
Abstract
Polypyrrole (PPy) was synthesized on graphene (GN) sheets which was reduced from graphene oxide by microwave irradiation in N-methyl-2-pyrrolidinone (NMP) and dispersed very well in β-naphthalene sulfonic acid solution. Homogeneous polypyrrole/graphene (PGN) composites had been successfully prepared via an in situ emulsion polymerization method and showed better electrochemical performance including high specific capacitance, rate characteristic and cycling stability compared with polypyrrole/graphene oxide (PGO) composites. The NMP molecules entered into GN which was helpful for PPy deposition on the GN sheets evenly. The supercapacitive behaviors of the corresponding electrodes were estimated with cyclic voltammetry and galvanostatic charge/discharge measurements in 1 M KCl electrolyte and the results showed that the specific capacitance of PGN composites was up to 906 F g−1 at the current density of 0.2 A g−1, much higher than that of the PGO (704 F g−1) and PPy (430 F g−1). Its specific capacitance retains more than 91 % of its initial capacitance after 1,000 cycles at a high current density of 5 A g−1. The good electrochemical performance of PGN composites is mainly attributed to its uniform structure, the N doping, and the synergistic effects between the two components.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Among the several new energy storage technologies available today, supercapacitors appear to be very promising choice due to their higher power density, longer cycle life than batteries and higher energy density than conventional capacitor. Generally, supercapacitors can be classified into two categories. One is the electrical double layer (EDL) capacitor and the other is the pseudo-capacitor [1, 2]. EDL capacitor stores energy through the adsorption of both anions and cations at the electrode–electrolyte interface [3, 4] and pseudo-capacitor does through fast surface redox reactions of the electro-active species [5, 6]. The most commonly used materials for EDL capacitor electrodes are carbon-based materials such as activated carbons (ACs), carbon nanotubes (CNTs), graphite oxide (GO) and graphene (GN) [1, 7, 8]. In recent years, GN has received extensive research interest, for its obvious advantages such as unique combination of electronic and mechanical properties, high conductivity, high surface area [9, 10]. Chemically reduced graphene oxide (RGO) is used as a substitution for pristine GN because of the low cost and facile processing of the chemical exfoliation [11]. However, the specific capacitance of graphene-based supercapacitors is not high enough and the toxicity accompanied with the chemical reduction limited their applications [12].
The most widely studied electrode materials for pseudo-capacitors are electrically conducting polymers [13–17]. Recently, polypyrrole (PPy) has been explored as one of the most competitive materials for pseudo-capacitors because of its relatively high conductivity, ease of synthesis and low cost [18]. However, the practical applications of PPy for pseudo-capacitors are still restricted because of its low rate capacitance resulting from the poor electrical conductivity and poor cycling stability due to the easily damaged structure of the materials during redox processes [19].
To resolve the above problems, PPy and its composites with GO or GN were designed to make use of the merits of both EDL capacitance and pseudo-capacitance. The specific capacitance of the binary composites may further be boosted by the synergetic effects of the two components. Ma [20] reported the enhanced capacitance and rate capability of GN/PPy composites under acid conditions and its specific capacitance of composites was as high as 482 F g−1 at a current density of 0.5 A g−1. Zhang prepared graphite oxide/polypyrrole composites (GPys) by in situ polymerization and reduced by NaBH4 to prepare reduced graphite oxide/polypyrrole composites (R-GPys). The specific capacitances of the composites before and after reduction (197 and 180 F g−1) were highly improved compared with that of pristine graphite oxide (11 F g−1) and PPy (112 F g−1), respectively [21]. Song et al. [22] synthesized PPy/GN composites doped with anion surfactants which exhibited enhanced specific capacitance of 351 F g−1 at current density of 1.0 A g−1 compared to the polymers deposited on pristine graphite foil. Although a lot of works had been done to improve the electrochemical performance of PPy and its composites as electrode, the capacitance was not high enough and the synthetic method was too complicated and not friendly to the environment.
Compared with the above results, GN was facilely reduced from GO in this paper by microwave irradiation in N-methyl-2-pyrrolidinone (NMP) which dispersed very well in β-naphthalene sulfonic acid (β-NSA) solution. More importantly, PGN was prepared firstly in β-NSA solution by in situ emulsion polymerization with homogeneous structure and the composites exhibited enhanced electrochemical performance. The specific capacitance of PGN composites was up to 906 F g−1 at the current density of 0.2 A g−1 in 1 M KCl electrolyte and the value was 590 F g−1 at the current density of 5 A g−1 which is much higher than the value reported previously. The cycling stability of PGN was excellent and its specific capacitance retained more than 91 % of its initial capacitance after 1,000 cycles at a current density of 5 A g−1.
2 Experimental
2.1 Chemicals
Graphite and β-NSA were supplied by Alfa Aesar and Shanghai Siyu Chemical Technology Co. Ltd., respectively. Pyrrole (Py) (98 %) was purchased from Aldrich and distilled under vacuum prior to use. All other reagents were of analytical grade and used as received without further purification.
2.2 Preparations of GO and GN
The GO was firstly prepared from natural graphite powder using a modified Hummers method [23]. GN was prepared from GO by a microwave irradiation method. 120 mg GO was dispersed in 40 mL NMP using magnetic stirring and ultrasonic treatment to form a homogeneous mixture. Then, the mixture was placed in a microwave oven for 2 min (power: 700 W) and then cooled to the room temperature to get a homogeneous GN suspension slurry. Finally, the suspension was filtered and the obtained black GN wad dried at 50 °C.
2.3 Preparation of PPy, PGO and PGN composites
PGN composites were prepared using an in situ emulsion polymerization method. In a typical experiment, firstly, 7.2 mmol β-NSA was dissolved in 25 mL distilled water in a three-neck flask. A certain amount of GN (the mass ratio of GN to Py was 10:90) was added into the β-NSA solution with ultrasonic treatment for 2 h and then 0.5 mL Py was added into the above solution with stirring for 30 min. Then, 0.39 g of FeCl3·6H2O was dispersed in 5 mL distilled water and slowly dropped into the reaction system. The reaction was carried out for 24 h under stirring at 0 °C. Finally, the reaction mixture was filtered, washed several times with distilled water and ethanol, and dried at 50 °C for 12 h to get the PGN composites.
By comparison, PGO composites were obtained successfully by replacing the GN with GO and repeating the above experiment. The pure PPy was prepared without GO and GN according to the same procedure.
2.4 Material characterization and electrochemical measurements
The microstructure and morphology of the samples were observed by scanning electron microscopy (SEM, Hitachi S-4800) with an accelerating voltage of 5 kV and transmission electron microscopy (TEM, JEM-2100) with an accelerating voltage of 200 kV, respectively. Fourier transformation infrared (FTIR) spectra were recorded on a TENSOR 27 FTIR spectrometer using KBr pellets. X-ray diffraction (XRD) patterns were obtained on a D8 ADVANCE (Bruker AXS, Germany) using Cu Kα radiation (λ = 1.54178 Å) radiation. Element analysis was carried out on an Elementar Vairo EL III (Germany). Two-probe technique X-ray photoelectron spectroscopy (XPS) measurements were made on an ESCALAB 250 spectrometer with an Mg Kα X-ray source (1,253.6 eV photons).
The three-electrode cell system was used to evaluate the electrochemical performance of the prepared electrode materials in 1 M KCl aqueous electrolyte. The working electrode was made by mixing 80 wt% active material, 10 wt% acetylene black, and 10 wt% poly (vinylidene fluoride) in NMP and the slurry was coated onto a 1 cm × 1 cm nickel foam current collector, and dried at 40 °C for 8 h to evaporate solvent. A platinum sheet and a standard calomel electrode (SCE) were used as counter electrode and reference electrode, respectively. Cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) were measured by a PARSTAT2263 electrochemical workstation. EIS was recorded in the frequency range from 105 to 0.1 Hz at open circuit potential with alternate current amplitude of 10 mV. The electrical conductivity was determined by a four-probe instrument (RTS-8) at room temperature. The samples were compacted into pellets of 15 mm in diameter and about 0.5 mm thickness under the pressure of 10 MPa.
3 Results and discussion
3.1 The morphology of the PPy, GO, GN, PGO and PGN composites
The morphology of the as-prepared PPy, GO, GN, PGO and PGN was investigated by SEM and TEM as shown in Fig. 1A, B, C, D, E and a, b, c, d, e, respectively. PPy synthesized by chemical oxidation in β-NSA solution is large lumps agglomerated with irregular particles which are shown in Fig. 1A, a. Figure 1B, b indicate that the morphology of GO has a typically wrinkled and scrolled loose layer structure. Figure 1C, c show GN crumpled sheets resemble into silk veil waves. Figure 1D revealed that most PPy deposited on the surface of GO and there were still a few small black blocks agglomerated with PPy between the layers of GO sheets as shown in Fig. 1d. Compared with PGO, PPy grew on the GN sheets more evenly than that on the GO and PGN composites exhibited PPy films attached to GN layer and looked like more homogeneous than PGO which was clear in Fig. 1E, e. The uniform coating of PPy on GN sheets may be beneficial to improve the electrochemical performance of PGN which is confirmed by the following testing results including CV, GCD and cycling stability.

The SEM and TEM images of the PPy, GO, GN, PGO and PGN composites (A, B, C, D, E and a, b, c, d, e are SEM and TEM images of the PPy, GO, GN, PGO and PGN composites, respectively)
3.2 The structure analysis of the PPy, GO, GN, PGO and PGN composites
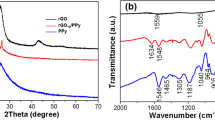
Figure 2 shows the FTIR spectra of the GO, GN, PPy, PGO and PGN composites. In Fig. 2a, the absorption peak for GO at 1,731 cm−1 was corresponded to the C=O stretching vibration, the vibration and deformation peaks of O–H groups were at 3,394 and 1,410 cm−1. The C–O (epoxy) stretching vibration peak was at 1,226 cm−1, and the C–O (alkoxy) stretching peak located at 1,050 cm−1. The above absorption peaks decreased strongly for GN, indicating that most oxygen in the GO was removed and the reduction of GO to GN by microwave irradiation [12, 24]. Nevertheless, the elimination of these functional groups is not complete, because the presence of the some peaks at about 1,731, 1,226 and 1,410 cm−1 imply that little fraction of C=O, C–O (epoxy) and O–H groups still remain in the GN. The π–π interactions between GN sheets and polymer backbone, as well as the hydrogen bonding between the functional groups of GN sheets and amino-groups in polymer chains would play an important role in polymer anchoring [19, 25].

The FTIR spectra of the samples (a) GO and GN, (b) PPy, PGO and PGN
Compared with GO and GN, it is clearly observed that the characteristic absorption peaks attributed to PPy appeared in the spectra of PGO and PGN as shown in Fig. 2b. The new peaks at 1,539, 1,446, and 3,441 cm−1 are correspond to the C–C, C–N, and N–H stretching vibration in the PPy ring [26]. Moreover, the absorption bands at 2,922 and 2,851 cm−1, which are due to the asymmetric stretching and symmetric vibration of CH2 of the β-NSA, were also observed [27, 28].
Figure 3 shows the XRD patterns of the GO, GN, PPy, PGO and PGN composites. The pattern of GO exhibited an intense, sharp peak centered at 2θ = 10.4o which is attributed to the (002) crystalline plane, corresponding to an average interlayer distance (d) of about 0.76 nm of the GO. This value is larger than the d-spacing (0.34 nm) of pristine graphite (2θ = 26.5°), resulting from the introduction of oxygenated functional groups on the carbon sheets [24]. As for GN, the diffraction peak at 10.4° was disappeared and a broad diffraction peak around 20.7° was observed, indicating that the interlayer distance was reduced to 0.43 nm [4, 29, 30]. For PGO and PGN composites, the diffractive peaks are almost as same as those of PPy and the peaks ascribed to GO and GN within the two samples disappeared, illustrating the complete coating of PPy on the surfaces of the layered GO and GN.

The XRD spectra of the GO, GN, PPy, PGO and PGN
3.3 Electrochemical performance of the samples
The electrochemical property of the PPy and its composites was characterized in detail in 1 M KCl electrolyte by means of a three-electrode cell system in Fig. 4. The CV, GCD, rate capacity and EIS curves of the GO and GN were given at the same time in order to compare the electrochemical characteristic of the PPy and its binary composites. Figure 4a shows the cyclic voltammetry (CV) behaviours of the PPy, PGO and PGN in the potential range between −0.2 and 0.6 V at scan rate of 5 mV s−1. The CV curve of the PGN composites exhibited the most close to rectangular, while the CV curve of pure PPy was the most distorted one. Compared with the CV curves of PPy and PGO composites, the CV curve of the PGN composites occupied much larger area, implying the higher capacitive behaviour. The GCD curves of electrodes in the potential range from −0.2 to 0.6 V at current densities of 0.2 A g−1 were shown in Fig. 4b and the corresponding specific capacitances (C g) are 430, 704 and 906 F g−1 for PPy, PGO and PGN composites according to the C g = I × Δt/(m × ΔV), respectively [3, 31]. Where C g is the specific capacitance (F g−1), I is the current (A), Δt is the discharge time (s), m is the mass of active materials in the electrode (g), andΔV is the potential window (V). The specific capacitance of the PGN composites was much higher than that of PPy and PGO composites at the same current density which might be related to its more uniform structure.

a CVs of GO, GN, PPy, PGO and PGN at a scan rate of 5 mV s−1. b GCD curves of GO, GN, PPy, PGO and PGN at current density of 0.2 A g−1. c The specific capacitances of GO, GN, PPy, PGO and PGN electrodes at different current densities. d Nyquist plots of GO, GN, PPy, PGO and PGN. The mass of sample is 3 mg
Increasing the current density from 0.5 to 1, 3, 5 A g−1, the C g of the samples tended to decrease gradually. When the current density was 5 A g−1, the C g of PGN was as high as 590 F g−1, retaining 65.0 % of its initial capacitance at 0.2 A g−1 as shown in Fig. 4c. In contrast, the C g of PGO and PPy composites retain 63.9 and 54.1 % respectively, indicating the best rate capacity of the PGN among the three samples.
The electrical conductivity of the PPy, PGO and PGN composites were measured by the four probe method and were listed in Table 1. The conductivity of pure PPy is 6.33 S cm−1, the value of PGO was decreased to 2.09 S cm−1 and that of PGN was reached up to 17.73 S cm−1. To further recognize the ion-transport kinetics and electrode conductivity, the Nyquist plots were given in Fig. 4d. In the low-frequency region, the slope of the plot of PGN is the largest and the pure PPy is the smallest, illustrating that PGN possesses the best capacitive behavior [32]. Meanwhile, in the high-frequency region (Fig. 4d inset), the real axis intercept is equivalent to series resistance (ESR) and the radius of the semicircle is corresponded to the charge-transfer resistance of the electrode samples [20, 33]. Among the three electrode materials, the ESR and the radius of the PGN is the smallest, implying that the PGN has the best supercapacitive characteristic which is consistent with the results of CV and GCD analysis.
In order to investigate the reason of the excellent capacitive characteristic of PGN composites comprehensively, the element analysis of the three samples was conducted and the results were shown in Table 1. The O content decreases dramatically after microwave irradiation which indicates that GO is reduced into GN with enhanced conductivity. The O content in PGN is slightly higher than that of PPy which shows that a few O atoms remain in GN which might be helpful to improve the capacitive characteristic because of the increased number of active sites during the charging/discharging process [34, 35]. The S content is higher in PGN than that in PGO which hints that more anions enter into the polymer chains for PGN composites and higher doping level of β-NSA [36]. The N content increases after microwave irradiation for PGN which is even higher than that of PPy, illustrating that the solvent NMP molecules or its oligomers attached onto the GN sheets which is helpful for the anchoring of the polymer chains on the surface of GN sheets. The lipophilic characteristic of NMP and its oligomers is beneficial to PPy to deposit on the GN sheets and get the uniform PGN composites according to its similar dissolving characteristic to PPy [37, 38].
Figure 5 shows the N 1s broad bands and the XPS survey scans of PPy, PGO and PGN. The N 1s broad band could be fitted into two Gaussian peaks for PPy as shown in Fig. 5a with binding energies of 399.4 (–NH-) and 402.2 eV (=N+–) and the two peaks for PGO in Fig. 5b were about 400.4 (-NH+-) and 403.4 eV (=N+–) [22, 39]. However, the N1s peak spectra could be resolved into three constituents for PGN as shown in Fig. 5c with binding energies of 398.0 (=N–), 400.0 (–NH–) and 403.0 eV (=N+–) which is related to the NMP attachment on the GN sheets [37, 40]. The XPS survey scans in Fig. 5d indicate that the N content in PGO is less than that of PPy and PGN obviously which is consistent with the results of element analysis.

N 1s XPS core level spectra of PPy (a), PGO (b), PGN (c) and (d) XPS spectra of the PPy, PGO, PGN
The cycling stability is another vital factor for the practical application of supercapacitors. Figure 6 exhibits the cycling performance of the three electrode samples by charging/discharging the capacitor for 1,000 cycles at a current density of 5 A g−1. The PGN electrode shows a good cycling stability and its specific capacitance still retains more than 91 % of its initial capacitance after 1,000 cycles. The conservation rate of PGO and PPy are 80 and 49 %, respectively. The N doping in GN, uniform structure accompanied with the strong synergistic effect between PPy and GN are the main reasons to explain the best cycling stability of the PGN among the three samples.

Cycle stability of PPy, PGO and PGN electrodes at a current density of 5 A g−1
4 Conclusions
Polypyrrole/graphene with homogenous structure and great electrochemical performance has been successfully prepared in β-NSA solution by an in situ emulsion polymerization method. The microwave irradiation in NMP not only reduces GO into GN but also introduces N into GN which is helpful for PPy deposit on GN sheets and further improves the electrochemical performance of PGN compared with PGO. PGN exhibits the highest specific capacitance, the best rate performance and cycling stability among the three samples and this facile method provides wider potential application of fabricating conducting polymer composites as supercapacitor electrodes.
References
T. Qian, C.F. Yu, S.S. Wu, J. Shen, J. Mater. Chem. A 1, 6539–6542 (2013)
P. Simon, Y. Gogotsi, Nat. Mater. 7, 845–854 (2008)
J. Li, H.Q. Xie, Y. Li, J. Power Sources 241, 388–395 (2013)
K. Zhang, L. Mao, L.L. Zhang, H.S.O. Chan, X.S. Zhao, J.S. Wu, J. Mater. Chem. 21, 7302–7307 (2011)
C.Z. Yuan, B. Gao, L.F. Shen, S.D. Yang, L. Hao, X.J. Lu, Nanoscale 3, 529–545 (2011)
X.J. Lu, F. Zhang, H. Dou, C.Z. Yuan, S.D. Yang, L. Hao, Electrochim. Acta 69, 160–166 (2012)
Y. Gogotsi, P. Simon, Science 334, 917–918 (2011)
T. Kuilla, S. Bhadra, D. Yao, N.H. Kim, S. Bose, J.H. Lee, Prog. Polym. Sci. 35, 1350–1375 (2010)
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, Science 306, 666–669 (2004)
D. Chen, L.H. Tang, J.H. Li, Chem. Soc. Rev. 39, 3157–3180 (2010)
S.F. Pei, H.M. Cheng, Carbon 50, 3210–3228 (2012)
C.Z. Zhu, S.J. Guo, Y.X. Fang, S.J. Dong, ACS Nano 4, 2429–2437 (2010)
D. Aradilla, F. Estrany, C. Alema´n. J. Phys. Chem. C 115, 8430–8438 (2011)
J. Yan, T. Wei, B. Shao, Z.J. Fan, W.Z. Qian, M.L. Zhang, Carbon 48, 487–493 (2010)
S. Chen, J.W. Zhu, X.D. Wu, Q.F. Han, X. Wang, ACS Nano 4, 2822–2830 (2010)
S. Chen, J.W. Zhu, H. Zhou, X. Wang, RSC Adv. 1, 484–489 (2011)
L. Sun, L. Wang, C.G. Tian, T.X. Tan, Y. Xie, K.Y. Shi, RSC Adv. 2, 4498–4506 (2012)
C. Bora, S.K. Dolui, Polymer 53, 923–932 (2012)
C.Z. Zhu, J.F. Zhai, D. Wen, S.J. Dong, J. Mater. Chem. 22, 6300–6306 (2012)
D.C. Zhang, X. Zhang, Y. Chen, P. Yu, C.H. Wang, Y.W. Ma, J. Power Sources 196, 5990–5996 (2011)
Y.Q. Han, L. Hao, X.G. Zhang, Synth. Met. 160, 2336–2340 (2010)
Y. Song, J.L. Xu, X.X. Liu, J. Power Sources 249, 48–58 (2014)
Y.G. Li, Y.Y. Wu, J. Am. Chem. Soc. 131, 5851–5857 (2009)
H.L. Guo, X.F. Wang, Q.Y. Qian, F.B. Wang, X.H. Xia, ACS Nano 3, 2653–2659 (2009)
J.P. Wang, Y.L. Xu, J.B. Zhu, P.G. Ren, J. Power Sources 208, 138–143 (2012)
G.J. Cho, B.M. Fung, D.T. Glatzhofer, J.S. Lee, Y.G. Shul, Langmuir 17, 456–461 (2001)
S. Bose, N.H. Kim, T. Kuila, K. T., Lau J. H. Lee. Nanotechnology 22, 295202–295210 (2011)
S. Bose, T. Kuila, M.E. Uddin, N.H. Kim, A.K.T. Lau, J.H. Lee, Polymer 51, 5921–5928 (2010)
H.F. Yang, C.S. Shan, F.H. Li, D.X. Han, Q.X. Zhang, L. Niu, Chem. Commun. 26, 3880–3882 (2009)
L.L. Zhang, S.Y. Zhao, X.N. Tian, X.S. Zhao, Langmuir 26, 17624–17628 (2010)
J. Zhang, Y. Yu, L. Liu, Y. Wu, Nanoscale 5, 3052–3057 (2013)
Y. Liu, Y. Ma, S.Y. Guang, H.Y. Xu, X.Y. Su, J. Mater. Chem. A 2, 813–823 (2014)
J.H. Liu, J.W. An, Y.X. Ma, M.L. Li, R.B. Ma, J. Electrochem. Soc. 159, A828–A833 (2012)
C.L. Wang, Y. Zhou, L. Sun, Q. Zhao, X. Zhang, P. Wan, J. Phys. Chem. C 117, 14912–14919 (2013)
L.F. Chen, X.D. Zhang, H.W. Liang, M.G. Kong, Q.F. Guan, P. Chen, ACS Nano 6, 7092–7102 (2012)
J. Liu, M.X. Wan, J. Polym. Sci. A Polym. Chem. 39, 997–1004 (2001)
S. Dubin, S. Gilje, K. Wang, V.C. Tung, K. Cha, A.S. Hall, ACS Nano 4, 3845–3852 (2010)
E.A. Ponzio, R. Echevarria, G.M. Morales, C. Barbero, Polym. Int. 50, 1180–1185 (2001)
J. Joo, J.K. Lee, S.Y. Lee, K.S. Jang, E.J. Oh, A.J. Epstein, Macromolecules 33, 5131–5136 (2000)
M.F. Cai, R.B. Smart, Energy Fuels 7, 52–56 (1993)
Acknowledgments
This study is supported by the College Scientific Plan Fund of Shandong Education Department (J10LD23) and the Doctoral Startup Foundation of Qilu University of Technology (12042826).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, M., Zhang, Y., Yang, L. et al. Excellent electrochemical performance of homogeneous polypyrrole/graphene composites as electrode material for supercapacitors. J Mater Sci: Mater Electron 26, 485–492 (2015). https://doi.org/10.1007/s10854-014-2425-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-014-2425-x