
Abstract
In this study, we developed and investigated nanoparticles of biologically-derived, biodegradable polyhydroxyalkanoates (PHAs) as carriers of a hydrophobic photosensitizer, 5,10,15,20-Tetrakis(4-hydroxy-phenyl)-21H, 23H-porphine (pTHPP) for photodynamic therapy (PDT). Three PHA variants; polyhydroxybutyrate, poly(hydroxybutyrate-co-hydroxyvalerate) or P(HB-HV) with 12 and 50 % HV were used to formulate pTHPP-loaded PHA nanoparticles by an emulsification-diffusion method, where we compared two different poly(vinyl alcohol) (PVA) stabilizers. The nanoparticles exhibited nano-scale spherical morphology under TEM and hydrodynamic diameters ranging from 169.0 to 211.2 nm with narrow size distribution. The amount of drug loaded and the drug entrapment efficiency were also investigated. The in vitro photocytotoxicity was evaluated using human colon adenocarcinoma cell line HT-29 and revealed time and concentration dependent cell death, consistent with a gradual release pattern of pTHPP over 24 h. This study is the first demonstration using bacterially derived P(HB-HV) copolymers for nanoparticle delivery of a hydrophobic photosensitizer drug and their potential application in PDT.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Photodynamic therapy (PDT) is an attractive and selective cancer treatment modality that is increasingly being recognized as an alternative to other conventional cancer treatments [1, 2]. Moreover, it does not require a long-term stay in hospital in contrast to weeks and months of surgery or chemotherapy [3, 4]. PDT is based on the combination of photosensitizer (PS), light, and tissue oxygen that can destroy abnormal tissue lesions without minimal damage to surrounding healthy tissues [5–7]. Upon local irradiation with a suitable wavelength of light, PS molecules accumulated in cells can undergo photo-excitation and transfer energy to molecular oxygen. This transfer leads to the production of reactive oxygen species (ROS), which can cause irreversible damage and ultimately cell death in the treated tissues [8, 9].
Although PDT has been established as a promising treatment for certain kinds of cancers, there are still some issues, especially with certain hydrophobic PS compounds, such as hematoporphyrin derivatives (i.e., Photofrin®), due to their poor water solubility [3]. Such PSs can easily aggregate in aqueous media, resulting in the impediment of their parenteral administration and the reduction of photoexcitation efficiency [10–12]. However, the hydrophobicity of the PS molecule is also an important property that can facilitate localization to cellular membranes, including mitochondrial membranes [13]. One approach to solve the PS solubility issue, improve blood circulation and reduce the systemic dose is to formulate the PS into polymeric nanoparticles (NPs) [14]. The accumulation of non-targeted NPs in tumors is mainly attributed to the enhanced permeation and retention effect (EPR) of vascular and lymphatic drainage [15, 16]. Studies show that NPs with size smaller than 200 nm could offer effective PDT by improving EPR and reducing uptake by phagocytic cells [17]. Several biocompatible and biodegradable aliphatic polyesters, such as poly(lactic acid) (PLA), poly(lactide-co-glycolide) (PLGA) and polycaprolactone (PCL) have already been investigated for PS formulation and administration [18–20].
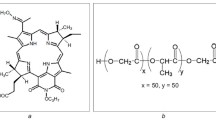
Polyhydroxyalkanoates (PHAs) is another interesting polyester system with great potential application in the biomedical field that has gained increasing attention over the last decade. PHAs are natural aliphatic polyesters accumulated as inclusion bodies in various bacteria and used as carbon and energy storage deposits under unfavorable growth condition [21, 22]. Bacterial PHAs are biodegradable with excellent biocompatibility, and often produced at high molecular weight (i.e., 105–106 g/mol) with low polydispersity index. In addition, they can be produced with different side chain compositions, making the PHA family useful in a wide range of applications [23]. Polyhydroxybutyrate or PHB is the most common type of PHA homopolymer and is relatively stiff and brittle. Incorporation of hydroxyvalerate (HV) monomers together with hydroxybutyrate (HB) to form a copolymer known as poly(hydroxybutyrate-co-hydroxyvalerate) or P(HB-HV) (Fig. 1a) can modify the PHA properties by reducing brittleness and melting temperature, while increasing the degradation rate [24]. Both PHB and P(HB-HV) have been successfully demonstrated for preparation of NPs that encapsulate and offer controlled release of various drugs [25–30].

a Chemical structure of poly(hydroxybutyrate-co-hydroxyvalerate) or P(HBx − HVy) and 1H-NMR spectrum of P(HB-50HV), where (b) and (s) stand for backbone and side chain, respectively. b Structural formula of 5,10,15,20-Tetrakis(4-hydroxy-phenyl)-21H, 23H-porphine (pTHPP). c Absorption spectrum of pTHPP dissolved in THF showing the characteristic of Soret peak at 422 nm. The insert shows the Q bands at 518, 554, 594 and 653 nm
Here, we prepare and characterize polymeric NPs of naturally derived PHAs and investigate their ability to encapsulate PS for PDT. We use the second generation PS, 5,10,15,20-Tetrakis(4-hydroxy-phenyl)-21H, 23H-porphine (pTHPP, Fig. 1b), which has demonstrated good potency to kill various tumors and is a good hydrophobic drug model for polymeric encapsulation studies [31, 32]. It has an absorption peak at 653 nm (Fig. 1c), which falls within the “therapeutic window” for improved tissue penetration [33–36]. Three naturally derived PHAs; PHB, P(HB-12HV) and P(HB-50HV) were investigated in this study, where P(HB-50HV) was prepared by bacterial cultivation “in house”. A modified emulsification-diffusion method with poly(vinyl alcohol) (PVA) as a stabilizer was used, where experimental conditions were tuned to yield sub-200 nm PHA NPs loaded with pTHPP. The NPs were characterized and compared with respect to size, drug loading, drug entrapment efficiency and drug release profile. The photocytotoxic effects of pTHPP-loaded NPs were also evaluated using HT-29 colon cancer cells. The results suggest the potential application of P(HB-HV) copolymer produced from bacteria as a novel NP delivery system of hydrophobic PSs in PDT for the treatment of cancers.
2 Materials and methods
2.1 Materials
Naturally derived polyhydroxybutyrate (PHB) (Mw = 3.50 × 105 g/mol), poly(hydroxybutyrate-co-hydroxyvalerate) with HV content 12 wt% P(HB-12HV) (Mw = 2.50 × 105 g/mol), PVA 87–89 % hydrolyzed including low Mw PVA (Mw = 13,000–23,000 g/mol) and high Mw PVA (Mw = 30,000–70,000 g/mol), thiazolyl blue tetrazolium bromide (MTT) and 5,10,15,20-tetrakis(4-hydroxy-phenyl)-21H, 23H-porphine (pTHPP), were obtained from Sigma-Aldrich (St. Louis, MO, USA). Chloroform, ethanol and tetrahydrofuran (THF) were purchased from RCI Labscan (Bangkok, Thailand). Dulbecco’s Modified Eagle’s Medium (DMEM), antibiotic–antimycotic solution and Fetal Bovine Serum (FBS) were supplied by Gibco (Grand Island, NY, USA). All other chemicals and solvents were of analytical grade and used without further purification. Milli-Q ultrapure water was used in all experiments.
2.2 Production and characterization of P(HB-50HV)
In this study, Cupriavidus necator H16 was used for polymer synthesis by two-stage cultivation according to a previously described protocol [37]. Sodium valerate was added to mineral salts medium as a carbon substrate in order to promote the biosynthesis of P(HB-50HV) [38]. Polymer-accumulating cells were centrifuged and washed twice with PBS and subsequently lyophilized for 24 h (Freeze zone plus 6, Labconco, USA). The methanolysis in chloroform of the lyophilized cells in the presence of 85 % (v/v) methanol, 15 % (v/v) sulfuric acid was carried out [39] prior to determining polymer composition through gas chromatography analysis (Model 6890 plus, Agilent Technology, USA) equipped with HP-INNOWAX 30.0 m, 0.25 mm, 0.25 μm capillary columns and flame deionization detector). To purify P(HB-50HV) copolymer, an established solvent extraction process was used following a previously described method [40]. In brief, a cell-chloroform mixture was refluxed for 24 h at 60 °C while stirring to extract the polymer. Next, the cell-chloroform mixture was filtered through a Whatman No. 1 paper to remove cellular debris. The polymer was precipitated with a tenfold volume of cold methanol. The % purity and the composition of the %HV monomer content of the purified polymer were also re-confirmed with 1H-NMR (Bruker Avance-500 MHz spectrometer, Bruker, Germany). The Mw of P(HB-50HV) was determined to be 1.69 × 106 g/mol.
2.3 Preparation of pTHPP-loaded PHA nanoparticles
In this study, NPs of three different PHA polymers; PHB, P(HB-12HV) and P(HB-50HV) and two types of PVA; low Mw PVA [41] and high Mw PVA [36, 42] were prepared using a modified emulsification-diffusion method [43, 44]. Briefly, PHA was dissolved in chloroform as a stock solution at 1 g/L. Next, the organic phase was prepared by blending the polymer stock solution with ethanol at 1:1 (v/v) to obtain a final volume of 3 mL. The PHA mixture was slowly added to 27 mL of an aqueous solution of 0.5 % w/v PVA under continuous stirring. To form NPs, this solution was subsequently ultrasonicated at 50 % power for 8 min using a titanium flat tip sonicator DT 2200 (Bandelin Sonopuls, Germany). Finally, 30 mL of water was added to the NP solution and continuously stirred at 500 rpm for 24 h to allow for organic solvent evaporation. To prepare pTHPP-loaded NPs, 0.75, 1.5 and 3 mg of pTHPP (i.e., corresponding to 5, 10 and 20 % theoretical drug loading, respectively) were dissolved in 50 μL THF and added to the organic phase prior to emulsification. All solutions were kept and protected from light during the preparation process. After solvent evaporation, the PHA NPs were filtered through a 0.45 μm syringe-filter prior to particle characterization and evaluation of photocytotoxicity.
2.4 Particle size analysis and zeta potential measurement
The mean size and polydispersity index (PDI) of all PHA NPs were measured after appropriate dilution in water to enable measurement using a Zetasizer dynamic light scattering (DLS) instrument (Nano ZS, Malvern instruments, UK), equipped with a He–Ne laser (λ = 633 nm) at a scattering angle of 173°. All size measurements were performed by collecting data over 15 runs at 10 s. The zeta potential (averaged over 100 sub-runs) was determined for PHA NPs after dilution in 0.1 M sodium acetate. All reported size and zeta potential values are averages from triplicate measurements.
2.5 Determination of drug loading and entrapment efficiency
Filtered drug-loaded NPs were collected from a 50 mL solution by centrifugation. The obtained pellet was washed twice with 0.25 % (w/v) PVA solution, followed by one wash with water. Freeze-drying of NPs was carried out without addition of any lyoprotectant. The freeze-dried samples were weighed and stored at 4 °C (in the dark) before performing drug loading analysis. To calculate % drug loading, a known mass of freeze-dried pTHPP-loaded PHA NPs was added to 1 mL THF and continuously stirred until completely dissolved. The concentration of pTHPP in the solution was measured at 422 nm using a spectrophotometer (Helios alpha, England), and compared to a standard curve of pTHPP dissolved in THF. The % drug loading and % entrapment efficiency in PHA NPs was determined according to following equations.
All extractions and measurements were performed in triplicate.
2.6 Transmission electron microscopy (TEM)
The morphology of pTHPP-loaded PHA NPs was observed by transmission electron microscopy imaging. For sample preparation, a few microliters of NP solution was added onto a 300-mesh carbon coated copper grid and allowed to air-dry. Next, the samples were negatively stained using 1.5 % phosphotungstic acid (PTA). The excess solution was blotted off using a filter paper. After additional air-drying, the NPs were visualized using a JEOL JEM-200CX transmission electron microscope (JEOL Ltd., Tokyo, Japan).
2.7 Drug release studies
The pTHPP release profile from PHA NPs was evaluated at physiological condition using a phosphate buffer saline (PBS) release medium containing 50 mg/mL bovine serum albumin (BSA) to enhance drug solubility. For comparison, an equivalent concentration of free pTHPP (50 μg/mL) was dispersed directly in 1 mL of the release medium. All solutions were protected from light and kept in a water bath under continuous shaking at 100 rpm and 37 °C. In vitro drug release was evaluated at fix time intervals (0, 1, 3, 8 and 24 h). Samples were taken from the water bath, centrifuged at 12,500 rpm for 15 min, where the amount of drug released into supernatant was measured spectrophotometrically at 422 nm. The amount of pTHPP released from each sample was calculated using a standard curve of pTHPP in PBS with dissolved BSA. All drug release experiments were carried out in triplicate. The data of in vitro release obtained were plotted as percentage cumulative of drug released over time.
2.8 Analysis of in vitro photocytotoxicity
The photocytotoxic effect of pTHPP-loaded PHA NPs was evaluated using human colon adenocarcinoma HT-29 cells. Typically, a monolayer culture of human colon adenocarcinoma cell line HT-29 was grown in DMEM supplemented with 10 % (v/v) FBS, 100 U/mL penicillin, 100 μg/mL streptomycin and 250 ng/mL amphotericin B under humidified atmosphere with 5 % (v/v) CO2 at 37 °C. Stock solutions of free pTHPP were prepared by first dissolving pTHPP in DMSO, followed by serial dilution using cell culture medium. The final DMSO concentration in all treatments of free pTHPP was kept at 0.2 %, which is considered to be non-toxic level to cells [45]. Briefly, 100 μL aliquot of 1 × 104 cells of HT-29 cells in culture medium was seeded into each well of a 96-well clear bottom black plate. The plate was incubated under 5 % CO2 atmosphere for 24 h at 37 °C to allow the cells to attach to the bottom of wells. Then, 100 μL of free pTHPP and pTHPP-loaded NPs were subsequently added to the medium in order to obtain final drug concentrations of 2, 4, and 8 μg/mL, respectively, and further incubated at 37 °C for different incubation time ranging from 1 to 24 h prior to light exposure. After the allotted incubation time, the culture plate was washed once with serum-free DMEM and 100 μL of fresh culture medium without the test samples was added into each well. Subsequently, the cells were exposed to red light for 12 min 45 s using a LED lamp (Acnelamp, Dima-Tech, Inc., USA) with a peak wavelength 653 nm. The specific light dose corresponds to 6 J/cm2. After exposure, the cells were incubated for an additional 24 h. Lastly, the viability of treated HT-29 cells was determined by using a standard cell viability assay (MTT assay) in order to measure the number of viable cells following a previously described protocol elsewhere [46]. In brief, the culture medium in each sample well was replaced with 100 μL of fresh culture medium containing 0.5 mg/mL MTT and incubated for 2 h. Subsequently, the MTT solution was replaced with 100 μL DMSO, followed by spectrophotometric measurement at 550 nm with a background subtraction at 650 nm using a microplate reader. The % cell viability from each condition was calculated and used to assess the cell activities comparing with the non-treated cells. An identical set of samples not exposed to light were also used as dark controls.
2.9 Statistical analysis
The data were subjected to a statistical analysis using SPSS 17.0 software package for windows. Statistical differences were evaluated using a one-way ANOVA with student’s t test. Significant differences were considered at a level of P ≤ 0.05.
3 Results and discussion
3.1 Preparation of PHA nanoparticles
To facilitate intravenous administration of polymeric NPs for drug delivery in cancers, the NP diameter should be around 200 nm or less. This size range has been suggested as an ideal NP size to facilitate prolonged circulation with reduced mononuclear phagocyte system clearance and accumulation in spleen or other organs [47]. The size of NPs can be manipulated by modulating polymer composition, polymer molecular weight, choice of particle stabilizer, solvent diffusion time, and also the presence of a surface tension reducer such as ethanol in the organic phase [48, 49]. Here, we prepared our pTHPP-loaded PHA NPs using an emulsification-diffusion method and tested two different PVA stabilizers. This process led to NPs having a hydrophilic PVA shell with the pTHPP molecules entrapped inside the hydrophobic PHA core.
3.2 Characterization of non-drug loaded PHA nanoparticles
In order to evaluate the influence of HV monomer composition and influence of different Mw PVA stabilizers (i.e., low vs. high Mw) on the size of our NPs, we prepared formulations of each PHAs and PVA, respectively.
The particle sizes, as measured by DLS, are summarized in Table 1, where all particles were in the range of 157.1–198.5 nm with PDI lower than 0.2. When prepared with low Mw PVA, P(HB-HV) copolymer NPs (i.e., P(HB-12HV) and P(HB-50HV)) had a larger mean size compared to the NPs made from homopolymer PHB with statistically significant at P ≤ 0.05. On the contrary, this phenomenon was not observed when using high Mw PVA. The slightly larger size of the P(HB-HV) NPs seen in the low Mw PVA system might be due to the bulkier side chain of the HV monomer that would pack less densely than the PHB homopolymer. The phenomenon is not seen for high Mw PVA, and could be due to different packing arrangement of the stabilizer, which might hide the subtle effect of core packing. In general, the high Mw PVA system yielded larger diameter NPs for each individual PHA system, compared to when prepared with the low Mw PVA. These findings are in accordance with a study performed by Shaffie et al. [50] showing that higher Mw of PVA stabilizer could lead to an increase of polymer particle size.
3.3 Characterization of pTHPP-loaded PHA nanoparticles
For NPs prepared using low Mw PVA stabilizer, adding more pTHPP to the formulation in general increased the particle size (P ≤ 0.05). The average diameter was about 169.0–211.2 nm. Table 1 shows trends that the particle size could be influenced by factors such as %HV monomer in the polymer chain, and the amount of pTHPP encapsulated. The pTHPP:PHB:low sample was the only one significantly different (P ≤ 0.05) compared to pTHPP:P(HB-12HV):low and pTHPP:P(HB-50HV):low even at relatively high pTHPP loading (i.e., 20 % theoretical drug loading). For the pTHPP-loaded PHA NPs prepared using high Mw PVA as a stabilizer, all sizes fall within a range of 185.8–208.3 nm. There was no statistical difference between the particle size at 5 % theoretical drug loading, while at 10 and 20 % theoretical drug loading, the observed smaller size of pTHPP:PHB:high was significantly different from those of the HV copolymer systems (P ≤ 0.05). Figure 2 shows a representative TEM micrograph of one of NP preparations.

TEM image of pTHPP:P(HB-50HV):low
All formulations of pTHPP-loaded NPs exhibited relatively low zeta-potential values ranging from −0.7 to −2.1 mV. No correlation between zeta potential values and monomer composition of PHAs were observed. A negative zeta potential value is expected due to the presence of carboxylic end groups from PHA polymers [51]. The relatively low values observed could be due to a shielding effect of surface charge by the PVA layer [52]. Another possibility might be due to the adsorption of hydrophobic drug onto the surface of the NPs, which could also shield the carboxylic end groups [53].
3.4 Characterization of drug loading and encapsulation efficiency
The drug loading and entrapment efficiency is an important aspect of NP drug formulation. To evaluate the amount of drug that could be incorporated into the NP system, we varied the theoretical loading amount of pTHPP in the formulation from 5 to 20 % (w/w).
When low Mw PVA was used as stabilizer, the % drug loading increased with increasing % theoretical loading (i.e., 5 to 20 %) for each formulation (i.e., 1.26 to 6.83, 1.53 to 9.33 and 1.11 to 8.67 % for pTHPP:PHB:low, pTHPP:P(HB-12HV):low and pTHPP:P(HB-50HV):low, respectively) with an corresponding increase in % entrapment efficiency (Table 2). The highest % drug loading and % entrapment efficiency were observed when using 20 % theoretical drug loading formulation. Likewise, for high Mw PVA, the % drug loading of pTHPP:PHB:high, pTHPP:P(HB-12HV):high and pTHPP:P(HB-50HV):high were found to be increased from 0.91 to 4.80, 1.49 to 6.78 and 1.22 to 5.24 %, respectively. The drug loading increased as more drug were added to the formulations, albeit the incorporation of drug was less when compared to the low Mw PVA system. However, the % entrapment efficiency for pTHPP:P(HB-12HV):high and pTHPP:P(HB-50HV):high were maximized (i.e., 41.04 and 41.71, respectively) when 10 % theoretical drug loading was used (Table 2). Overall, for both PVA systems, P(HB-HV) copolymers tend to produce sub-200 nm NPs with higher % drug loading and % entrapment efficiency compared to the PHB homopolymer. This might be due to the fact that P(HB-HV) generally have more amorphous (i.e., less crystalline) structure than PHB because of the presence of ethyl side chain (i.e., HV portion), thus allowing the polymer chains to be loosely packed and able to entrap more hydrophobic drug molecules [54, 55]. In this study, we observed similar behavior of both the P(HB-12HV) and the P(HB-50HV) copolymer systems.
Besides the effect of HV, when comparing between the effect of low Mw and high Mw PVA systems at 20 % theoretical drug loading, % entrapment efficiency of NPs made with the low Mw PVA was found to be higher for each PHA type (P ≤ 0.05), suggesting that using the low Mw PVA as NP stabilizer would facilitate the entrapment of pTHPP drugs within the PHA NPs better than the high Mw PVA. Investigation of this behavior was beyond the scope of this study and additional experiments are needed to further elucidate the effect of PVA stabilizer on the NP formation and drug loading. Nonetheless, there was an earlier study reporting the effect of PVA Mw on NP loading, in which they found that the optimal PLGA NPs loaded with plasmid DNA had higher % DNA loading when the lower Mw PVA was used [56].
3.5 Drug release characteristics
To characterize the drug release and PDT characteristics of PHA NPs, we choose to use the NP formulations with 10 % (w/w) theoretical drug loading for both PVA systems as they had similar % drug loading. Measurements to obtain the release profile of pTHPP-loaded NPs were performed over 24 h in PBS buffer pH 7.4 at 37 °C. To improve the solubility of the released pTHPP, BSA was added to the releasing medium since it is an abundant plasma protein that binds non-specifically to many hydrophobic molecules [57]. The release profiles of pTHPP from various PHA NP formulations showed similar patterns (Fig. 3). During the first 3 h, a relatively fast release of pTHPP was found with a cumulative release around 60 %, followed by a slow release phase until reaching nearly 100 % after 24 h. The early release data was fitted to the Korsmeyer–Peppas model using non-linear regression fitting (Graph Prism Plus 4.0);
where F is the amount of drug released in time t, K is release rate constant, n is the release exponent which is characteristic of the release mechanism and t is the time in hours [58–60]. The release exponents and goodness of fit (R2) values is shown in Table 3. Values of n, 0.43 < n < 0.85, indicate anomalous transport (relaxational and diffusional drug transport). In our case, all drug release profiles show values of n < 0.43, indicating a diffusion-based drug release. No influence of either polymer compositions or types of PVA stabilizer on the release profile of pTHPP-loaded NPs could be observed.

Drug release profiles of pTHPP from PHA nanoparticles in PBS (pH 7.4 containing BSA) at 37 °C. Values represent mean ± SD of three batches (Some data points had a small standard deviation and theses error bars were not included in the graph)
3.6 Photocytotoxicity of free pTHPP
Firstly, we evaluated free pTHPP in solution as a photocytotoxic agent for in vitro killing of cancer cells. The HT-29 cells were treated with 8 μg/mL free pTHPP at different incubation times (1, 3, 6, 12 and 24 h), then pTHPP was removed, new media added, with subsequent irradiation of the cells with 6 J/cm2 of red light. The dark cytotoxicity (no light) and photocytotoxicity (light irradiation) were evaluated by MTT test at 24 h post irradiation. It should be noted that the light irradiation alone did not cause a significant reduction in percentage of cell viability under all experimental conditions used (Data not shown).
As shown in Fig. 4, pTHPP with light irradiation could lead to about 40 % cell death after incubation for 1 h, whereas more than 90 % cell death was observed after 6 h incubation. These results indicate a significant accumulation of pTHPP within the cells after 1–3 h. In contrast, <10 % cell death was observed in the dark controls up to 6 h. However, after 24 h incubation, the dark cytotoxicity could cause cell death up to 40 %, a value similar to a previous report [61].

Percentage of cell viability of HT-29 cells treated with free pTHPP at 8 μg/mL for 1–24 h, with or without 6 J/cm2 light irradiation. MTT assay was performed at 24 h after light irradiation. Values represent mean ± SD of three independent sets of experiments
3.7 Photocytotoxicity of pTHPP-loaded PHA nanoparticles
According to the drug release profiles, the pTHPP released from six different NP formulations did not show any significant differences. So, considering the potential for higher drug loading in the P(HB-HV) copolymer systems, we decided to focus on P(HB-HV) NP formulations prepared from both PVA systems in order to compare between the commercially available copolymer; P(HB-12HV), and one that we prepared “in house”; P(HB-50HV), for further evaluation of in vitro photocytotoxicity.
The in vitro photocytotoxicity of pTHPP-loaded NPs against HT-29 colon cancer cells was evaluated by treating the cells with different formulations of drug-loaded NPs at increasing pTHPP concentration (equivalent to 2, 4 and 8 μg/mL) and incubation times (1, 3, 6, 12 and 24 h) before light irradiation.
After irradiation at a dose of 6 J/cm2, the increase in % cell death induced by pTHPP-loaded NPs was found to be time- and concentration-dependent, and a similar pattern of cell death was observed in all the formulations tested (Fig. 5a–d). The photocytotoxicity of all formulations gradually increased as the incubation time increased. These results correlate well with the release profile of pTHPP (Fig. 3). The highest photocytotoxic effect of all the test formulations, resulting in 93–94 % cell death, was observed at 8 μg/mL equivalent pTHPP concentration and after 24 h incubation before light irradiation. The delayed photocytotoxicity of pTHPP-loaded NPs compared with free pTHPP could be explained by the difference between passive diffusion of free pTHPP aided by adding DMSO, and the requirement for cellular uptake process of NPs that requires time to allow degradation of polymers for releasing the loaded pTHPP into cytoplasm. Previous studies also demonstrated the delayed cytotoxicity of drug-loaded NPs, particularly made with PHA polymers, compared with the free drug. For instance, death of HeLa cells treated with free doxorubicin were observed at 24 h, while the comparable cytotoxicity of doxorubicin encapsulated in the P(HB-6.5HV) particles was found at 72 h [62]. Similarly, a 2 day delay of cytotoxic effect of paclitaxel-containing P(HB-12HV) NPs (1 μM paclitaxel) was observed in Ishikawa cells to free drug [36].

Percentage of cell viability of HT-29 cells treated with various formulations of pTHPP-loaded PHA nanoparticles; a pTHPP:P(HB-12HV):low, b pTHPP:P(HB-50HV):low, c pTHPP:P(HB-12HV):high and d pTHPP:P(HB-50HV):high, at equivalent drug dose of 2, 4 and 8 μg/mL. Cells were incubated the pTHPP-loaded nanoparticles for 1–24 h before light irradiation. MTT assay was performed at 24 h after light irradiation at a dose of 6 J/cm2. Values represent mean ± SD of three independent sets of experiments
Interestingly, there might be a difference in terms of the delayed cytotoxic effect in PHA drug delivery system when compared to PLGA polymer system. While our PHA NPs show a delayed effect, the previously study reported that pTHPP-loaded PLGA NPs required relatively short incubation time (i.e., 30–60 min) to induce satisfactory photodynamic damage on EMT-6 tumor cells [63]. One possible explanation can be the slower in vitro degradation of our PHAs over the PLGA polymer systems that might be due to the molecular weight differences [64, 65]. The molecular weight of the PHAs used here (i.e., 105–106 g/mol) is higher than other aliphatic polyesters, such as PLGA or PLA (i.e., 103–104 g/mol), which are often used to prepare drug-loaded NPs with faster release rates [66, 67]. Still, the delayed PDT effect of PHA NP system might offer an advantage for using the NPs in vivo, since they might prevent too early release and subsequent degradation/clearance of the anti-cancer drug before reaching the tumor sites, thus preventing the drug from interacting with normal tissues, and reducing systemic side effects.
4 Conclusion
Here, we successfully formulated and characterized different PHA polymeric NPs for delivery of a hydrophobic PS, pTHPP for PDT. A modified emulsification-solvent diffusion method was employed to prepare pTHPP-loaded PHA NPs in the range of 169.0–211.2 nm with different % drug loading. An effect of HV monomer content on the mean particle size and drug loading could be observed, however only when formulated with low Mw PVA. The release profiles of PHA NPs were evaluated using the Korsmeyer–Peppas model and found to follow diffusion-based drug release. The pTHPP-loaded PHA NPs showed high photocytotoxicity against HT-29 cancer cells, especially after longer incubation time (i.e., 24 h). Our results show PHA NPs as promising vehicles for delivery of hydrophobic PS drugs with potential application in PDT.
References
Schuitmaker JJ, Baas P, Leengoed HLLM, Meulen FW, Star WM, Zandwijk N. Photodynamic therapy: a promising new modality for the treatment of cancer. J Photochem Photobiol B, Biol. 1996;34:3–12.
Hopper C. Photodynamic therapy: a clinical reality in the treatment of cancer. Lancet Oncol. 2000;1:212–9.
Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497–508.
Hopper C, Kubler A, Lewis H, Tan IB, Putnam G. mTHPC-mediated photodynamic therapy for early oral squamous cell carcinoma. Int J Cancer. 2004;111:138–46.
Molinari A, et al. m-THPC-mediated photodynamic therapy of malignant gliomas: assessment of a new transfection strategy. Int J Cancer. 2007;121:1149–55.
Zimmermann A, Ritsch-Marte M, Kostron H. mTHPC-mediated photodynamic diagnosis of malignant brain tumors. Photochem Photobiol. 2001;74:611–6.
O’Connor AE, Gallagher WM, Byrne AT. Porphyrin and nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy. Photochem Photobiol. 2009;85:1053–74.
Henderson BW, Dougherty TJ. How does photodynamic therapy work? Photochem Photobiol. 1992;55:145–57.
Berg K, et al. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J Microsc. 2005;218:133–47.
Kostron H, Bellnier DA, Lin CW, Swartz MR, Martuza RL. Distribution, retention, and phototoxicity of hematoporphyrin derivative in a rat glioma. J Neurosurg. 1986;64:768–74.
Taquet JP, Frochot C, Manneville V, Barberi-Heyob M. Phthalocyanines covalently bound to biomolecules for a targeted photodynamic therapy. Curr Med Chem. 2007;14:1673–87.
Bechet D, Couleaud P, Frochot C, Viriot ML, Guillemin F, Barberi-Heyob M. Nanoparticles as vehicles for delivery of photodynamic therapy agents. Trends Biotechnol. 2008;26:612–21.
Pavani C, Uchoa AF, Oliveira CS, Iamamoto Y, Baptista MS. Effect of zinc insertion and hydrophobicity on the membrane interactions and PDT activity of porphyrin photosensitizers. Photochem Photobiol Sci. 2009;8:233–40.
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47.
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92.
Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–6.
Konan-Kouakou YN, Boch R, Gurny R, Allemann E. In vitro and in vivo activities of verteporfin-loaded nanoparticles. J Control Release. 2005;103:83–91.
Zeisser-Labouebe M, Lange N, Gurny R, Delie F. Hypericin-loaded nanoparticles for the photodynamic treatment of ovarian cancer. Int J Pharm. 2006;326:174–81.
Hu Z, Pan Y, Wang J, Chen J, Li J, Ren L. Meso-tetra (carboxyphenyl) porphyrin (TCPP) nanoparticles were internalized by SW480 cells by a clathrin-mediated endocytosis pathway to induce high photocytotoxicity. Biomed Pharmacother. 2009;63:155–64.
Ungun B, Prud’homme RK, Budijon SJ, Shan J, Lim SF, Ju Y, Austin R. Nanofabricated upconversion nanoparticles for photodynamic therapy. Opt Express. 2009;17:80–6.
Anderson AJ, Dawes EA. Occurrence, metabolism, metabolic role, and industrial use of bacterial polyhydroxyalkanoates. Microbiol Rev. 1990;54:450–72.
Lee EY, Kang SH, Choi CY. Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by newly isolated Agrobacterium sp. SH-1 and GW-014 from structurally unrelated single carbon substrates. J Ferment Bioeng. 1995;79:328–34.
Verlinden RAJ, Hill DJ, Kenward MA, Williams CD, Radecka I. Bacterial synthesis of biodegradable polyhydroxyalkanoates. J Appl Microbiol. 2007;102:1437–49.
Chanprateep S. Current trends in biodegradable polyhydroxyalkanoates. J Biosci Bioeng. 2010;110:621–32.
Duran N, Alvarenga MA, Da Silva EC, Melo PS, Marcato PD. Archives of microencapsulation of antibiotic rifampicin in poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Arch Pharm Res. 2008;31:1509–16.
Vilos C, Constandil L, Herrera N, Solar P, Escobar-Fica J, Velasquez L. Ceftiofur-loaded PHBV microparticles: a potential formulation for a long-acting antibiotic to treat animal infections. Electron J Biotechnol. 2012;15:1–13.
Grillo R, et al. Controlled release system for ametryn using polymer microspheres: preparation, characterization and release kinetics in water. J Hazard Mater. 2011;186:1645–51.
Lee J, Jung SG, Park CS, Kim HY, Batt CA, Kim YR. Tumor-specific hybrid polyhydroxybutyrate nanoparticle: surface modification of nanoparticle by enzymatically synthesized functional block copolymer. Bioorg Med Chem Lett. 2011;21:2941–4.
Masood F, Chen P, Yasin T, Fatima N, Hasan F, Hameed A. Encapsulation of Ellipticine in poly-(3-hydroxybutyrate-co-3-hydroxyvalerate) based nanoparticles and its in vitro application. Mater Sci Eng C Mater. 2013;33:1054–60.
Vilos C, et al. Paclitaxel-PHBV nanoparticles and their toxicity to endometrial and primary ovarian cancer cells. Biomaterials. 2013;34:4098–108.
Bonnett R, McGarvey DJ, Harriman A, Land EJ, Truscott TG, Winfield UJ. Photophysical properties of meso-tetraphenylporphyrin and some meso-tetra (hydroxyphenyl) porphyrins. Photochem Photobiol. 1988;48:271–6.
Nawalany K, et al. Novel nanostructural photosensitizers for photodynamic therapy: in vitro studies. Int J Pharm. 2012;430:129–40.
Berenbaum MC, Akande SL, Bonnett R, Kaur H, Ioannou S, White RD, Winfield UJ. meso-Tetra (hydroxyphenyl) porphyrins, a new class of potent tumour photosensitisers with favourable selectivity. Br J Cancer. 1986;54:717–25.
Bonnett R, White RD, Winfield UJ, Berenbaum MC. Hydroporphyrins of the meso-tetra (hydroxyphenyl) porphyrin series as tumour photosensitizers. Biochem J. 1989;261:277–80.
Gomer CJ. Preclinical examination of first and second generation photosensitizers used in photodynamic therapy. Photochem Photobiol. 1991;54:1093–107.
Kepczynski M, Nawalany K, Kumorek M, Kobierska A, Jachimska B, Nowakowska M. Which physical and structural factors of liposome carriers control their drug-loading efficiency? Chem Phys Lipids. 2008;155:7–15.
Shang L, Yim SC, Park HG, Chang HN. Sequential feeding of glucose and valerate in a fed-batch culture of Ralstonia eutropha for production of poly(hydroxybutyrate-co-hydroxyvalerate) with high 3-hydroxyvalerate fraction. Biotechnol Prog. 2004;20:140–4.
Loo CY, Sudesh K. Biosynthesis and native granule characteristics of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in Delftia acidovorans. Int J Biol Macromol. 2007;40:466–71.
Huijberts GNM, van der Wal H, Wilkinson C, Eggink G. Gas-chromatographic analysis of poly(3-hydroxyalkanoates) in bacteria. Biotechnol Tech. 1994;8:187–92.
Jacquel N, Lo CW, Wei YH, Wu HS, Wang SS. Isolation and purification of bacterial poly(3-hydroxyalkanoates). Biochem Eng J. 2008;39:15–27.
de Mello VA, Ricci-Junior E. Encapsulation of naproxen in nanostructured system: structural characterization and in vitro release studies. Quim Nova. 2011;34:933–9.
Averineni RK, et al. PLGA 50:50 nanoparticles of paclitaxel: development, in vitro anti-tumor activity in BT-549 cells and in vivo evaluation. Bull Mater Sci. 2012;35:319–26.
Konan YN, Gurny R, Allemann E. Preparation and characterization of sterile and freeze-dried sub-200nm nanoparticles. Int J Pharm. 2002;233:239–52.
Konan YN, Cerny R, Favet J, Berton M, Gurny R, Allemann E. Preparation and characterization of sterile sub-200 nm meso-tetra(4-hydroxylphenyl)porphyrin-loaded nanoparticles for photodynamic therapy. Eur J Pharm Biopharm. 2003;55:115–24.
Yoshimoto AN, et al. Hedgehog pathway signaling regulates human colon carcinoma HT-29 epithelial cell line apoptosis and cytokine secretion. PLoS One. 2012;7:e45332.
Lirdprapamongkol K, Kramb JP, Suthiphongchai T, Surarit R, Srisomsap C, Dannhardt G, Svasti J. Vanillin suppresses metastatic potential of human cancer cells through PI3K inhibition and decreases angiogenesis in vivo. J Agric Food Chem. 2009;57:3055–63.
Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharm Rev. 2001;53:283–318.
Quintanar-Guerrero D, Fessi H, Allemann E, Doelker E. Influence of stabilizing agents and preparative variables on the formation of poly(d,l-lactic acid) nanoparticles by an emulsification-diffusion technique. Int J Pharm. 1996;143:133–41.
Poletto FS, Fiel LA, Donida B, Re MI, Guterres SS, Pohlmann AR. Controlling the size of poly(hydroxybutyrate-co-hydroxyvalerate) nanoparticles prepared by emulsification-diffusion technique using ethanol as surface agent. Colloids Surf Physicochem Eng Asp. 2008;324:105–12.
Shaffie KA, Moustafa AB, Saleh NH, Nasr HE. Effect of polyvinyl alcohol of different molecular weights as protective colloids on the kinetics of the emulsion polymerization of vinyl acetate. J Am Sci. 2010;6:1202–12.
Manangana T, Shawaphuna S. Quantitative extraction and determination of polyhydroxyalkanoate accumulated in Alcaligenes latus dry cells. Sci Asia. 2010;36:199–203.
Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82:105–14.
Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, Puglisi G. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm. 2006;325:172–9.
Choi GG, Kim HW, Rhee YH. Enzymatic and non-enzymatic degradation of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) copolyesters produced by Alcaligenes sp. MT-16. J Microbiol. 2004;42:346–52.
Asrar J, Valentin HE, Berger PA, Tran M, Padgette SR, Garbow JR. Biosynthesis and properties of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) polymers. Biomacromolecules. 2002;3:1006–12.
Prabha S, Labhasetwar V. Critical determinants in PLGA/PLA nanoparticle-mediated gene expression. Pharm Res. 2004;21:354–64.
Zunszain PA, Ghuman J, Komatsu T, Tsuchida E, Curry S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct Biol. 2003;7:3–6.
Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5:23–36.
Siepmann J, Peppas NA. Higuchi equation: derivation, applications, use and misuse. Int J Pharm. 2011;418:6–12.
Bhosale UV, Devi K, Choudhary S. Development and in vitro-in vivo evaluation of oral drug delivery system of acyclovir loaded PLGA nanoparticles. Int J Drug Deliv. 2013;5:331–43.
Nawalany K, et al. Comparison of photodynamic efficacy of tetraarylporphyrin pegylated or encapsulated in liposomes: in vitro studies. J Photochem Photobiol B Biol. 2009;97:8–17.
Murueva AV, Shishatskaya EI, Kuzmina AM, Volova TG, Sinskey AJ. Microparticles prepared from biodegradable polyhydroxyalkanoates as matrix for encapsulation of cytostatic drug. J Mater Sci Mater Med. 2013;24:1905–15.
Konan YN, Berton M, Gurny R, Allemann E. Enhanced photodynamic activity of meso-tetra(4-hydroxyphenyl)porphyrin by incorporation into sub-200nm nanoparticles. Eur J Pharm Sci. 2003;18:241–9.
Pouton CW, Majid MIA, Natarianni LJ. Degradation of polyhydroxbutyrate and related copolymers. Proc Int Symp Control Release Bioact Mater. 1988;15:181–3.
Holland SJ, Yasin M, Tighe B. Polymers for biodegradable medical devices VII. Hydroxy butyrate-hydroxyvalerate copolymers: degradation of copolymers and their blends with polysaccharides under in vitro physiological conditions. Biomaterials. 1990;11:206–15.
Vargas A, et al. Improved photodynamic activity of porphyrin loaded into nanoparticles: an in vivo evaluation using chick embryos. Int J Pharm. 2004;286:131–45.
Lee DJ, et al. Multifunctional poly (lactide-co-glycolide) nanoparticles for luminescence/magnetic resonance imaging and photodynamic therapy. Int J Pharm. 2012;434:257–63.
Acknowledgments
This work was supported by Thailand Research Fund (TRF) - MRG5380110 in cooperation with Office of the Higher Education Commission, Science Achievement Scholarship of Thailand (SAST) and Faculty of Science, Mahidol University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pramual, S., Assavanig, A., Bergkvist, M. et al. Development and characterization of bio-derived polyhydroxyalkanoate nanoparticles as a delivery system for hydrophobic photodynamic therapy agents. J Mater Sci: Mater Med 27, 40 (2016). https://doi.org/10.1007/s10856-015-5655-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-015-5655-4