
Abstract
The phase equilibria and diffusivity of Co–Fe–Mn system were investigated using alloy equilibrium and the diffusion couple technique. Furthermore, thermodynamic properties and diffusion mobilities were assessed using the CALPHAD approach. Isothermal sections of the ternary phase diagrams of the Co–Fe–Mn alloy at 800, 900, and 1000 °C were obtained. The phase boundaries between face-centered cubic (fcc)/A13 were experimentally determined for the first time, whereas those between fcc/body-centered cubic were similar to those reported in previous studies. The thermodynamic parameters of the A13 phase were assessed based on these results. The phase diagrams obtained using the thermodynamic interaction parameters in this study are in accordance with the experimental results. The diffusion paths of the fcc Co–Fe–Mn ternary systems at 900, 1000, and 1100 °C were experimentally determined, and the interdiffusivities were evaluated from the composition-penetration profiles using the Whittle–Green method. The interdiffusion coefficients and penetration profiles were calculated using the assessed atomic mobility parameters. The calculated interdiffusion coefficients and penetration profiles agreed with the experimental ones, validating the values of the optimized atomic mobility parameters.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Most alloys used in daily or industrial applications comprise a solvent metallic element and various constituent elements that tailor the properties of the materials [1]. In 1995, the concept of high-entropy alloys (HEAs) was introduced by Yeh et al. [2]. HEAs can be defined as alloys with five or more elements, each with an atomic percentage ranging from 5 to 35% and an equiatomic size [3, 4]. Although HEAs possess excellent properties, such as continuous steady strain hardening at low temperatures [5], high strength and ductility [6,7,8], excellent thermal stability [8], and high oxidation and corrosion resistance [9, 10], disadvantages like lack of bulk castability owing to the presence of multiple phases and that of experimental data on processing techniques like melting, homogenization, and thermomechanical processing limit the industrial application of HEAs [11, 12]. The time consumed by the trial and error method performed with various compositions of constituent elements to obtain stable HEAs for industrial applications can be reduced by predicting the thermodynamic properties and phase diagrams using the CALPHAD method [13]. Among the four core effects of HEAs, viz. high configurational entropy, severe lattice distortion, cocktail effect, and sluggish diffusion [14], "high configurational entropy" favors the formation of single-phase HEAs; however, it has been proven to be insufficient for overcoming the high enthalpy effect due to the strong interactions between the elements [4, 15]. In contrast, “sluggish diffusion” has garnered increasing attention in recent years because it contributes to high hardness and strength [16], excellent corrosion resistance, and outstanding thermal stability [8]. Sluggish diffusion is an effect of reduced diffusion kinetics compared with those observed for conventional alloys and pure metals. Pickering et al. [17] reported that there remains a lack of clear evidence of sluggish diffusion, and that the existence of the core effect “sluggish diffusion” in HEAs is doubtful. The thermodynamic properties and atomic mobility parameters of HEAs are essential for understanding their phase stabilities and diffusion kinetics.
Accurate information on the thermodynamic and kinetic parameters of suballoys plays a crucial role in predicting the properties of HEAs [18]. There is a lack of experimental information to determine the accurate thermodynamic and kinetic parameters of the Co–Fe–Mn system. The main objective of our study is to create an atomic mobility database for HEAs. This study mainly focused on (1) obtaining accurate thermodynamic parameters by determining the ternary phase diagrams at 800, 900, and 1000 °C, which facilitates diffusion mobility assessment, and (2) determining the ternary atomic mobility parameters in the Co–Fe–Mn system by optimizing the composition-penetration curves of the diffusion couples in the fcc phase region.
Literature review
Co–Mn system
A comprehensive phase diagram was compiled in [19], and a CALPHAD-type assessment of the system was performed by Kaufman et al. [20]. Hasebe et al. [21] assessed this system by considering the magnetic contributions to the Gibbs energy and predicted a two-phase separation in the fcc phase at approximately the Curie temperature. Huang et al. [22] presented a new thermodynamic analysis using the Hillert and Jarl’s modification [23] of the Inden model [24] to determine the magnetic contribution. The thermodynamic parameters assessed by Huang et al. [22] were used in this study.
Ijima [25] and Neumeier et al. [26] performed experimental evaluations to determine the interdiffusivities of Co–Mn alloys using the diffusion couple method. Diffusion couples with concentrations Co/Co-30.28 at.% Mn and Co/Co-51.76 at.% Mn were used to determine the interdiffusivities at temperatures ranging from 860 to 1150 °C. Iijima et al. [27] determined the impurity diffusion coefficients of Mn54 in Co-5.22 at.% Mn and Co-10.24 at.% Mn alloys. Later, Liu et al. [28] fabricated two diffusion couples Co/Co-43.8 at.% Mn annealed at 800 °C for 336 h and Co/Co-49.2 at.% Mn annealed at 1000 °C for 60 h, to verify the existence of a miscibility gap in the Co–Mn phase diagram described by Huang et al. [22]. The interdiffusivities calculated by Liu et al. [28] at 1000 °C and others [26, 27] were used to determine the optimized mobility parameters.
Fe–Mn system
The CALPHAD-type thermodynamic assessment of the Fe–Mn system was performed by several researchers [29,30,31], and experimental information for the phase diagram was compiled in their studies. The thermodynamic parameters assessed by Huang et al. [29] are used in this study as they produce better optimized mobility parameters.
Liu et al. [32] used the empirical expression proposed by Vignes et al. [33] to verify the reliability of diffusivity values. The tracer diffusion coefficients of Fe reported by Million et al. [34] increase with the concentration of Mn \((x_{{{\text{Co}}}}\)) when \(x_{{{\text{Co}}}} > 0.1\). Because the diffusivity of Mn is higher than that of Fe, the interdiffusivities obtained using the tracer diffusion coefficients are higher than those of the experimentally determined values when \(x_{{{\text{Co}}}} > 0.1\). Liu et al. [32] determined the binary mobility parameters in an Fe–Mn system by considering the tracer diffusion coefficients of Fe for \(x_{{{\text{Co}}}} < 0.1\) obtained by Million et al. [33], and all diffusivity values reported by Nohara and Hirano et al. [35].
Co–Fe system
Phase diagrams were experimentally determined, and thermodynamic parameters were assessed by several authors [36,37,38]. The reasons for the further improvement in the phase diagram are described in [39]. The latest thermodynamic parameters assessed by Wang et al. [39] were used in this study. The atomic mobility parameters determined by Gong et al. [40] were validated by comparing the calculated results with experimental data from several sources including [41, 42] and are thus suitable for use in this study.
Co–Fe–Mn system
The experimental phase diagrams were reported only by Köster and Speidel et al. [43]. They determined the phase boundaries of the fcc/body-centered cubic (bcc) two-phase region at 600, 700, and 800 °C by observing the microstructure. However, the experimental data are limited to the Co–Fe side. Huang et al. [44] assessed the thermodynamics of the system based on experimental data obtained by Köster and Speidel et al. [43]. There is no experimental phase diagram information for the entire region, thereby questioning the reliability of the existing thermodynamic parameters. The experimental information and CALPHAD-type assessment of the diffusion mobilities of this system have not been reported thus far.
Experimental procedure
Phase diagram
The alloy compositions listed in Table 1 were used for phase diagram investigation. The raw materials, viz. Co, Fe, and Mn, used to prepare the alloys were 99.9 wt% pure. The alloys were prepared using an arc equipped with two Cu–W electrode bars in a water-cooled Cu hearth under Ar atmosphere. The alloys were remelted more than five times in the hearth to ensure their homogeneity. Alloys with high Mn content were melted in an induction furnace under an Ar atmosphere to suppress the evaporation of Mn. The ingots were cut into blocks with dimensions of 5 × 5 × 8 mm using a disk cutter and sealed in quartz capsules that were evacuated and introduced into an Ar atmosphere. The specimens in the quartz capsules were heat-treated at 800 °C for 1080 h, 900 °C for 576 h, and 1000 °C for 480 h, followed by quenching in ice water. The equilibrium composition of each specimen was measured using an energy-dispersive spectrometer (EDS) after polishing the surface. The microstructures were observed using field-emission scanning electron microscopy.
Diffusion couple
The alloys used for the diffusion couples listed in Table 2 were cast using an induction furnace. Thereafter, the ingots were cut into cylindrical samples with dimensions of φ16 × 5 mm using a disk cutter. Subsequently, the alloys were polished using SiC sandpaper from 80# to 2000# and 0.3 µm diamond particles. A clean surface ensures sound atomic diffusion in the diffusion couple and straight diffusion interface. Diffusion couples were made by compressing the samples with mirror-finished surfaces facing each other at the same temperature as heat-treatment temperature and a ‘zero’ force for approximately 6000 s using Thermecmastor-Z (a dynamic thermomechanical simulation testing machine). Diffusion couples sealed in quartz tubes under an Ar atmosphere were heat-treated at 900 °C for 336 h, 1000 °C for 170 h, and 1100 °C for 72 h. The compositions of the diffusion couples were selected such that their penetration profiles along the diffusion direction, measured by an electron probe microanalyzer (EPMA, JXA-8530F, JEOL), were in the fcc single-phase region and intersected the isothermal sections of the ternary phase diagram.
Model description
Thermodynamic model
Thermo-Calc software [45] was used to perform all thermodynamic calculations and optimizations in this study. The thermodynamic data of the pure elements Co, Fe, and Mn were adopted from the Scientific Group Thermodata Europe (SGTE) data for pure elements [46]. The phases present in the Co–Fe–Mn system in the range of 800–1000 °C were fcc, bcc, and A13. According to the sub-regular solution model, the molar Gibbs energies of the fcc, bcc, and A13 phases are expressed as follows:
where \(X_{i}\) denotes the molar fraction of the element ‘\(i\)’, R is the gas constant, and T is the temperature. The term \({}_{ }^{0} G_{i}^{\phi }\) denotes the molar Gibbs free energy of the pure element ‘\(i\)’ in the ‘\(\phi\)’ phase. The term \({}_{ }^{{{\text{ex}}}} G_{m}^{\phi }\) represents the molar Gibbs excess energy contributed by the nonideal interactions between the elements, and it can be expanded as follows:
where \({}_{ }^{r} L_{i,j}^{\phi }\) denotes the binary thermodynamic interaction parameter.
Mobility and diffusivity
In a ternary system with 1, 2 and 3 as components, the flux of element ‘\(i\)’ (\(i = 1, 2\)) can be computed using Onsager’s derivation of Fick’s second law, as follows [47]:
where \(J_{i}\) denotes the flux of element \(i\). Terms \(D_{11}^{3} {\text{and }}D_{22}^{3}\) represent the main components, whereas \(D_{12}^{3} {\text{and}} D_{21}^{3}\) represent the cross-interdiffusivities of the ternary interdiffusivity matrix. Here, 3 is considered as the dependent concentration variable [48]. The term \(C_{i}\) denotes the molar concentration of element \(i,\) and the units are mol/m3. The term \(x\) denotes the diffusion distance. Assuming that the partial molar volumes of the elements in the fcc phase are constant [49, 50], the relationship between the interdiffusion coefficient (\(D_{ij}^{3}\)) and mobility can be expressed as follows:
where \(\delta_{pi}\) denotes the Kronecker delta (\(\delta_{pi} = 1\) when p = i; otherwise, \(\delta_{pi} = 0\)), \(X_{i}\) and \(\mu_{i}\) are the mole fraction and chemical potential of element \(i\) (i = 1, 2, 3), respectively, and \(\frac{{\partial \mu_{p} }}{{\partial X_{j} }}\) is the partial derivative of the chemical potential of element i with respect to the molar fraction of element j (j = 1, 2, 3), which can be easily computed [51] from the thermodynamic data of the system. The second term in Eq. (4) represents the vacancy-wind effect [52]: s = (0, 1). When s = 1, the vacancy-wind effect was considered, whereas when s = 0, the effect was ignored. The value of \(A_{0}\) depends on the crystal structure and is equal to 7.15 for fcc crystal structures [53]. The vacancy-wind effect was considered in this study at all temperatures while optimizing the mobility interaction parameters. The term \(M_{i}\) represents the mobility of element i, and according to the absolute rate theory, it can be expressed as follows:
where \(M_{i}^{0}\) represents the effect of the atomic jump distance and frequency, which are assumed to depend exponentially on the composition; R is the gas constant, and T is the temperature. The term \(Q_{i}\) represents the diffusion activation energy of component \(i\). The temperature and concentration-dependent \(\varphi_{i}\) values are given by \(\varphi_{i} = Q_{i} - {\text{R}}T\ln M_{i}^{0}\). The direct relationship between the tracer diffusivity (\(D_{i}^{*}\)) and mobility (\(M_{i}\)) is expressed as follows:
Ågren et al. [54,55,56] expressed the composition and temperature dependence of \(\varphi_{i} \) using the Redlich–Kister polynomial in Eq. (7) as follows [57]:
where \(\varphi_{i}^{l} {\text{and }}{}_{ }^{r} \varphi_{i}^{p,q}\) denote the mobility parameters, which exhibit a notable linear relationship with temperature.
The mobility parameters can be determined by minimizing the error between the calculated and experimentally obtained diffusion data, such as interdiffusivities, tracer diffusivities, and composition profiles. In this study, the atomic mobility parameters in a ternary Co–Fe–Mn system were determined by minimizing the variance between the experimental and calculated fluxes.
The experimental interdiffusion flux can be obtained from the experimental data and the Whittle–Green relation as follows [58]:
where \(J_{i}^{{{\text{exp}}}}\) denotes the experimental interdiffusion flux of element \(i\) at position \(x\) obtained from the experimental diffusion profiles. The term t represents the diffusion time, whereas \(C_{i}^{ + }\) and \(C_{i}^{ - }\) denote the molar concentrations of element i at the right and left ends of the semi-infinite diffusion couple, respectively. Whittle and Green et al. [58] introduced a normalized concentration variable (\(Y_{i}\)), as expressed in Eq. (9), to reduce or avoid errors caused by using the position of the Matano plane in the Boltzmann–Matano method:
The four interdiffusivities (\(D_{{{\text{CoCo}}}}^{{{\text{Mn}}}} , D_{{{\text{CoFe}}}}^{{{\text{Mn}}}} , D_{{{\text{FeFe}}}}^{{{\text{Mn}}}} , \,{\text{and}} \,D_{{{\text{FeCo}}}}^{{{\text{Mn}}}}\)) at the cross-point of the composition profiles of the ternary isothermal section of the Co–Fe–Mn system were determined using the Whittle–Green method [58]. The following two equations can be solved for two intersecting diffusion couples to obtain the values of the four interdiffusion coefficients:
Results and discussion
Microstructure observation and thermodynamic assessment of the Co–Fe–Mn system
The equilibrium compositions of the annealed alloys and their respective phases are summarized in Table 1. The backscattered electron (BSE) images of the Co7.0Fe30.1Mn62.9, and Co46.6Fe48.9Mn4.5 alloys annealed at 800 °C for 1080 h are shown in Fig. 1 as typical microstructures of two-phase alloys in the Co–Fe–Mn system. EDS analysis indicated that the light gray, dark gray, and black regions in Fig. 1a represent the A13, fcc, and oxide phases, respectively. The oxides were due to the high Mn content in the fcc and A13 two-phase regions. These oxides have no influence on the equilibrium of the phases because the solubilities of oxygen in the fcc and A13 phases are almost zero according to the Mn–O phase diagram [59]. In Fig. 1b, the darker and lighter regions are identified as the bcc and fcc phases, respectively, from the EDS analysis. The white regions along the boundaries of the fcc region also correspond to the fcc phases with different orientations. Figure 2 shows the isothermal cross sections of the ternary phase diagram at 800, 900, and 1000 °C. The dashed lines in Fig. 2 indicate the phase boundaries obtained by using the selected binary assessments [22, 29, 39] in Thermo-Calc. In the Mn-rich corner, the calculated phase boundaries of fcc/A13 deviated from the experimental data, whereas those of fcc/bcc were consistent with the experimental data. According to these results, the thermodynamic parameters of the bcc and fcc phases do not require new ternary parameters; only those of A13 were assessed.

Typical equilibrium microstructures (BSE images) of; (a) Co7.0Fe30.1Mn62.9 alloy with A13 and fcc phases, and (b) Co46.6Fe48.9Mn4.5 alloy with bcc and fcc phases at 800 °C

Isothermal sections of the ternary phase diagram of the Co–Fe–Mn system at (a) 800, (b) 900, and (c) 1000 °C. Dashed lines represent the phase boundaries that were obtained from the selected binary thermodynamic assessments [22, 29, 39], whereas phase boundaries in black that can be observed agreeing with the experimental data were calculated with the optimized thermodynamic parameter as \({}_{ }^{0} L_{{{\text{Co,}}\,{\text{Fe}}}}^{A13} = - 13365 + 5\,{\text{T}}\)
The optimized parameters are listed in Table 3. The thermodynamic interaction parameters of the binary subsystems Co–Fe, Co–Mn, and Fe–Mn were acquired from previous studies by Wang [39] and Huang et al. [22, 29]. The effect of the ternary interactions between Co, Fe, and Mn is considered negligible because these three elements are adjacent to each other on the periodic table and exhibit almost equal atomic sizes. Huang et al. [44] estimated the binary interaction parameter between Co and Fe in the A13 phase as − 10 kJ/mol. However, this interaction parameter could not reproduce the experimental results. Thus, using the experimental results of this study, the thermodynamic interaction parameter of Co–Fe in the A13 phase was modified as \({}_{ }^{0} L_{{{\text{CoFe}}}}^{{{\text{A1}}3}} = - 13365 + 5T\) to adjust the equilibrium region in the Mn-rich region. The calculated phase diagrams obtained after modifying the binary interaction parameter (\({}_{ }^{0} L_{{{\text{CoFe}}}}^{{{\text{A}}13}}\)) are represented by solid lines in Fig. 2. The excellent agreement between the results of this study and the experimental data validates the values of the thermodynamic parameters of the ternary Co–Fe–Mn system.
Results of diffusion couples and discussion on the atomic mobility assessments
Interdiffusivities obtained from experimental concentration profiles.
The interdiffusivities were evaluated from the experimental concentration profiles measured by EPMA. Fitting the experimental concentration profiles before determining the interdiffusivities ensures accurate determination of the slope [60] in Eqs. (10, 11). The measured experimental composition profiles of the diffusion couples are shown in Fig. 7. These were fitted using a cubic spline function [61,62,63,64]. After fitting, four interdiffusivities were calculated using the Whittle–Green method, as described in “Mobility and diffusivity” section, at the intersection points of the composition profiles at three different temperatures. The obtained interdiffusivities are listed in Table 4. According to Kirkaldy [65], the values of the four interdiffusivities in a ternary system can be validated using the three thermodynamic constraints mentioned in Eqs. (12–14).
It was observed that all the ternary interdiffusivities calculated in this study satisfied these three constraints.
Assessment method of mobility parameters
The self-atomic mobility parameters of fcc Co, Fe, and Mn determined by Zhang [66], Jönsson [67], and Liu et al. [32], respectively, were adopted in this study. The impurity atomic mobility parameters of Co in fcc-Fe [68], Co in fcc-Mn [28], Fe in fcc-Co [40], Fe in fcc-Mn [32], Mn in fcc-Co [28], and Mn in fcc-Fe [32] obtained from previous studies were also utilized in this study to determine the interaction parameters of mobility in fcc Co–Fe–Mn ternary systems. In this study, ternary \(\varphi_{{{\text{Co}}}}^{{\text{Fe,Mn}}} , \varphi_{{{\text{Fe}}}}^{{\text{Co,Mn}}} , \,\,{\text{and}}\,\, \varphi_{{{\text{Mn}}}}^{{\text{Co,Fe}}} \) mobility parameters were determined to provide excellent agreement between the ternary Co–Fe–Mn experimental and calculated concentration profiles. The binary \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}} \,\,{\text{ and}} \,\,\varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) parameters were also evaluated because the parameters in [28] could not reasonably reproduce the ternary concentration profiles. We used only flux to evaluate the mobility parameters. A customized Python program was developed to optimize the mobility parameters. The optimized atomic mobilities in the fcc Co–Fe–Mn system were obtained by minimizing the error of the flux, that is, the sum of the square of the differences between the experimental (\(J_{{{\text{exp}}}} ) \,{\text{and}}\,{\text{calculated}}\,\left( {J_{{{\text{calc}}}} } \right)\) \({\text{fluxes}}\) at each point along the diffusion distance, as expressed in the following equation:
where \(J_{{{\text{exp}}}}\) is calculated using Eq. (8). The term \(J_{{{\text{calc}}}}\) was calculated using Eqs. (3–7), where the composition gradients were obtained from the diffusion profiles and the chemical potential was calculated using the parameters listed in Table 3. Other than flux, interdiffusion coefficients [69] or calculated concentration profiles are usually used to determine the mobility parameters [52]. One of the advantages of solving this optimization problem is that the flux at each point of the experimental diffusion profiles can be used for optimization, unlike interdiffusion coefficients, which use only the data at the intersection points of the experimental concentration profiles. Also, concentration profiles measured by EPMA were scattered and did not provide reliable values of calculated interdiffusion coefficients. Integral of the concentration profile, which is used to calculate flux, is less scattered than the slope. Another advantage is that the time required for this optimization is shorter than that required to fit the calculated diffusion profiles. The availability of abundant experimental data increased the accuracy of the optimized parameters. Hence, the flux values were selected to solve the optimization problem using the Nelder–Mead method [70, 71]. The optimized mobility interaction parameters were established by adjusting the initial simplex, considering the error function values after minimization, and comparing the calculated diffusion profiles with experimental composition curves.
Assessment results: atomic mobility parameters
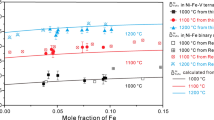
The optimized mobility parameters of the fcc Co–Fe–Mn ternary system are listed in Table 5. It is observed from Table 5 that previously reported values of binary \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}} \,\,{\text{and}}\,\, \varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) mobility parameters were modified, whereas the ternary \(\varphi_{{{\text{Co}}}}^{{\text{Fe,Mn}}} , \varphi_{{{\text{Fe}}}}^{{\text{Co,Mn}}} , \,{\text{and}}\, \varphi_{{{\text{Mn}}}}^{{\text{Co,Fe}}} \) mobility parameters were determined in this study. The modified binary \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}} \,\,\,{\text{and}}\,\, \varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) mobility parameters listed in Table 5 were verified by comparing the experimental Co–Mn diffusion profiles [28] with the concentration profiles calculated by DICTRA using the modified parameters, as shown in Fig. 3. The modified parameters accurately reproduced concentration profiles. Figure 4 illustrates a comparison between the calculated interdiffusivities in a binary Co–Mn system and the experimental results obtained by Iijima et al. [25]. It is observed that at low Mn contents, there is excellent agreement between the experimental and calculated results. With an increase in Mn content, there was a slight deviation from the experimental data. \(\varphi_{{{\text{Fe}}}}^{{{\text{Mn}}}}\) was empirically calculated by Liu et al. and \(\varphi_{{{\text{Mn}}}}^{{{\text{Mn}}}}\) was assumed to be equal with \( \varphi_{{{\text{Fe}}}}^{{{\text{Mn}}}}\) [32]. Considering the fact that Mn diffuses faster than Fe at Mn concentration around 0.38 [32], the assumption is not valid and \(\varphi_{{{\text{Mn}}}}^{{{\text{Mn}}}}\) should be higher than reported value around Mn concentration of 0.38. Since fcc-Mn is metastable, self and impurity diffusion in fcc-Mn are not yet reported clearly. The deviations in Fig. 4 around and above Mn concentration of 0.3 might be due to the lower estimated value of \(\varphi_{{{\text{Mn}}}}^{{{\text{Mn}}}}\). But our assessed parameters are still valid at Mn concentration lower than 0.3. In Fig. 5, the experimentally determined tracer diffusivities [27] are compared with the calculated tracer diffusivities obtained in this study and a previous study [27]. The tracer diffusivities calculated using the values from the previous study significantly deviated from the experimental data, whereas those calculated using the present study agreed with the experimental data. Liu et al. [28] determined the binary \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}} { }\,\,{\text{and}}\,\, \varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) mobility parameters using the values of interdiffusivities [25,26,27] in the PARROT module of the DICTRA software package. A linear relationship between the atomic mobility parameters \(\varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) and temperature was considered, whereas \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}}\)’s was not [28]. As mentioned earlier, the interdiffusivities depend on the slopes of the composition curves at the point of interest, and the slopes are related to the annealing time. The analysis of more binary fcc Co–Mn diffusion couples with longer heat-treatment times is expected to provide accurate temperature-dependent atomic mobility parameters. Considering that the experimental composition profiles in this study were analyzed at three temperatures with considerable heat-treatment periods, and the composition profiles of the Co–Mn binary diffusion couples were reproduced well using the mobility parameters obtained in this study, the values of the binary \(\varphi_{{{\text{Co}}}}^{{\text{Co,Mn}}} \,\,{\text{and}}\,\, \varphi_{{{\text{Mn}}}}^{{\text{Mn,Co}}}\) mobility parameters obtained in this study are thus acceptable.

Comparison between model-calculated concentration profiles and experimental data [28] in diffusion couples with varying concentrations; (a) Co/Co-43.8 Mn (at.%) annealed at 800 °C for 336 h, and (b) Co/Co-49.2 Mn (at.%) annealed at 1000 °C for 60 h

Comparison between the experimentally determined interdiffusivities [25] in binary Co–Mn alloy and calculated interdiffusivities using modified Co–Mn binary mobility interaction parameters of this study

The values of the ternary \(\varphi_{{{\text{Co}}}}^{{\text{Fe,Mn}}} ,\, \varphi_{{{\text{Fe}}}}^{{\text{Co,Mn}}} , \,\,{\text{and}} \,\,\varphi_{{{\text{Mn}}}}^{{\text{Co,Fe}}} \) mobility parameters listed in Table 5 can be validated by comparing the numerically simulated concentration profiles with the experimental concentration profiles and diffusion paths on the ternary Co–Fe–Mn diffusion couples. All the ternary calculated profiles in this study were determined by solving the partial differential equations (PDEs) in Eq. (3), that is, Onsager’s formula in a customized Python program. This method of calculating the concentration profile across the diffusion distance by solving PDEs is known as forward method [51]. Figure 6 shows the comparison of composition profiles calculated by considering and not considering vacancy-wind effect with the experimental profiles. Solid black lines and dashed black lines indicated the calculated concentration profiles with and without considering vacancy-wind, respectively. Comparison between concentration profiles calculated by with and without considering vacancy-wind effect is shown in Fig. 6 to explain the notable deviation between the experimental and simulated results for the Fe–Mn side. In particular, as shown in Fig. 6c, the diffusion couples DC2 and DC4 deviate significantly from the experimental data on the Fe–Mn side of the isothermal section of the phase diagram at 1100 °C. The experimental diffusion paths sharply curved after following the Fe–Mn edge, in relation to the calculated lines. These results suggest that the diffusivity of Mn in the Fe–Mn alloy was significantly higher than the evaluated value. Nohara and Hirano et al. [34] reported the interdiffusivities and self-diffusivities of the Fe–Mn system. The interdiffusivities obtained from the diffusion couple were higher than those estimated from Darken equations [72] using self-diffusivities. They concluded that the origin of the difference was the vacancy-wind effect from an analysis using Manning’s equation [53]. The diffusivity parameters reported by Liu et al. [32] for the Fe–Mn system adopted in this study were determined from the self-diffusivity data reported by Nohara and Hirano [35]. Although the vacancy-wind effect was considered in our study to determine the mobility parameters, calculated profiles cannot fit well with the experimental profiles on the Fe–Mn side. This is due to the lower number of experimental data on the Fe–Mn side available for optimization. As can be seen from Fig. 6a, b, deviation between the two calculated profiles increase with increase in ‘Mn’ content in the diffusion couple. By considering the vacancy-wind effect in the ternary Co–Fe–Mn system, the agreement between the experimental and calculated concentration profiles has considerably increased at 900 and 1000 °C. In Fig. 6c, the calculated concentration profiles with and without considering vacancy-wind effect are almost overlapping. Therefore, to increase the alignment with experimental data on the Fe–Mn side of the Co–Fe–Mn ternary phase diagram in Fig. 6c, further investigation of the kinetics of the binary Fe–Mn system is required.

Comparison between the concentration profiles obtained by solving the forward problem with and without considering vacancy-wind effect and the experimental diffusion profiles (obtained in this study) on isothermal sections of the ternary Co–Fe–Mn phase diagram at (a) 900, (b) 1000, and (c) 1100 °C
Excellent agreement between the experimental and numerically simulated concentration penetration profiles at three temperature levels (900, 1000, and 1100 °C) across the diffusion distance is observed in Fig. 7, supporting the reliability of the atomic mobility parameter values obtained in this study.

Comparison between the concentration profiles obtained by solving the forward problem and the experimental diffusion profiles (obtained in this study) in (a) DC1, (b) DC2, and (c) DC3 annealed at 900 °C for 336 h, (d) DC1, (e) DC2, and (f) DC3 annealed at 1000 °C for 170 h, and (g) DC1, (h) DC2, and (i) DC4 annealed at 1100 °C for 72 h. Black lines indicate the calculated concentration profiles, whereas the colored scatter plot represents the experimental data obtained in this study
Furthermore, the interdiffusivities determined by Eq. (4), using the optimized mobility parameters, were compared with the interdiffusivities obtained by the Whittle–Green method using the experimental diffusion profiles shown in Fig. 8. All the values were close to the x = y line, indicating the validity of the obtained parameters.

Comparison between interdiffusion coefficients (y-axis) and experimental interdiffusion coefficients (x-axis)
Conclusions
-
(a)
The phase equilibria of the Co–Fe–Mn system at 800, 900, and 1000 °C were experimentally determined through EDS analysis of the equilibrated two-phase alloys. The Mn-rich corner is experimentally verified for the first time. The obtained experimental phase diagrams were different for the Mn-rich corner compared with the calculated phase diagram using the parameters reported by Huang et al. [44].
-
(b)
The thermodynamic parameters of the ternary Co–Fe–Mn system were assessed using the experimental data. The phase diagram obtained using the new set of parameters is consistent with the experimental data.
-
(c)
The interdiffusivities and diffusion paths at 900, 1000, and 1100 °C in the fcc single-phase region of the Co–Fe–Mn system were determined using the diffusion couple technique for the first time. Furthermore, atomic mobility assessment of the ternary Co–Fe–Mn system was performed using the diffusion flux determined from the experimental concentration profiles across the diffusion interface. The atomic mobility parameters obtained in this study were verified by comparing calculated and experimental concentration profiles. The concentration profiles of the binary Co–Mn system were well reproduced using the modified mobility parameters adopted in this study. The interdiffusivities in the ternary Co–Fe–Mn system were obtained using the diffusion couple and Whittle–Green methods, and they agreed well with the calculated interdiffusivities using the assessed mobility parameters, which also indicated the reliability of the assessed parameters.
The thermodynamic and kinetic assessments in this study will facilitate the development of thermodynamic and kinetic databases for HEAs.
References
Davis JR (Ed.) (1990) Metals Handbook. In: 10th ed., ASM International, Metals Park
Yeh J-W (2006) Recent progress in high-entropy alloys. Ann Chim Sci Des Matériaux 31:633–648. https://doi.org/10.3166/acsm.31.633-648
Yeh JW, Chen SK, Lin SJ, Gan JY, Chin TS, Shun TT, Tsau CH, Chang SY (2004) Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv Eng Mater 6:299–303. https://doi.org/10.1002/adem.200300567
Murty SRBS, Yeh JW (2014) High-entropy alloys. Butterworth-Heinemann, Oxford. https://doi.org/10.1016/C2013-0-14235-3
Li Z, Zhao S, Ritchie RO, Meyers MA (2019) Mechanical properties of high-entropy alloys with emphasis on face-centered cubic alloys. Prog Mater Sci 102:296–345. https://doi.org/10.1016/j.pmatsci.2018.12.003
Li D, Li C, Feng T, Zhang Y, Sha G, Lewandowski JJ, Liaw PK, Zhang Y (2017) High-entropy Al0.3CoCrFeNi alloy fibers with high tensile strength and ductility at ambient and cryogenic temperatures. Acta Mater 123:285–294. https://doi.org/10.1016/j.actamat.2016.10.038
Liu WH, Lu ZP, He JY, Luan JH, Wang ZJ, Liu B, Liu Y, Chen MW, Liu CT (2016) Ductile CoCrFeNiMox high entropy alloys strengthened by hard intermetallic phases. Acta Mater 116:332–342. https://doi.org/10.1016/j.actamat.2016.06.063
Zhao YY, Chen HW, Lu ZP, Nieh TG (2018) Thermal stability and coarsening of coherent particles in a precipitation-hardened (NiCoFeCr)94Ti2Al4 high-entropy alloy. Acta Mater 147:184–194. https://doi.org/10.1016/j.actamat.2018.01.049
Raza A, Abdulahad S, Kang B, Ryu HJ, Hong SH (2019) Corrosion resistance of weight reduced AlxCrFeMoV high entropy alloys. Appl Surf Sci 485:368–374. https://doi.org/10.1016/j.apsusc.2019.03.173
Liu YY, Chen Z, Shi JC, Wang ZY, Zhang JY (2019) The effect of Al content on microstructures and comprehensive properties in AlxCoCrCuFeNi high entropy alloys. Vacuum 161:143–149. https://doi.org/10.1016/j.vacuum.2018.12.009
Jablonski PD, Licavoli JJ, Gao MC, Hawk JA (2015) Manufacturing of high entropy alloys. Jom 67:2278–2287. https://doi.org/10.1007/s11837-015-1540-3
Lu Y, Dong Y, Jiang H, Wang Z, Cao Z, Guo S, Wang T, Li T, Liaw PK (2020) Promising properties and future trend of eutectic high entropy alloys. Scr Mater 187:202–209. https://doi.org/10.1016/j.scriptamat.2020.06.022
Mao H, Chen HL, Chen Q (2017) TCHEA1: a thermodynamic database not limited for “high entropy” alloys. J Phase Equilibria Diffus 38:353–368. https://doi.org/10.1007/s11669-017-0570-7
Miracle DB (2017) High-entropy alloys: a current evaluation of founding ideas and core effects and exploring “nonlinear alloys.” Jom 69:2130–2136. https://doi.org/10.1007/s11837-017-2527-z
Senkov ON, Miller JD, Miracle DB, Woodward C (2015) Accelerated exploration of multi-principal element alloys with solid solution phases. Nat Commun 6:1–10. https://doi.org/10.1038/ncomms7529
Tong CJ, Chen MR, Chen SK, Yeh JW, Shun TT, Lin SJ, Chang SY (2005) Chang, Mechanical performance of the AlxCoCrCuFeNi high-entropy alloy system with multiprincipal elements. Metall Mater Trans A Phys Metall Mater Sci 36:1263–1271. https://doi.org/10.1007/s11661-005-0218-9
Pickering EJ, Jones NG (2016) High-entropy alloys: a critical assessment of their founding principles and future prospects. Int Mater Rev 61:183–202. https://doi.org/10.1080/09506608.2016.1180020
Abrahams K, Zomorodpoosh S, Khorasgani AR, Roslyakova I, Steinbach I, Kundin J (2021) Automated assessment of a kinetic database for fcc Co-Cr-Fe-Mn-Ni high entropy alloys. Model Simul Mater Sci Eng. https://doi.org/10.1088/1361-651X/abf62b
Ishida K, Nishizawa T (1990) The Co-Mn (Cobalt-Manganese) system. Bull Alloy Phase Diagr 11:125–137. https://doi.org/10.1007/BF02841695
Kaufman L (1979) Coupled phase diagrams and thermochemical data for transition metal binary systems-VI. Calphad 3:45–76. https://doi.org/10.1016/0364-5916(79)90020-8
Hasebe M, Oikawa K, Nishizawa T (1982) Computer calculation of phase diagrams of Co-Cr and Co-Mn systems. J Jpn Inst Met 46:577–583. https://doi.org/10.2320/jinstmet1952.46.6_577
Huang W (1989) An assessment of the Co-Mn system. Calphad 13:231–242. https://doi.org/10.1016/0364-5916(89)90003-5
Hillert M, Jarl M (1978) A model for alloying in ferromagnetic metals. Calphad 2:227–238. https://doi.org/10.1016/0364-5916(78)90011-1
Inden G (1976) Project meeting calphad V, Ch. 111. 4: 1–13
Iijima H, Taguchi O, Hirano KI (1977) Interdiffusion in Co-Mn alloys. Metall Trans A 8:991–995. https://doi.org/10.1007/BF02661584
Neumeier S, Rehman HU, Neuner J, Zenk CH, Michel S, Schuwalow S, Rogal J, Drautz R, Göken M (2016) Diffusion of solutes in FCC Cobalt investigated by diffusion couples and first principles kinetic Monte Carlo. Acta Mater 106:304–312. https://doi.org/10.1016/j.actamat.2016.01.028
Iijima Y, Hirano K-I, Taguchi O (1977) Diffusion of manganese in cobalt and cobalt-manganese alloys. Philos Mag 35:229–244. https://doi.org/10.1080/14786437708235985
Liu H, Liu Y, Du Y, Min Q, Zhang J, Liu S (2019) Atomic mobilities and diffusivities in fcc Co–X (X = Mn, Pt and Re) alloys. Calphad Comput Coupling Phase Diagrams Thermochem 64:306–312. https://doi.org/10.1016/j.calphad.2019.01.003
Huang W (1989) An assessment of the Fe-Mn system. Calphad 13:243–252. https://doi.org/10.1016/0364-5916(89)90003-5
Witusiewicz VT, Sommer F, Mittemeijer EJ (2004) Reevaluation of the Fe-Mn phase diagram. J Phase Equilibria Diffus 25:346–354. https://doi.org/10.1361/15477030420115
Li L, Hsu TY (1997) Gibbs free energy evaluation of the fcc(γ) and hcp(ε) phases in Fe-Mn-Si alloys. Calphad 21:443–448. https://doi.org/10.1016/S0364-5916(97)00044-8
Liu Y, Zhang L, Du Y, Yu D, Liang D (2009) Atomic mobilities, uphill diffusion and proeutectic ferrite growth in Fe–Mn–C alloys. Calphad 33:614–623. https://doi.org/10.1016/j.calphad.2009.07.002
Vignes A, Birchenall C (1968) Concentration dependence of the interdiffusion coefficient in binary metallic solid solution. Acta Metall 16:1117–1125. https://doi.org/10.1016/0001-6160(68)90047-3
Million B, Rüzickova J, Kucera J (1993) Volume Self-diffusion of Fe 59 in Face-centered Cubic Fe–Mn Alloys/Volumenselbstdiffusion von Fe 59 in kubisch flächenzentrierten Fe–Mn-Legierungen. Int J Mater Res 84:687–689. https://doi.org/10.1515/ijmr-1993-841006
Nohara K, Hirano K (1973) Self-diffusion and interdiffusion in γ solid solutions of the iron-manganese system. J Japan Inst Met 37:51–61. https://doi.org/10.2320/jinstmet1952.37.1_51
Guillermet AF (1987) Critical evaluation of the thermodynamic properties of the iron-cobalt system. High Temp Press 19:477–499
Ohnuma I, Enoki H, Ikeda O, Kainuma R, Ohtani H, Sundman B, Ishida K (2002) Phase equilibria in the Fe-Co binary system. Acta Mater 50:379–393. https://doi.org/10.1016/S1359-6454(01)00337-8
Turchanin MA, Dreval LA, Abdulov AR, Agraval PG (2011) Mixing enthalpies of liquid alloys and thermodynamic assessment of the Cu-Fe-Co system. Powder Metall Met Ceram 50:98–116. https://doi.org/10.1007/s11106-011-9307-z
Wang J, Lu XG, Zhu N, Zheng W (2017) Thermodynamic and diffusion kinetic studies of the Fe-Co system. Calphad Comput Coupling Phase Diagrams Thermochem 58:82–100. https://doi.org/10.1016/j.calphad.2017.06.001
Gong XM, Lu XG, He YL (2014) Study on atomic mobility and molar volume for FCC Fe-Co phase. Adv Mater Res 936:1201–1208. https://doi.org/10.4028/www.scientific.net/AMR.936.1201
Badia M, Vignes A (1969) Interdiffusion and the Kirkendall effect in binary alloys. Mem Sci Rev Met 66:915–927
Hirano K, Iijima Y, Araki K, Homma H (1977) Interdiffusion in iron-cobalt alloys. Trans Iron Steel Inst Japan 17:194–203. https://doi.org/10.2355/isijinternational1966.17.194
Köster W, Speidel M (1962) Über die Gleichgewichtseinstellung im Dreistoffsystem Eisen-Kobalt-Mangan. Arch Für Das Eisenhüttenwes 33:873–876. https://doi.org/10.1002/srin.196203407
Huang W (1990) Thermodynamics of the Co-Fe-Mn system. Calphad 14:11–22. https://doi.org/10.1016/0364-5916(90)90035-X
Andersson J-O, Helander T, Höglund L, Shi P, Sundman B (2002) Thermo-Calc & DICTRA, computational tools for materials science. Calphad 26:273–312. https://doi.org/10.1016/S0364-5916(02)00037-8
Dinsdale AT (1991) SGTE data for pure elements. Calphad 15:317–425. https://doi.org/10.1016/0364-5916(91)90030-N
Onsager L (1931) Reciprocal relations in irreversible processes: I. Phys Rev 37:405–426. https://doi.org/10.1103/PhysRev.37.405
Dayananda MA (1990) 6.2 Solutions of diffusion equations for constant ternary interdiffusion coefficients. In: Mehrer H (ed) Diffusion in solid metals and alloys. Springer, Berlin, pp 372–375. https://doi.org/10.1007/10390457_64
Kattner UR, Campbell CE (2009) Invited review: Modelling of thermodynamics and diffusion in multicomponent systems. Mater Sci Technol 25:443–459. https://doi.org/10.1179/174328408X372001
Andersson J, Ågren J (1992) Models for numerical treatment of multicomponent diffusion in simple phases. J Appl Phys 72:1350–1355. https://doi.org/10.1063/1.351745
Zhong J, Chen W, Zhang L (2018) HitDIC: a free-accessible code for high-throughput determination of interdiffusion coefficients in single solution phase. Calphad Comput Coupling Phase Diagrams Thermochem 60:177–190. https://doi.org/10.1016/j.calphad.2017.12.004
Chen W, Zhang L, Du Y, Tang C, Huang B (2014) A pragmatic method to determine the composition-dependent interdiffusivities in ternary systems by using a single diffusion couple. Scr Mater 90–91:53–56. https://doi.org/10.1016/j.scriptamat.2014.07.016
Manning JR (1967) Diffusion and the Kirkendall shift in binary alloys. Acta Metall 15:817–826. https://doi.org/10.1016/0001-6160(67)90363-X
Ågren J (1982) Numerical treatment of diffusional reactions in multicomponent alloys. J Phys Chem Solids 43:385–391. https://doi.org/10.1016/0022-3697(82)90209-8
Ågren J (1982) Computer simulations of the austenite/ferrite diffusional transformations in low alloyed steels. Acta Metall 30:841–851. https://doi.org/10.1016/0001-6160(82)90082-7
Ågren J (1992) Computer simulations of diffusional reactions in complex steels. ISIJ Int 32:291–296. https://doi.org/10.2355/isijinternational.32.291
Redlich O, Kister AT (1948) Algebraic representation of thermodynamic properties and the classification of solutions. Ind Eng Chem 40:345–348. https://doi.org/10.1021/ie50458a036
Whittle DP, Green A (1974) The measurement of diffusion coefficients in ternary systems. Scr Metall 8:883–884. https://doi.org/10.1016/0036-9748(74)90311-1
Wang M, Sundman B (1992) Thermodynamic assessment of the Mn-O system. Metall Trans B 23:821–831. https://doi.org/10.1007/BF02656461
Wei M, Zhang L (2018) Application of distribution functions in accurate determination of interdiffusion coefficients. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-22992-5
Dierckx P (1975) An algorithm for smoothing, differentiation and integration of experimental data using spline functions. J Comput Appl Math 1:165–184. https://doi.org/10.1016/0771-050X(75)90034-0
Dierckx P (1981) An improved algorithm for curve fitting with spline functions. TW Reports
Dierckx P (1982) A fast algorithm for smoothing data on a rectangular grid while using spline functions. SIAM J Numer Anal 19:1286–1304. https://doi.org/10.1137/0719093
Dierckx P (1995) Curve and surface fitting with splines. Oxford University Press, Oxford
Kirkaldy JS, Weichert D, Haq Z-U-H (1963) Diffusion in multicomponent metallic systems: Vi. Some thermodynamic properties of the d matrix and the corresponding solutions of the diffusion equations. Can J Phys 41:2166–2173. https://doi.org/10.1139/p63-211
Zhang L, Du Y, Ouyang Y, Xu H, Lu XG, Liu Y, Kong Y, Wang J (2008) Atomic mobilities, diffusivities and simulation of diffusion growth in the Co-Si system. Acta Mater 56:3940–3950. https://doi.org/10.1016/j.actamat.2008.04.017
Jönsson B (1994) Mobilities in Fe-Ni alloys: assessment of the mobilities of Fe and Ni in fcc Fe-Ni alloys. Scand J Metall 23:201–208
Badia M, Vignes A (1969) Iron, nickel and cobalt diffusion in transition metals of iron group. Acta Metall 17:177–187. https://doi.org/10.1016/0001-6160(69)90138-2
Cui YW, Jiang M, Ohnuma I, Oikawa K, Kainuma R, Ishida K (2008) Computational study of atomic mobility for fcc phase of Co-Fe and Co-Ni binaries. J Phase Equilibria Diffus 29:2–10. https://doi.org/10.1007/s11669-007-9238-z
Nelder JA, Mead R (1965) A simplex method for function minimization. Comput J 7:308–313. https://doi.org/10.1093/comjnl/7.4.308
Gao F, Han L (2012) Implementing the Nelder-Mead simplex algorithm with adaptive parameters. Comput Optim Appl 51:259–277. https://doi.org/10.1007/s10589-010-9329-3
Darken LS (1948) Diffusion, mobility and their interrelation through free energy in binary metallic systems. Trans Aime 175:184–201
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “High Entropy Alloys—Science of New Class of Materials Based on Elemental Multiplicity and Heterogeneity” (JSPS KAKENHI Grant Number 18H05454) and a Grant-in-Aid for Scientific Research (B) “Interfacial control of Co-based superalloy for new forging process” (JSPS KAKENHI Grant Number 18H01742). We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Contributions
SPP contributed to methodology, investigation, software, validation, visualization, formal analysis, and writing—original draft. NU contributed to formal analysis, resources, and writing—review and editing. KO contributed to conceptualization, funding acquisition, methodology, resources, writing—review and editing, and supervision. YT contributed to formal analysis and writing—review and editing. TK contributed to formal analysis, funding acquisition, resources, and writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling Editor: P. Nash.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pendem, S.P., Ueshima, N., Oikawa, K. et al. Thermodynamic and atomic mobility assessment of the Co–Fe–Mn system. J Mater Sci 57, 15999–16015 (2022). https://doi.org/10.1007/s10853-022-07612-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-07612-y