
Abstract
Near-infrared downconversion phenomenon has been demonstrated in Lu2O3: Tm3+/Yb3+ phosphor upon direct excitation of Tm3+:1G4 level at 463 nm. The efficient energy transfer from Tm3+: 1G4 → Yb: 2F5/2 has been elucidated by the excitation spectra, the visible and NIR spectra as well as the decay curves of Tm: 1G4 state. The mechanism of downconversion in Lu2O3:Tm3+/Yb3+ has been discussed in detail. According to analysis of the dependence of the initial transfer rate over Yb3+ ion concentration, it could be included that energy transfer (ET) from Tm3+ to Yb3+ is a single-step ET process instead of a cooperative one. By varying the Yb3+ concentrations, we obtain the Lu2O3: 0.2%Tm3+/30%Yb3+ sample with theoretical quantum efficiency as high as 148.2%. Because the excited state of Yb3+ just above the band edge of crystalline silicon, it suggested that Lu2O3: Tm3+/Yb3+ sample will be beneficial to improve the conversion efficiency of c-Si solar cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The long-run demand to exploit renewable, sustainable and clean energy sources continues to stimulate new approaches to manufacture of efficient and low-cost photovoltaic devices [1, 2]. At present, photovoltaic devices fabricated from silicon wafers dominating the marketplace possess a quite low efficiency of only 18% [3]. This is attributed to the large spectrum mismatch between solar radiation spectrum and the response spectrum of solar cells [4]. To circumvent this difficulty, either improvements in structure and composition of solar cells or modification of solar radiation spectrum has been suggested [5–8]. The spectral distribution of sunlight spectrum covers from 300 to 2500 nm at Air Mass 1.5 global (AM 1.5G), but only a small fraction around 800–1100 nm could be effectively utilized by silicon solar cells with 1.05 eV bandgap [9–11]. This is due to the higher reflection and absorption by the antireflection coatings optimized for longer wavelengths of solar cells [12]. To take advantage of high-energy photons with short wavelength, downconversion (DC) is considered as the most promising candidate for converting the incident high-energy photons into efficient near-infrared (NIR) photons to realize spectrum modification so far [13–15]. DC has been realized in RE3+/Yb3+ couples, such as Er3+/Yb3+, Pr3+/Yb3+, Ce3+/Yb3+, Ho3+/Yb3+ and Tm3+/Yb3+ [16–20]. Yb3+ is suitable as an acceptor and emitter for the reason that the Yb3+ ion has an only excited state approximately 10,000 cm−1 just above the band edge of crystalline silicon [21]. Herein, we mainly focused on Tm3+/Yb3+ couples which could convert blue photons around 463 nm into NIR photons around 1000 nm by efficient energy transfer from Tm3+ to Yb3+. The wavelength at 1000 nm could be effectively utilized by solar cells.
The cubic rare earth sesquioxide Lu2O3 is selected as a host because of its chemically and environmentally stable properties [22]. On the contrary, the use of fluoride materials is restricted by their toxicity owing to the fluorine-containing species unlike the oxides [23]. It could also achieve a high rare-earth doping level in Lu2O3 [24]. Besides, Lu2O3 possesses a larger thermal conductivity (12.5 W/mk) [25] than YAG(10.7 W/mk) [26] making it more desirable for device manufacture. Recently, it has been reported that Lu2O3 is an excellent host for phosphors with efficient upconversion luminescence properties between Tm3+/Yb3+, Er3+/Yb3+, Ho3+/Yb3+ couples [22, 27]. Moreover, strong downconversion luminescence in Tb3+ and Yb3+ codoped Lu2O3 has also been investigated [28]. However, little researches refer to downconversion between Tm3+/Yb3+ combinations in Lu2O3 host and the transfer mechanism has not been demonstrated yet.
In this study, we report the efficient NIR DC in Lu2O3: Tm3+/Yb3+ phosphor and analyze the energy transfer(ET) mechanism in Tm3+/Yb3+ couples. We systematically demonstrate the DC process between Tm3+/Yb3+ couples is a single-step ET process instead of a cooperative ET process assumed in other materials [29]. Visible and NIR emission spectra, decay time and energy transfer efficiency have been investigated in detail with varying Yb3+ concentration. Besides, the theoretical quantum efficiency as high as 148.2% has been obtained, well over the efficiency limit (29%) estimated by Shockley and Queisser [30]. Therefore, results show that Lu2O3 doped with Tm3+/Yb3+ offers the opportunity for application in solar cells.
2 Experimental
2.1 Sample preparation
The series of samples investigated in this work with the general formula Lu2−0.2%−x%O3: 0.2%Tm3+, x%Yb3+ (x = 0, 1, 5, 10, 20, 30) were prepared by a solid-state reaction. The oxides Lu2O3(4 N), Yb2O3(4 N), and Tm2O3(4 N) were employed as the raw materials, which were mixed homogeneously by an agate mortar for 30 min, placed in a crucible with a lid, then sintered at 1500 °C for 4 h.
2.2 Measurements and characterization
Powder X-ray diffraction (XRD) data was collected using Cu-Kα radiation (λ = 1.54056 Å) on a Bruker D8 advance diffractometer equipped with a linear position-sensitive detector (PSD-50m, M. Braun), operating at 40 kV and 40 mA with a step size of 0.01° (2θ) in the range of 10°–80°. The steady state excitation and emission spectra under direct excitation were measured using an FLS920 spectrometer (Edinburgh Instruments, U.K.). In energy level lifetime measurements, an OPO was used as an excitation source, and the signals were detected using a Tektronix digital oscilloscope (TDS 3052). The lifetimes were calculated by integrating the area under corresponding lifetime curves with the normalized initial intensity.
3 Results and discussion
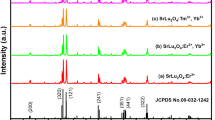
The structures of Lu2O3 samples with nominal compositions 0.2% Tm3+/ x% Yb3+ (x = 0, 1, 5, 10, 20, 30) were examined by typical powder X-ray diffraction pattern, as shown in Fig. 1. The positions and relative intensity of diffraction peaks for the powder samples can be well indexed to the standard cards of JPCD cards 12–0728. According to Fig. 1, there is no other phase in all XRD patterns. It demonstrates that Tm3+ and Yb3+ substitutions for Lu3+ sites have no effect on the phase structure.

The standard XRD data of Lu2O3 and the XRD patterns of the samples doped with different concentrations of Yb3+
In the UV region, high energy photons might be absorbed by either the Lu2O3 host or the rare-earth ions through charge transfer and f–d transitions. The possibility of the occurrence of DC is then clarified experimentally. To demonstrate the existence of DC from Tm: 1G4 to Yb: 2F5/2 in Lu2O3: Tm3+/Yb3+ host, excitation spectra (PLE) was measured to be a direct evidence of DC as shown in Fig. 2. The intense excitation band centered at 463 nm is ascribed to 1G4 → 3H6 transition when monitoring the Tm3+: 1G4 → 3F4, 1G4 → 3H5, 1G4 → 3H4 emission, respectively. Similarly, when monitoring the Yb3+: 2F5/2 → 2F7/2 infrared emission at 980 nm, the excitation band could also be observed. This indicates the presence of direct ET from Tm3+ to Yb3+. The weak excitation intensity for monitoring 980 nm emission of Yb3+ and 1200 nm emission of Tm3+: 1G4 → 3H4 is because of lack of signal at infrared wavelength region for InGaAs detector used in present work.

Excitation spectra of Tm3+: 1G4 → 3F4, 1G4 → 3HS, 1G4 → 3H4 emission in Lu2O3: 0.2%Tm3+/5%Yb3+ sample
As another direct evidence of Tm3+ → Yb3+ ET, the emission spectra covering from visible and NIR range of Lu2O3 samples with varying Yb3+ concentrations are depicted in Fig. 3. The emission peaked at 654 nm is originated from Tm3+: 1G4 → 3F4 transition. The group of emission lines at around 800 nm is corresponding to Tm3+: 1G4→3H5 transition. In the NIR region (900–1300 nm), the strong emission band peaked at 1024 nm is observed for the Tm3+ and Yb3+ codoped samples, which is responsible for Yb3+: 2F5/2 → 2F7/2 transition under 463 nm Tm3+ excitation. The appearance of Yb3+ emission upon Tm3+: 1G4 excitation demonstrates the effective energy transfer from Tm3+ to Yb3+. The emission band in the range of 1100–1300 nm is assigned to Tm3+: 1G4 → 3H4 transition.

Comparison of visible and NIR emission spectra of Lu2O3 samples with different Yb3+ doping levels and fixed 0.2%Tm3+ concentration under 463 nm excitation
As depicted in Fig. 3, it can be concluded that the intensities of 1G4 → 3F4, 1G4 → 3H5, 1G4 → 3H4 transition of Tm3+ have shown a markedly fast decrease with Yb3+ concentration increasing. Meanwhile, the NIR emission of Yb3+ increases rapidly when raising Yb3+ doping levels. This is attributed to the energy transfer process described as Tm3+: 1G4 → Yb3+: 2F5/2 to convert blue photons into NIR photons. It is noticed that when the concentration of Yb3+ is over 5%, the intensity of Yb3+ emission decreases gradually, which is due to concentration quenching between Yb3+ ions. Therefore, 5 mol% denotes the optimal Yb3+ doped concentration in this series samples.
The PLE and PL spectra prove the energy transfer from Tm3+ to Yb3+ does exist in Lu2O3 host. For better understanding the energy transfer mechanism, the schematic thulium–ytterbium energy level diagram under 463 nm excitation with involved ET processes is depicted in Fig. 4. Upon 463 nm excitation, the Tm3+ ion in the ground state 3H4 is populated to the upper 1G4 level. The Tm3+ ions in the 1G4 level may undergo two possible energy transfer routes: (1) Cooperative ET, as indicated in Fig. 4(a). As the excited energy level of Tm3+ donor ion is located at approximately twice the energy of that of Yb3+, the DC mechanism was supposed to be a second-order cooperative ET process before [31]. A Tm3+ ion in the 1G4 level transfers energy simultaneously to two nearby Yb3+ ions in the ground state and excites them to 2F5/2 level, resulting in two emitting photons of Yb3+ with a wavelength around 1000 nm. (2)Single-step energy transfer, as shown in Fig. 4(b). A Tm3+ ion at 1G4 state transfers energy to one Yb3+ nearby through a phonon-assisted ET process, leading to one NIR emitting photon of Yb3+. Meanwhile, it populates the 3H5 state of Tm3+ and results in mid-NIR emission from 3F4 state through fast nonradiative relaxation from 3H5. It is known that cooperative ET is hard to occur because of the less probability of a second order ET comparing with a first-order one. In order to determine the real energy transfer process in Lu2O3: Tm3+/Yb3+ system, the dependence of energy transfer rate upon Yb3+ ion concentration has been discussed. For the single-step energy transfer situation, the transfer rate of a Tm3+ ion at site 0 with a Yb3+ surrounding at site i can be described as.

Schematic thulium-ytterbium energy level diagram under 463 nm excitation including possible ET processes
x denotes the concentration of the acceptor Yb3+ and thus could be representative of the probability of a Yb3+ at site i. X0 is the radiative decay rate of Tm3+:1G4 state in Lu2O3. As a consequence, for all Tm3+ ions with Yb3+ neighboring at distances R0i, the transfer rate for single-step energy transfer can be written as
It leads to
In this way, the energy transfer rate of single-step energy transfer is proportional to the acceptor concentration.
For the cooperative ET situation, one Tm3+ transfers its energy simultaneously to two Yb3+ adjacent at site i and j (i < j). According to Eq. (1), the transfer rate could be obtained as
Taking account into all the pairs of Yb3+ ions, the energy transfer rate of cooperative ET is
Thus, it could be deduced that
That is to say, the energy transfer rate of cooperative ET is proportional to the square of acceptor concentration.
The energy transfer rates are obtained from the decay curves of Tm3+: 1G4 level in Lu2O3: 0.2%Tm3+/ x%Yb3+ (x = 0, 1, 5, 10, 20, 30) depicted in Fig. 5. A rapid decline in decay curves has been observed due to the existence of extra decay pathways with Yb3+ concentration increasing. Considerable energy transfer of Tm3+: 1G4 → Yb3+: 2F5/2 accelerates the depopulation of Tm3+: 1G4 state and leads to a variety of ET rates with different Yb3+ doping concentrations. All decay curves exhibit nonexponential characteristics. This result suggests that the radiative decay of Tm3+ might be accompanied by both multiphonon assisted relaxation and ET to adjacent Yb3+ ions. We could calculate the lifetime by integrating the area under the corresponding decay curves with the normalized initial intensity at different Yb3+ doping levels. The lifetime results are presented in Table 1. The decay rate can be generally expressed by W = Wr + Wnr + WET, where Wr, Wnr, and WET are the radiative decay, non-radiative decay, and the ET rates. Accordingly, the energy transfer rate WET can be defined as

Decay curves of Tm3+:1G4 state in Lu2O3: 0.2%Tm3+/x%Yb3+ (x = 0, 1, 5, 10, 20, 30) under 463 nm excitation
where \({{\tau }_{0}}\) and \(\tau\) denote the average lifetimes of Tm3+: 1G4 state in the absence and presence of energy transfer acceptors (Yb3+ ions). Consequently, we obtained the Yb3+ ion concentration dependence of the energy transfer rates and plotted in a double-logarithmic diagram, as shown in Fig. 6. When the concentration of Yb3+ ions changes from 1 to 30%, the slope is found to be around 1. It indicates that the Tm3+-Yb3+ ET system is dominated by the single-step ET process rather than a cooperative ET process according to the analysis above. The possibility is quite low for Tm3+→Yb3+ energy transfer to achieve DC with one-to-two photon emission in Lu2O3 material.

Plot (log–log) of the energy transfer rate versus the Yb3+ concentration in Lu2O3: 0.2%Tm3+/x%Yb3+ (x = 0, 1, 5, 10, 20, 30)
Based on the decay curves shown in Fig. 5, the energy transfer efficiency (η ETE ) and theoretical quantum efficiency (η TQE ) can be determined. The η ETE is defined as the ratio of Tm3+ depopulated by energy transfer to Yb3+ over the total number of excited Tm3+ ions. Hence, the η ETE could be expressed as a function of Yb3+ concentration by lifetimes
where \(\tau\) denotes the decay time of Tm3+: 1G4 level with various concentrations of Yb3+, the same as Eq. (7). For the samples doped with x mol% Yb3+ ions (x = 1, 5, 10, 20, 30), the ET from Tm3+ to Yb3+ occurs by single-step ET process. In this way, one infrared photons is emitted by Yb3+ ions per absorbed one blue photon (around 463 nm) by Tm3+ ions in theory. Correspondingly, theoretical quantum efficiency (η TQE ) is defined as the ratio for the number of emitted photons over the number of absorbed photons. The relation between the energy transfer efficiency and the theoretical quantum efficiency is linear and can be expressed as
where \({{\eta }_{Tm}}\) and \({{\eta }_{Yb}}\) correspond to the quantum efficiencies of Tm3+ and Yb3+ ions, respectively. By assuming that all excited ions decay radiatively, their values are set to 1. This means nonradiative processes such as phonon-assisted relaxation, cross-relaxation between Tm3+ and Yb3+ as well as energy back transfer from Yb3+ to Tm3+ are all eliminated. The assumption can reach the upper limit of the theoretical quantum efficiency, the Eq. (9) thus can be simplified as
The \({{\eta }_{ETE}}\) and \({{\eta }_{TQE}}\) are listed in Table 1 based on the formulas mentioned above. The estimated \({{\eta }_{ETE}}\) exhibits an obvious increase from 3.9 to 48.2% with increasing Yb3+ concentration. It is worth noticed that \({{\eta }_{TQE}}\) reaches 148.2% for the Lu2O3: 0.2%Tm3+/30% Yb3+ sample.
4 Conclusion
In this study, we demonstrate the efficient NIR downconversion in Tm3+ and Yb3+ codoped Lu2O3 samples. By measuring the excitation and emission spectra as well as the lifetime of 1G4 level, effective energy transfer from Tm3+: 1G4 to Yb3+: 2F5/2 has been proved under 463 nm excitation. The dependence of the initial transfer rate upon Yb3+ concentration has been discussed to determine the downconversion process. It demonstrates that the ET from Tm3+ to Yb3+ occurs by single-step ET process. With raising Yb3+ doping level, a continuous increasing in theoretical quantum efficiency has been obtained by carefully calculation. The estimated maximum theoretical quantum efficiency reaches 148.2% in the Lu2O3: 0.2%Tm3+/30%Yb3+ sample. As the main emission peak of Yb3+ in Lu2O3 around 1000 nm matches better with the optimal spectral response of the c-Si solar cell, it indicates that Lu2O3: Tm3+/Yb3+ is promising DC material for application in solar cells.
References
S.E. Habas, HAS Platt, MFAM van Hest et al., Low-cost inorganic solar cells: from ink to printed device. Chem. Rev. 110(11), 6571–6594 (2010)
K. Lee, J. Lee, B.A. Mazor et al., Transforming the cost of solar-to-electrical energy conversion: Integrating thin-film GaAs solar cells with non-tracking mini-concentrators. Light 4(5), e288 (2015)
K. Yu, J. Chen, Enhancing solar cell efficiencies through 1-D nanostructures. Nanoscale Res. Lett. 4(1), 1 (2008)
B.M. van der Ende, L. Aarts, A. Meijerink, Near-infrared quantum cutting for photovoltaics. Adv. Mater. 21(30), 3073–3077 (2009)
J. Peet, J.Y. Kim, N.E. Coates et al., Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 6(7), 497–500 (2007)
M. Al-Ibrahim, O. Ambacher, S. Sensfuss et al., Effects of solvent and annealing on the improved performance of solar cells based on poly (3-hexylthiophene): fullerene. Appl. Phys. Lett. 86(20), 201120 (2005)
X. Huang, S. Han, W. Huang et al., Enhancing solar cell efficiency: the search for luminescent materials as spectral converters. Chem. Soc. Rev. 42(1), 173–201 (2013)
E.D. Kosten, J.H. Atwater, J. Parsons et al., Highly efficient GaAs solar cells by limiting light emission angle. Light. Sci. Appl. 2(1), e45 (2013)
J.C. Arvesen, R.N. Griffin, B.D. Pearson, Determination of extraterrestrial solar spectral irradiance from a research aircraft. Appl. Optics 8(11), 2215–2232 (1969)
B.S. Richards, Enhancing the performance of silicon solar cells via the application of passive luminescence conversion layers. Sol. Energy Mater. Sol. cells 90(15), 2329–2337 (2006)
B.M. van der Ende, L. Aarts, A. Meijerink, Lanthanide ions as spectral converters for solar cells. Phys. Chem. Chem. Phys. 11(47), 11081–11095 (2009)
C. Strümpel, M. McCann, G. Beaucarne et al., Modifying the solar spectrum to enhance silicon solar cell efficiency—an overview of available materials. Sol. Energy Mater. Sol. Cells 91(4), 238–249 (2007)
Q.Y. Zhang, X.Y. Huang, Recent progress in quantum cutting phosphors. Prog. Mater. Sci. 55(5), 353–427 (2010)
B.S. Richards, Luminescent layers for enhanced silicon solar cell performance: down-conversion. Sol. Energy Mater. Solar cells 90(9), 1189–1207 (2006)
D.C. Yu, R. Martín-Rodríguez, Q.Y. Zhang et al., Multi-photon quantum cutting in Gd2O2S: Tm3+ to enhance the photo-response of solar cells. Light. Sci. Appl. 4(10), e344 (2015)
L. Aarts, B.M. Van der Ende, A. Meijerink, Downconversion for solar cells in NaYF4: Er, Yb. J. Appl. Phys. 106(2), 023522 (2009)
G. Xiang, J. Zhang, Z. Hao et al., The energy transfer mechanism in Pr3+ and Yb3+ codoped β-NaLuF4 nanocrystals. Phys. Chem. Chem. Phys. 16(20), 9289–9293 (2014)
J. Li, L. Chen, Z. Hao et al., Efficient near-infrared downconversion and energy transfer mechanism of Ce3+/Yb3+ codoped calcium scandate phosphor. Inorg. Chem. 54(10), 4806–4810 (2015)
K. Deng, T. Gong, L. Hu et al., Efficient near-infrared quantum cutting in NaYF4: Ho3+, Yb3+ for solar photovoltaics. Opt. Express 19(3), 1749–1754 (2011)
H. Lin, S. Zhou, X. Hou et al., Down-conversion from blue to near infrared in Tm–Yb codoped YO transparent ceramics. IEEE Photon. Technol. Lett. 22(12), 866–868 (2010)
P. Vergeer, TJH Vlugt, MHF Kox et al., Quantum cutting by cooperative energy transfer in YbxY1– x PO4: Tb3+. Phys. Rev. B 71(1), 014119 (2005)
J. Yang, C. Zhang, C. Peng et al., Controllable red, green, blue (RGB) and bright white upconversion luminescence of Lu2O3: Yb3+/Er3+/Tm3+ nanocrystals through single laser excitation at 980 nm. Chemistry 15(18), 4649–4655 (2009)
A. Ulas, K.K. Kuo, C. Gotzmer, Ignition and combustion of boron particles in fluorine-containing environments. Combust. Flame 127(1), 1935–1957 (2001)
R. Peters, C. Kränkel, K. Petermann et al., Crystal growth by the heat exchanger method, spectroscopic characterization and laser operation of high-purity Yb: Lu2O3. J. Cryst. Growth 310(7), 1934–1938 (2008)
U. Griebner, V. Petrov, K. Petermann et al., Passively mode-locked Yb: Lu2O3 laser. Opt. Express 12(14), 3125–3130 (2004)
A.I. Zagumennyi, G.B. Lutts, P.A. Popov et al., The thermal conductivity of YAG and YSAG laser crystals. Laser Phys. 3(5), 1064–1065 (1993)
L. An, J. Zhang, M. Liu et al., Preparation and upconversion properties of Yb3+, Ho3+: Lu2O3 nanocrystalline powders. J. Am. Ceram. Soc. 88(4), 1010–1012 (2005)
Li Li, Xiantao Wei, C. Yonghu et al., Energy transfer in Tb3+, Yb3+ codoped Lu2O3 near-infrared downconversion nanophosphors. J. Rare Earths 30(3), 197–201 (2012)
J. Li, J. Zhang, Z. Hao et al., Intense upconversion luminescence and origin study in Tm3+/Yb3+ codoped calcium scandate. Appl. Phys. Lett. 101(12), 121905 (2012)
W. Shockley, H.J. Queisser, Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 32(3), 510–519 (1961)
J.M. Meijer, L. Aarts, B.M. van der Ende et al., Downconversion for solar cells in YF3: Nd3+, Yb3+. Phys. Rev. B 81(3), 035107 (2010)
Acknowledgements
This work was partially supported by the National Key Research and Development Program of China (Grant No. 2016YFB0701003, 2016YFB0400605), National Natural Science Foundation of China (Grant No. 61275055, 11274007, 51402284 and 11604330), Natural Science Foundation of Jilin province (Grant No. 20140101169JC, 20150520022JH and 20160520171JH), and the prior sci-tech program of innovation and entrepreneurship of oversea Chinese talent of Jilin province.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, W., Zhang, J., Hao, Z. et al. Near-infrared quantum cutting and energy transfer mechanism in Lu2O3: Tm3+/Yb3+ phosphor for high-efficiency photovoltaics. J Mater Sci: Mater Electron 28, 8017–8022 (2017). https://doi.org/10.1007/s10854-017-6506-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-6506-5