
Abstract
Properties of the quantum-cutting phosphors are dependent on various factors such as dopant concentration, crystallinity, homogeneity, particle size and surface morphology. Effective control of the above parameters can enhance the quantum-cutting ability of the phosphor material. Nano-sized particles of Y2O3:Tb3+,Yb3+ were prepared with a solution-based co-precipitation method and subsequent calcination. Effective control of the reaction parameters and doping concentration helped to produce uniform nanostructures with high quantum-cutting efficiency up to 181.1 %. The energy transfer mechanism between Tb3+ and Yb3+ was studied by considering their spectroscopic properties and time-resolved spectroscopy. The high efficiency and small particle size of the quantum-cutting phosphor Y2O3:Tb3+,Yb3+ make it a suitable candidate for its application in solar cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Witnessing significant development of the photovoltaic (PV) industry over the past few decades, researchers are still struggling for an efficient and cost-effective conversion of solar energy into electricity through PV cells. Quantum cutting through down-conversion is able to split one incident photon into two (or more) lower-energy photons with conversion efficiency exceeding 100 % [1]. With incorporation of quantum-cutting phosphors, the loss of energy caused due to thermalization of hot charge carriers after the absorption of the high-energy photons can be avoided by the absorption of high-energy photons by the solar cell through down-conversion phosphors. Trupke et al. [2] showed that incorporation of quantum-cutting phosphors in solar cells can effectively enhance its conversion efficiency by up to 38.6 %.
Solar cells are typically composed of special light-absorbing semiconducting materials. Incorporation of wavelength-converting phosphors in solar cell assemblies for up- and down-conversion of the solar spectrum is in large demand. Since quantum-cutting phosphors have ability to split one incident high-energy photon into two (or more) lower-energy photons with a conversion efficiency higher than 100 %, applying quantum-cutting phosphors that show emission near the band gap of a solar cell can significantly improve the cell efficiency over the Shockley–Queisser limits. When applied to a solar cell, those quantum-cutting materials are supposed to be placed on top of the solar cell (facing the Sun) so that the high-energy photons (wavelength < 400 nm) can be converted into lower-energy photons [visible or near-infrared (NIR) light] before they reach the solar cell. Therefore, the ideal design for quantum-cutting phosphors in solar cell is their incorporation in a thin transparent layer on top of the solar cell. To realize the transparent layer, the size of quantum-cutting phosphors needs to be controlled, with dimensions smaller than ~150 nm to avoid light scattering [3]. There have been several research papers published for the development of quantum-cutting phosphors for solar cells but only a few considered their size aspects [4, 5].
In this work, we focus on the synthesis of nano-sized, quantum-cutting phosphors with a higher energy transfer rate and external quantum efficiency. Their properties are dependent on various factors such as dopant concentration, crystallinity, homogeneity, particle size and surface morphology. Effective control of the above parameters can enhance the quantum-cutting ability of the phosphor material. The high quantum-cutting efficiency of the down-converting Y2O3:Tb3+,Yb3+ nanophosphors makes it a suitable candidate for a transparent layer of solar cells.
Experimental
Materials
Yttrium (III) chloride hexahydrate (YCl3·6H2O, Chemical Pure, Daejung Chemicals, Korea), terbium (III) chloride (TbCl3, 99.9 %, Sigma-Aldrich, MO, USA), ytterbium (III) chloride hexahydrate (YbCl3·6H2O, 99.9 %, Sigma-Aldrich, MO, USA), oleic acid (C18H34O2, Daejung Chemicals, Korea), sodium chloride (NaCl, Junsei Chemicals, Japan) and sodium carbonate hydrate (Na2CO3·H2O, extra pure, Daejung Chemicals, Korea) were used as received without further purification.
Synthesis
The synthesis of Yb- and Tb-doped yttrium oxide was carried out using a co-precipitation method reported previously [6]. Powder samples of Y2O3 were prepared by the stoichiometric ratio of Y1.9−x O3:Tb0.1Yb x % (x = 0–0.2). The precursors were weighed carefully in respective reaction flasks and dissolved in 50 mL of aqueous solution. Oleic acid (1.0 mL, 3.2 mmol) was added to the aqueous solution and NaCl was added to make a 0.15 M-concentration. The emulsion was stirred at room temperature for 1 h and then the pH of the solution was adjusted to 7.0 by the dropwise addition of sodium carbonate aqueous solution (Na2CO3·H2O, 0.3 M). A white-colored precipitate formed after addition of the sodium carbonate, indicating the formation of carbonate complexes, as indicated in the Eq. 1.
The stirring was continued for 2 h at room temperature and the precipitate was then separated by centrifugation at 10,000 rpm for 10 min. The obtained solid was washed thoroughly using deionized (DI) water for 3 times and then washed with ethanol twice to remove organic impurities, resulting in a white solid. The solid was dried in an oven at 120 °C for 4 h and calcinated at 800 °C for 2 h under an ambient condition. The carbonate complexes turned to oxide complexes by sequential decarboxylation during the calcination, as indicated in the Eq. 2.
Characterizations
The powder X-ray diffraction (XRD) patterns of the annealed samples were recorded using CuKα radiation (λ = 1.54056 Å) on a Bruker D8 Advanced diffractometer operating at 40 kV and 40 mA at a scanning rate of 05° per step in the 2θ range of 10° ≤ 2θ ≤ 120°. The reference data for the comparison of obtained diffraction patterns were obtained from JCPDS cards. Particle size and morphology of the calcinated nanophosphors were detected by field emission scanning electron microscopy (FE-SEM, model FEI-XL30 FEG [field emission gun], accelerating voltage = 15 kV; FEI Co., Hillsboro, OR, USA) and transmittance electron microscopy (TEM, model Tecnai G2, accelerating voltage = 200 kV; FEI Co., Hillsboro, OR, USA). The photoluminescence spectroscopy of the quantum-cutting phosphor was measured by a Hitachi F-7000 fluorescence spectrophotometer with a 200-W xenon lamp source (Hitachi, Japan).
Results and discussion
As represented in Fig. 1, the crystalline Y2O3 has a cubic bixbyite structure with an Ia3 space group, containing 16 formula units in each primitive cell [7]. Among the 32 six-fold coordinated cations in its primitive cell, 8 are centrosymmetric with C 3i symmetry, and 24 are non-centrosymmetric with C 2 symmetry. While the point symmetries of S 6 and C 3i have an inversion center and smaller crystal field, disallowing electron–dipole transitions in the absence of lattice distortion (thermal or due to impurity incorporation), the C 2 sites are non-centrosymmetric, i.e. have no center of inversion allowing electron–dipole transitions and, therefore, being predominantly responsible for the luminescent emissions when occupied by transition metal ions [8]. The cubic phase of yttrium oxide shows the presence of Y3+ in two different environments (C 2 or S 6) with six oxygen atoms surrounding it. The isomorphic substitution of terbium and ytterbium ions in a Y2O3 lattice will usually occupy these symmetry sites (low-symmetry C 2 and high-symmetry S 6). As the number of C 2 sites is about three times higher than number of S 6 sites, the dopant ions Tb and Yb occupy more C 2 positions than S 6 positions.

Cubic bixbyite structure of Y2O3 with C 2 and C 3i/S 6 symmetry sites
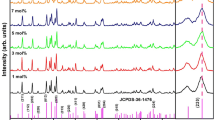
Figure 2 represents XRD patterns of Y2O3 phosphors with various concentrations of Yb (0–10 mol%) with a fixed amount of Tb (5 mol%). All of the Bragg reflections in the diffraction patterns were indexed by comparing with standard diffraction peaks of the body-centered cubic structure (space group I a3) of Y2O3 (JCPDS # 83-0927). No additional secondary phase, impurities or any traces of amorphous nature were observed, indicating the structural purity of the synthesized compounds. This result also indicates that high lanthanide doping levels up to 15 mol% in respect to Y3+ can be used in Y2O3 crystal without altering the basic structure of the host under the synthesis conditions. The respective changes in lattice parameters and unit cell volume due to increasing amount of dopants are summarized in Table 1. Doping with 5-mol% terbium ions, the unit cell volume increased by 13.04 Å3 in respect with the standard Y2O3 sample. With increasing dopant concentration of ytterbium ions to Y1.9O3:Tb0.1, the unit cell volume showed minimal deviation from that of the Y1.9O3:Tb0.1 sample. Those changes in the unit cell volume are affected by the differences in the ionic radii of the Y3+, Tb3+ and Yb3+ ions [r(Y3+) = 1.019 Å, r(Tb3+) = 1.040 Å and r(Yb3+) = 0.985 Å; for the coordination number (CN) 8] [9].

Comparison of XRD patterns of Y1.9−x O3:Tb0.1,Yb x (x = 0, 0.02, 0.06, 0.10, 0.14, 0.20) prepared by a co-precipitation method and calcined at 800 °C for 2 h
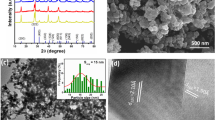
The size and morphology of the Y2O3 phosphors were analyzed by SEM and TEM images. Figure 3 shows SEM images of Y1.9−x O3:Tb0.1,Yb x (x = 0, 0.02, 0.06, 0.10, 0.14, and 0.20) in series. When the doping concentration of ytterbium ion was below 3 mol%, the particle size was below 40 nm and the particle morphology was more or less spherical and homogeneous. However, when the doping levels were increased more than 5 mol%, the grain size grew bigger, reaching 60–70 nm with 10-mol% Yb doping. The particle morphology also changed to rough and irregular and agglomeration was found in some portion. To study the microstructure and crystallinity of the Y2O3 phosphors, a TEM image of Y1.84O3:Tb0.1,Yb0.06 phosphor was examined and shown in Fig. 4. The particle morphology was more or less spherical with a little facet on the surface, and the crystallinity appeared to be very high.

Comparison of SEM images of Y1.9−x O3:Tb0.1,Yb x (x = 0, 0.02, 0.06, 0.10, 0.14, and 0.20) by the doping concentration of ytterbium ions

TEM image of Y1.84O3:Tb0.1,Yb0.06 nanophosphor
The spectroscopic properties of the Y2O3 phosphors were examined with photoluminescence spectroscopy and the results are shown in Figs. 5 and 6. Under the excitation of UV light (λ exc. = 304 nm), both visible light emission within 470–700 nm and NIR light emission within 950–1050 nm were observed. Several lines in the visible range correspond to the multiplet electronic transition of 5D4–7F J (J = 6, 5, 4, 3, 2) of Tb3+, whereas the NIR light emission corresponds to electronic transition between Stark levels of 2F5/2 and 2F7/2 of Yb3+. The luminescence excitation spectrum through emission at 545 nm showed broad absorption in the range of 200–330 nm. Van Pieterson et al. [10] explained that the charge transfer absorption of O2−–Yb3+ in yttrium oxide is found in the region of λ < 250 nm; thus, it is clear that the observed absorption band relates to electronic transition of Tb3+(7F6) → Tb3+(4f75d1). As the Yb3+ concentration was increased from 0 to 10 mol%, the visible light emission decreased gradually, as shown in Fig. 5; whereas, the NIR emission was increased along with Yb3+ concentration, as indicated in Fig. 6. The NIR emission of Yb3+ originates from the energy transfer of Tb3+ to Yb3+ since the intra-ion relaxation rate of Tb3+(5D4) → Tb3+(7F J ) is relatively slower than the inter-ion transition of Tb3+(5D4) → 2Yb3+ (2F5/2). With absorption of UV light, Tb3+ ions are excited up to its 4f75d1 energy level and successively follow fast intra-ion relaxation to a 5D4 level, denoted as a green line in Fig. 7. The relaxed photon at 5D4 gives spontaneous 5D4–7F J (J = 0–6) emission and cooperative energy transfer between Tb3+(5D4) → 2Yb3+(2F5/2) with some degree of deactivation by quenching, denoted as red and dotted lines, respectively, in Fig. 7 [11]. In an effective doping system, a single cooperative acceptor of the Yb–Yb type is formed where two Yb3+ ions occupy the nearest neighboring coordination sphere of Tb3+ [12].

Excitation and emission spectra of Y1.9−x O3:Tb0.1,Yb x (x = 0, 0.02, 0.06, 0.10, 0.14, and 0.20)

NIR emission spectra of Y1.9−x O3:Tb0.1,Yb x (x = 0, 0.02, 0.06, 0.10, 0.14, and 0.20)

Quantum cutting through cooperative energy transfer between Tb3+ → Yb3+
NIR quantum cutting for Tb3+–Yb3+ co-doped systems was first studied and reported by Vergeer et al. [11] on YPO4 phosphors. They proposed a cooperative quantum-cutting mechanism by analyzing the time-resolved spectroscopic study of Tb3+ as a function of Yb3+ doping concentration. For the Tb3+–Yb3+ couple, the transition of Tb3+ (5D4 → 7F J ) is approximately twice the energy of 2F5/2 → 2F7/2 transition of Yb3+. The energy transfer and quantum efficiencies were calculated from the luminescence decay curves using the following equations,
where I denotes intensity, x %Yb stands for Yb concentration and η Tb represents quantum efficiency (QE) for Tb3+ and is set to 1 [11].
Following the calculation method suggested by Vergeer et al., quantum efficiencies of our Y2O3 phosphors were obtained from the respective decay time of Tb3+: 2.28, 1.86, 1.84, 1.82, 1.80 and 1.74 ms for samples prepared with 0, 1, 3, 5, 7 and 10 mol% of Yb3+ , respectively (Fig. 8). By taking the upper limited value of the cooperative energy transfer efficiency as assuming the Tb3+(5D4) → 2Yb3+ (2F5/2) cooperative transfer is 100 %, the theoretical quantum efficiency is calculated to be 67.3, 139.1, 164.5, 169.5, and 181.1 % for samples prepared with 1, 3, 5, 7 and 10 mol% of Yb3+, respectively. However, there is a limitation to providing further insight on the QE results because there is much assumption applied in their calculation. On the other hand, the measurement of internal and external QEs was difficult as photon detectors used for visible and NIR regions were different.

Time-resolved luminescence spectroscopy of Tb3+ 5D4–7F5 emission at 545 nm for various concentrations of Yb3+ under an excitation of 303 nm
Conclusion
Rare-earth-doped Y2O3 nanocrystals were synthesized by a co-precipitation method and subsequent calcination at 800 °C. The nanocrystals were found to be uniform in size and highly crystalline with an average diameter of ~50 nm. With Tb3+ and Yb3+ co-doped Y2O3, the energy transfer mechanisms in the visible and NIR region were studied by considering their photoluminescence spectrums and time-resolved luminescence techniques. Photons excited by UV to a Tb3+ 4f75d1 energy level and subsequently relaxed to a 5D4 level appear to transfer to Yb3+ 2F5/2 level, resulting in NIR emission. Quantum efficiencies of Y1.9−x O3:Tb0.1,Yb x were calculated following the method suggested by Vergeer et al. [11] and a QE as high as 181.1 % was obtained with 10-mol% Yb doping. Since these Y1.9−x O3:Tb0.1,Yb x phosphors were designed to be nano-sized in order to avoid light scattering, they can make good candidates for applications in solar cell devices. Efforts to make them into a transparent layer for silicon solar cell panels are in progress.
References
Q.Y. Zhang, X.Y. Huang, Prog. Mater. Sci. 55, 353 (2010)
T. Trupke, M.A. Green, P. Wurfel, J. Appl. Phys. 92, 1668 (2002)
W.G.J.H.M. van Sark, A. Meijerink, R.E.I. Schropp in Solar Spectrum Conversion for Photovoltaics Using Nanoparticles, Third Generation Photovoltaics, ed. by V. Fthenakis (In Tech, 2012), p. 1
H. Lin, D.Q. Chen, Y.L. Yu, A.P. Yang, Y.S. Wang, Opt. Lett. 36, 876 (2011)
Q.Y. Zhang, C.H. Yang, Z.H. Jiang, X.H. Ji, Appl. Phys. Lett. 90, 061914 (2007)
A.P. Jadhav, A. Pawar, C.W. Kim, H.G. Cha, U. Pal, Y.S. Kang, J. Phys. Chem. C 113, 16652 (2009)
F. Hanic, M. Hartmanova, G.G. Knab, A.A. Urusovskaya, K.S. Bagdasarov, Acta Crystallogr. B 40, 76 (1984)
M. Klintenberg, S. Edvardsson, J.O. Thomas, J. Alloys Compd. 275, 174 (1998)
P. Ayyub, V.R. Palkar, S. Chattopadhyay, M. Multani, Phys. Rev. B 51, 6135 (1995)
L. van Pieterson, M. Heeroma, E.D. Heer, A. Meijerink, J. Lumin. 91, 177 (2000)
P. Vergeer, T.J.H. Vlugt, M.H.F. Kox, M.I. den Hertog, J.P.J.M. van der Eerden, A. Meijerink, Phys. Rev. B 71, 014119 (2005)
J.L. Yuan, X.Y. Zeng, J.T. Zhao, Z.J. Zhang, H.H. Chen, X.X. Yang, J. Phys. D Appl. Phys. 41, 105406 (2008)
Acknowledgments
We thank the financial support from the Korea Institute of Science and Technology (KIST) institutional funding (Project No. 2E26120) and the international cooperation program managed by the National Research Foundation of Korea (NRF-2014K1A3A1A09063246).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jadhav, A.P., Khan, S., Kim, S.J. et al. Near-infrared quantum cutting in Tb3+ and Yb3+-doped Y2O3 nanophosphors. Res Chem Intermed 43, 3463–3471 (2017). https://doi.org/10.1007/s11164-016-2427-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2427-9