can deteriorate the accuracy of the rates constants measured using a Hahn-echo based 15N R2 experiments and also for low CPMG-frequency data points (< 100 Hz) in CPMG relaxation dispersion experiments. For CMPG frequencies > 100 Hz as well as for 15N R1ρ experiments that apply a high-power spin-lock RF amplitude, of e.g. 2 kHz, the modulation of the 15N chemical shift tensor by deuterium isotope effect due to amide exchange between N-H and N-D is suppressed and will not lead to artificial Rex contributions, even in the presence of larger amounts of D2O in the sample buffer. Therefore, the discussed effect is uncritical for standard 15N R2 experiments that aim at the characterization of ps-ns dynamics. For Hahn-echo based 15N R2 measurement or CPMG-based 15N R2 relaxation dispersion experiments at near physiological conditions that aim at the characterization of µs-ms dynamics, we however recommend the use of a very low D2O content in the sample buffer, as low as 1% molar fraction or, alternatively, the use of an external deuterium reference. This applies both to in vitro or in-cell NMR experiments (Hansel et al. 2014; Reckel et al. 2007; Luchinat et al. 2014; Plitzko et al. 2017; Lippens et al. 2018; Theillet et al. 2016) and is most important for intrinsically disordered proteins that are characterized by low 15N R2 rate constants and where even small Rex contributions can lead to large changes in the measured 15N R2 rate constant.
References
Abyzov A et al (2016) Identification of dynamic modes in an intrinsically disordered protein using temperature-dependent NMR relaxation. J Am Chem Soc 138:6240–6251
Arai M, Sugase K, Dyson HJ, Wright PE (2015) Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc Natl Acad Sci USA 112:9614–9619
Bah A et al (2015) Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519:106–240
Bai YW, Milne JS, Mayne L, Englander SW (1993) Primary structure effects on peptide group hydrogen-exchange. Protein Struct Funct Genet 17:75–86
Bouvignies G et al (2005) Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc Natl Acad Sci USA 102:13885–13890
Camilloni C, Vendruscolo M (2012) NMR chemical shifts and protein dynamics. FEBS J 279:529–529
Campioni S et al (2014) The presence of an air-water interface affects formation and elongation of alpha-Synuclein fibrils. J Am Chem Soc 136:2866–2875
Case DA (2013) Chemical shifts in biomolecules. Curr Opin Struct Biol 23:172–176
Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ (2007) Protein NMR spectroscopy. Academic Press, New York
Charlier C et al (2013) Nanosecond time scale motions in proteins revealed by high-resolution NMR relaxometry. J Am Chem Soc 135:18665–18672
Charlier C, Cousin SF, Ferrage F (2016) Protein dynamics from nuclear magnetic relaxation. Chem Soc Rev 45:2410–2422
Charlier C et al (2017) Structure and dynamics of an intrinsically disordered protein region that partially folds upon binding by chemical-exchange NMR. J Am Chem Soc 139:12219–12227
Connelly GP, Bai YW, Jeng MF, Englander SW (1993) Isotope effects in peptide group hydrogen-exchange. Protein Struct Funct Genet 17:87–92
Croke RL, Sallum CO, Watson E, Watt ED, Alexandrescu AT (2008) Hydrogen exchange of monomeric alpha-synuclein shows unfolded structure persists at physiological temperature and is independent of molecular crowding in Escherichia coli. Prot Sci 17:1434–1445
Delaforge E et al (2018) Deciphering the dynamic interaction profile of an intrinsically disordered protein by NMR exchange spectroscopy. J Am Chem Soc 140:1148–1158
Dempsey CE (2001) Hydrogen exchange in peptides and proteins using NMR-spectroscopy. Prog Nucl Magn Reson Spectrosc 39:135–170
Farrow NA et al (1994) Backbone dynamics of a free and a phosphopeptide-complexed Src homology-2 domain studied by N-15 Nmr relaxation. Biochemistry 33:5984–6003
Favier A, Brutscher B (2011) Recovering lost magnetization: polarization enhancement in biomolecular NMR. J Biomol NMR 49:9–15
Fawzi NL, Ying JF, Ghirlando R, Torchia DA, Clore GM (2011) Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature 480:268–361
Fenwick RB et al (2011) Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J Am Chem Soc 133:10336–10339
Gianni S et al (2005) The kinetics of PDZ domain-ligand interactions and implications for the binding mechanism. J Biol Chem 280:34805–34812
Hansel R, Luh LM, Corbeski I, Trantirek L, Dotsch V (2014) In-Cell NMR and EPR Spectroscopy of Biomacromolecules. Angew Chem Int Ed Engl 53:10300–10314
Henry GD, Weiner JH, Sykes BD (1987) Backbone dynamics of a model membrane-protein—measurement of individual amide hydrogen-exchange rates in detergent-Solubilized M13 coat protein using C-13 Nmr hydrogen-deuterium isotope shifts. Biochemistry 26:3626–3634
Huang C, Ren G, Zhou H, Wang CC (2005) A new method for purification of recombinant human alpha-synuclein in Escherichia coli. Protein Expr Purif 42:173–177
Ishima R, Torchia DA (1999) Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J Biomol NMR 14:369–372
Iwahara J, Clore GM (2006) Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature 440:1227–1230
Johnson M, Coulton AT, Geeves MA, Mulvihill DP (2010) Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS One 5:e15801
Kannan A, Camilloni C, Sahakyan AB, Cavalli A, Vendruscolo MA (2014) Conformational ensemble derived using nmr methyl chemical shifts reveals a mechanical clamping transition that gates the binding of the HU protein to DNA. J Am Chem Soc 136:2204–2207
Kateb F, Pelupessy P, Bodenhausen G (2007) Measuring fast hydrogen exchange rates by NMR spectroscopy. J Magn Reson 184:108–113
Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by N-15 inverse detected heteronuclear NMR-spectroscopy—application to staphylococcal nuclease. Biochemistry 28:8972–8979
Kay LE, Keifer P, Saarinen T (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114:10663–10665
Khare D, Alexander P, Orban J (1999) Hydrogen bonding and equilibrium protium-deuterium fractionation factors in the immunoglobulin G binding domain of protein G. Biochemistry 38:3918–3925
Kim S, Wu KP, Baum J (2013a) Fast hydrogen exchange affects (1)(5)N relaxation measurements in intrinsically disordered proteins. J Biomol NMR 55:249–256
Kim S, Wu KP, Baum J (2013b) Fast hydrogen exchange affects N-15 relaxation measurements in intrinsically disordered proteins. J Biomol NMR 55:249–256
Kiteyski-LeBlanc JL et al (2018) Investigating the dynamics of destabilized nucleosomes using methyl-TROSY NMR. J Am Chem Soc 140:4774–4777
Kurzbach D, Kontaxis G, Coudevylle N, Konrat R (2015) NMR spectroscopic studies of the conformational ensembles of intrinsically disordered proteins. Intrinsically Disordered Proteins Stud Nmr Spectrosc 870:149–185
Lakomek NA, Ying JF, Bax A (2012) Measurement of N-15 relaxation rates in perdeuterated proteins by TROSY-based methods. J Biomol NMR 53:209–221
Lakomek NA et al (2013) Internal dynamics of the homotrimeric HIV-1 viral coat protein gp41 on multiple time scales. Angew Chem Int Ed Engl 52:3911–3915
Lakomek NA, Draycheva A, Bornemann T, Wintermeyer W (2016) Electrostatics and intrinsic disorder drive translocon binding of the SRP receptor FtsY. Angew Chem Int Ed Engl 55:9544–9547
Lange OF et al (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320:1471–1475
Lindorff-Larsen K, Best RB, DePristo MA, Dobson CM, Vendruscolo M (2005) Simultaneous determination of protein structure and dynamics. Nature 433:128–132
Lippens G et al (2018) In-cell NMR: from metabolites to macromolecules. Analyst 143:620–629
Lopez J, Schneider R, Cantrelle FX, Huvent I, Lippens G (2016) studying intrinsically disordered proteins under true in vivo conditions by combined cross-polarization and carbonyl-detection NMR spectroscopy. Angew Chem Int Ed Engl 55:7418–7422
Loria JP, Rance M, Palmer AG (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc 121:2331–2332
Luchinat E et al. (2014) In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat Commun 5:5502
Luginbuhl P, Wuthrich K (2002) Semi-classical nuclear spin relaxation theory revisited for use with biological macromolecules. Prog Nucl Magn Reson Spectrosc 40:199–247
Maltsev AS, Chen J, Levine RL, Bax A (2013) Site-specific interaction between alpha-synuclein and membranes probed by NMR-observed methionine oxidation rates. J Am Chem Soc 135:2943–2946
Markwick PRL, Showalter SA, Bouvignies G, Bruschweiler R, Blackledge M (2009) Structural dynamics of protein backbone phi angles: extended molecular dynamics simulations versus experimental (3) J scalar couplings. J Biomol NMR 45:17–21
Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG (2000) The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc 122:2867–2877
Mittermaier A, Kay LE (2006) Review—new tools provide new insights in NMR studies of protein dynamics. Science 312:224–228
Mittermaier AK, Kay LE (2009) Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci 34:601–611
Mulder FAA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE (2001) Measurement of slow (mu s-ms) time scale dynamics in protein side chains by N-15 relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc 123:967–975
Palmer AG (2004) NMR characterization of the dynamics of biomacromolecules. Chem Rev 104:3623–3640
Palmer AG (2015) Enzyme dynamics from NMR spectroscopy. Acc Chem Res 48:457–465
Palmer AG, Kroenke CD, Loria JP (2001) Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Nuclear Magn Reson Biol Macromol Pt B 339:204–238
Pelupessy P, Ravindranathan S, Bodenhausen G (2003) Correlated motions of successive amide N-H bonds in proteins. J Biomol NMR 25:265–280
Pervushin K, Riek R, Wider G, Wuthrich K, Attenuated T-2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94, 12366–12371 (1997)
Pervushin KV, Wider G, Wuthrich K (1998) Single transition-to-single transition polarization transfer (ST2-PT) in [N-15,H-1]-TROSY. J Biomol NMR 12:345–348
Peti W, Meiler J, Bruschweiler R, Griesinger C (2002) Model-free analysis of protein backbone motion from residual dipolar couplings. J Am Chem Soc 124:5822–5833
Pintacuda G, Otting G (2002) Identification of protein surfaces by NMR measurements with a paramagnetic Gd(III) chelate. J Am Chem Soc 124:372–373
Plitzko JM, Schuler B, Selenko P (2017) Structural biology outside the box—inside the cell. Curr Opin Struct Biol 46:110–121
Reckel S, Hansel R, Lohr F, Dotsch V (2007) In-cell NMR spectroscopy. Prog Nucl Magn Reson Spectrosc 51:91–101
Rezaei-Ghaleh N, Blackledge M, Zweckstetter M (2012) Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery. Chembiochem 13:930–950
Salvi N, Ulzega S, Ferrage F, Bodenhausen G (2012) Time scales of slow motions in ubiquitin explored by heteronuclear double resonance. J Am Chem Soc 134:2481–2484
Salvi N, Abyzov A, Blackledge M (2017) Atomic resolution conformational dynamics of intrinsically disordered proteins from NMR spin relaxation. Prog Nucl Magn Reson Spectrosc 102:43–60
Schneider R et al (2015) Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion. NMR J Am Chem Soc 137:1220–1229
Showalter SA, Bruschweiler R (2007) Validation of molecular dynamics simulations of biomolecules using NMR spin relaxation as benchmarks: application to the AMBER99SB force field. J Chem Theory Comput 3:961–975
Skrynnikov NR, Ernst RR (1999) Detection of intermolecular chemical exchange through decorrelation of two-spin order. J Magn Reson 137:276–280
Sugase K, Dyson HJ, Wright PE (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447:1021–1025
Theillet FX et al (2016) Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530:45–50
Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH (2001) Structural and dynamic analysis of residual dipolar coupling data for proteins. J Am Chem Soc 123:1416–1424
Tompa P (2002) Intrinsically unstructured proteins. Trends Biochem Sci 27:527–533
Tugarinov V (2014) Indirect use of deuterium in solution NMR studies of protein structure and hydrogen bonding. Prog Nucl Magn Reson Spectrosc 77:49–68
Tugarinov V, Kay LE (2005) Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. Chembiochem 6:1567–1577
Vallurupalli P, Bouvignies G, Kay LE (2012) Studying “Invisible” excited protein states in slow exchange with a major state conformation. J Am Chem Soc 134:8148–8161
Vasos PR, Hall JB, Kummerle R, Fushman D (2006) Measurement of N-15 relaxation in deuterated amide groups in proteins using direct nitrogen detection. J Biomol NMR 36:27–36
Vogeli B (2017) Cross-correlated relaxation rates between protein backbone H-X dipolar interactions. J Biomol NMR 67:211–232
Vogeli B, Yao LS (2009) Correlated dynamics between protein HN and HC bonds observed by NMR cross relaxation. J Am Chem Soc 131:3668–3678
Vogeli B, Kazemi S, Guntert P, Riek R (2012) Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs. Nat Struct Mol Biol 19:1053–1110
Vogeli B et al (2014) Towards a true protein movie: a perspective on the potential impact of the ensemble-based structure determination using exact NOEs. J Magn Reson 241:53–59
Wagner G, Wuthrich K (1979) Structural interpretation of the amide proton-exchange in the basic pancreatic trypsin-inhibitor and related proteins. J Mol Biol 134:75–94
Wang C, Rance M, Palmer AG 3rd (2003) Mapping chemical exchange in proteins with MW> 50 kD. J Am Chem Soc 125:8968–8969
Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 293:321–331
Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16:18–29
Xu J, Millet O, Kay LE, Skrynnikov NR (2005) New spin probe of protein dynamics: Nitrogen relaxation in N-15-H-2 amide groups. J Am Chem Soc 127:3220–3229
Xu XF et al (2008) Dynamics in a pure encounter complex of two proteins studied by solution scattering and paramagnetic NMR spectroscopy. J Am Chem Soc 130:6395–6403
Yuwen T, Skrynnikov NR (2014) CP-HISQC: a better version of HSQC experiment for intrinsically disordered proteins under physiological conditions. J Biomol NMR 58:175–192
Yuwen T, Skrynnikov NR, Proton-decoupled CPMG (2014) A better experiment for measuring N-15 R-2 relaxation in disordered proteins. J Magn Reson 241:155–169
Zweckstetter M (2016) Intrinsically disordered proteins in neurodegeneration Markus Zweckstetter. Biophys J 110:2a
Author information
Authors and Affiliations
Corresponding authors
Additional information
This manuscript is dedicated to Alexander Sobol.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumari, P., Frey, L., Sobol, A. et al. 15N transverse relaxation measurements for the characterization of µs–ms dynamics are deteriorated by the deuterium isotope effect on 15N resulting from solvent exchange. J Biomol NMR 72, 125–137 (2018). https://doi.org/10.1007/s10858-018-0211-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-018-0211-4