
Abstract
Nanocrystalline metals are generally unstable due to a large volume fraction of high-energy grain boundaries associated with a small grain size. Preferential dopant segregation to the high-energy grain boundaries is observed to enhance the stability of the material’s microstructure by minimizing its energy. Nanocrystalline aluminum-dopant systems were evaluated for thermodynamic stability against grain growth and phase precipitation via the mechanism of grain boundary segregation according to a modified regular nanocrystalline solution model. Fifty-one potential dopant elements have been evaluated for their efficacy in stabilizing nanostructures with three potential candidates, magnesium, lanthanum, and silicon, identified possessing the characteristics to promote grain boundary segregation and a state of thermodynamic stability in aluminum’s nanocrystalline regime. The minimum dopant content required to achieve nanocrystalline microstructure stability is identified for each of the three candidate elements. Beyond this minimum content, further addition of the dopant elements decreased the final microstructure’s stability with no effects on the existence of a stable nanocrystalline state.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanocrystalline metals are comprised of ultra-fine grains with diameters of typically less than 100 nm [1, 2]. Due to the small size of the constituent grains, large fractions of atoms are positioned at or near grain boundaries, compared to conventional polycrystalline metals with coarse grains [2, 3]. As a result of this structure, nanocrystalline metals exhibit superior mechanical properties as compared to their polycrystalline counterparts. The extraordinary mechanical properties of nanocrystalline metals include high ductility at room temperature, high hardness and high strength [3,4,5]. The hardness of nanocrystalline metals has been observed to be up to seven times higher than in coarse-grained materials [4]. Additionally, the yield strength of nanocrystalline metals can be up to ten times higher than of coarse-grained materials [6,7,8,9]. These unique qualities are attributed to the large fraction of interfacial materials present within the grain boundaries. It is thus apparent that the nanoscale grain size in nanocrystalline metals brings about a significant increase in the strength of the materials.
Despite the impressive mechanical properties, nanocrystalline metals are restricted to use in real application, largely due to the microstructural instability. Retaining the morphology and size of the constituent grains in nanocrystalline metals is a major challenge to successfully use in the many applications over a long period of time. The compromised stability of nanocrystalline metals originates from the excess energy due to the high-volume fraction of grain boundaries in these ultra-fine grained structures [10,11,12,13]. This complexity is associated with both the highly degenerate nature of grain boundary structures as well as the interconnected nature of the boundary network within nanocrystalline microstructure. In the grain boundary region, atoms are typically shifted from their regular lattice sites to accommodate the mismatch between the adjacent grains causing high-energy configurations [1]. As a result, grain boundaries are associated with an excess stored energy compared to the grain interior. This excess stored energy provides a driving force for grain coarsening–a detrimental microstructural phenomenon which is characterized by the growth of the large grains at the expense of smaller ones. A coarse-grained structure is thermodynamically favored since it has less excess stored energy due to a smaller grain boundary volume as compared to a nanocrystalline structure. Due to this excess stored energy, nanocrystalline metals coarsen rapidly even at low temperatures and lose their extraordinary mechanical properties [7]. This highlights that the grain size, as widely accepted, is not the only structural feature of interest for nanocrystalline materials. The grain boundary state considerably alters the thermodynamic and mechanical stability of the materials. Therefore, it must be the focus when designing nanocrystalline materials.
To retain the properties of nanocrystalline materials in bulk components for structural applications, stabilization of the nanostructure with respect to coarsening is highly desirable. An ideal metallic nanostructure is one in which the as-manufactured grain morphologies and sizes are preserved, regardless of exposure conditions [7]. It is imperative that the characteristic nanocrystalline grain size, and high-volume fraction of grain boundaries be retained in the long-term in order to preserve the material’s extraordinary properties. To enhance the stability of the nanocrystalline metals, the excess stored energy in the grain boundaries should be reduced [1]. The preferential decoration of grain boundaries with dopants provides a potentially promising approach to stabilize the NC structure [10, 11]. Recent theories have suggested that through the addition of dopant elements in small amounts to pure nanocrystalline metals, the interfacial free energy associated with a high-volume fraction of grain boundaries in the material can be reduced. In some cases, the interfacial energy can be reduced below the free energy of the bulk solution, thereby eliminating the driving force required for grain growth, which results in a stable segregated nanocrystalline state [8,9,10]. The precise mechanism by which this happens is still a subject of conjecture. Several mechanisms have been proposed to explain how nanocrystalline metals are strengthened by grain boundary segregation. In one of these mechanisms, the partitioning of dopants to the grain boundaries in nanocrystalline metals increases the atomic registry at the boundaries, which shifts the energy of the grain boundary atoms to lower value [14]. Along with a reduction in the grain boundary energy [11,12,13], a contribution to the impediment of grain boundary motion from Zener drag exists [14, 15]. Via this mechanism, dopants at the grain boundaries have also been suggested to mechanically restrict the migration of grain boundaries significantly, thus reducing coarsening.
Precipitation of secondary intermetallic phases due to the presence of dopants is one of the processing challenges which can disrupt the necessary segregation required for grain stability and strength. When the concentration of the added dopant is in a supersaturated state in the nanocrystalline matrix, such thermodynamic conditions will promote second phase precipitates to nucleate and grow in the grain interiors. In general, the second phase precipitates in metallic materials are often considered to serve as obstacles to dislocation glide and cause hardening of the material [15]. This notion, however, fails to explain recent discoveries of high-strength and large-ductility materials with a high density of these precipitates, as obstacles to dislocation glide leading to high stress concentration and even microcracks. Most often these deleterious secondary intermetallic phases are brittle in nature which negatively affects the mechanical properties of the material. Furthermore, the formation of the coarse, incoherent precipitates along grain boundaries leads to softening and substantially reduces the fracture resistance of these materials [16,17,18]. To achieve the best possible effect from grain boundary stabilization through the addition of segregation of dopants, it is necessary to identify the stable nanocrystalline thermodynamic states that do not form precipitates.
To realize this, an understanding of the principles of thermodynamics, and how these principles can be harnessed to achieve stable nanocrystalline structure, is required. In order to model the thermodynamics of both grain boundary segregation and the formation of solid solutions, the thermodynamics of the solid state can be employed. The primary concept from which thermodynamic stabilization mechanisms of nanocrystalline solids is built is that nature trends toward minimizing the energy state of a natural system. This concept is tied to the concept of entropy or disorder, is generated during any thermodynamic process. An ideal system may generate no disorder, i.e., its entropy generation is zero, but entropy may never be annihilated by a thermodynamic process [19]. To this end, if a thermodynamic formulation that describes the variation in the energy state of a system can be established, it can be said that the point at which this energy is minimized will be the stable state of the system, toward which it will trend. Additionally, the energy itself is neither created nor destroyed, it is only conserved and converted between forms. This provides the concept of enthalpy, or the energy which is necessary for a process to occur [19]. These process enthalpies are the driving forces for thermodynamically driven change; in the specific case of this research, these enthalpies will be the driving forces for the segregation of atoms and for the formation of solid solutions.
Traditionally, thermodynamic modeling of nanocrystalline materials has evolved from the basis of describing solute segregation energetically [20]. Weissmuller was one of the first to quantitatively describe that a reduction in grain boundary energy can be achieved through the segregation of solute atoms to a nanocrystalline material’s grain boundaries [21]. This concept was formulated from the Gibbs adsorption isotherm, and the works of Birringer [22], who was the first to propose the theory. From Weissmuller’s description of solute partitioning utilizing the Langmuir-McLean segregation equation, Liu and Kirchheim were able to formulate a relationship between the bulk and grain boundary solute contents that specifically described solute segregation for binary nanocrystalline alloys [23]. From this relationship, Kirchheim described the conditions that would lead to metastability of nanocrystalline materials [23]. Weissmuller expanded upon Kirchheim’s criteria to create a regular solution model for a binary polycrystalline alloy system in terms of bond energies and number of bonds in each of the grain boundary, intercrystalline, and transition regions of the material [21]. It is important to note that Kirchheim’s work neglected the elastic and mechanical effects of solute segregation. This shortcoming was in part rectified by Trelewicz and Schuh [23], and then fully described by Saber and Chookajorn [25, 26]. The resulting models were applied to binary systems with a positive enthalpy of mixing by Murdoch and Schuh [27], to ternary alloy systems by Saber [28], and in a modified state to binary systems with both positive and negative enthalpies of mixing in this work.
The objective of this work is to establish the conditions to stabilize the grain structure of nanocrystalline aluminum (NC Al). Based on thermodynamic principles, a highly efficient dopant design framework has been used to identify the specific dopants that interact with high-energy grain boundaries in NC Al to release the excess free energy in the boundaries. It is of interest to improve the stability of the NC Al not only against grain growth, but also against phase separation. The dopant design framework is based on a modified regular solution model to describe the free energy state of the nanocrystalline structure in terms of bond energies and thermodynamic parameters.
In Section Thermodynamic framework, the thermodynamic framework and analytical methods used in this work are presented. Section Results and discussion describes the results and discussion before concluding in Section Conclusion.
Thermodynamic framework
To assess the efficacy of various dopants in stabilizing NC Al, a standard regular solution model for binary nanocrystalline alloys is adopted [27]. The nanocrystalline structure is modeled as two physically and chemically distinct regions in nanocrystalline metals namely: the ordered crystalline bulk \((c)\) and the disordered grain boundary \((gb)\). The energetics of the system is described by a mixing free energy which is formulated with separate energetic interactions for the two regions in the nanostructure. The crystalline bulk and the grain boundary are, however, not treated as separate phases, per se, but are considered to be geometrically connected to one another such that the global dopant content of the nanocrystalline structure satisfies the mass balance
where \({X}_{gb}\) is the concentration of a dopant species in the grain boundary region, \({X}_{c}\) is the concentration of a dopant species in the crystalline bulk. Treating the two regions as geometrically connected will allow such that a reduction in grain size, d, will cause an increase in the grain boundary volume fraction,\({V}_{gb}\), which follows a cubic scaling
where \(\xi\) is the width of the transition region which is smeared over a finite distance and is composed of bonds between atoms in the crystalline bulk and in the grain boundary. The width of the transition region will be taken as 0.5 nm [27] in all the following. The geometry of the nanocrystalline metal is postulated as a distribution of atomic bonds between the crystalline bulk, grain boundary and transition region. The energies associated with such a geometry is contained in the final free energy function of explicit form
This expression is a solution model for a binary system, in which grain size is a state variable and grain boundary segregation contribute strongly to the energetics of the system. The subscripts denote the crystalline bulk (c) and grain boundary (gb), and the superscripts denote the two chemical species, A (Al solvent) and B (dopant). \({\gamma }^{A}\mathrm{ and} {\gamma }^{B}\) are the interfacial energies of pure Al and the dopant, respectively. The other terms are associated with the geometrical way in which those two regions interact. The parameters \(z,\) \(\Omega\) and \(v\) are the coordination number of the bulk material, the atomic volume and the fraction of atoms contributing bonds to the transitional bonding region, respectively. The interaction parameters are related to the enthalpy terms that describe the tendency of the solute atoms to mix into the solvent as a solid solution or segregate to the grain boundary, respectively, with positive values of the corresponding parameter indicating a higher preference for each tendency. The interaction parameter in the grain boundaries (\({\omega }_{gb}\)) is related to the enthalpy of segregation (\(\Delta H_{seg}\)) as
This captures the thermodynamics of the grain boundary environment, which incorporates chemical interactions, elastic mismatch, and the mismatch in interfacial energies. The interaction parameter in the bulk grain (\({\omega }_{c}\)) is related to the enthalpy of mixing (\(\Delta H_{mix}\)) following
The free energy function in Eq. (3) is essentially in the form a modified regular solution model in the limit of infinite grain size and will also reproduce a grain boundary energy in the proper limit. The estimate values of the enthalpy of mixing for fifty-one aluminum-dopant binary systems are obtained using [29],
where \({X}_{A}\) and \({X}_{B}\) are the compositions of the dopant and the Al solvent, respectively, and \({X}_{A}^{s}\) and \({X}_{B}^{s}\) are effective fractions of the surface of dopant atoms in contact with Al atoms and vice versa. This describes the difference in interaction between a B atom surrounded by A atoms, and one surrounded by some mixture of A and B atoms, dictated by composition \(\Delta H_{SS}^{structural}\) is the enthalpy of the formation of a solid solution of the dopant element in the matrix of the solvent. \(\Delta H_{AinB}^{elastic}\) accounts for the elastic strain effects caused by solute atoms expanding or contracting the lattice of the solvent, if they are larger or smaller than the solvent atoms, respectively. Chemical interactions are captured by the terms of form \(\Delta H_{BinA}^{int}\) which describe, e.g., the enthalpy of a B atom completely surrounded by A atoms. Furthermore, grain boundary segregation enthalpies are calculated as [30],
the term \(c_{0} \gamma_{A}^{S} V_{A}^{\frac{2}{3}}\) is the surface enthalpy of a pure metal where \(c_{0} = 4.5 \times 10^{8}\) is a dimensionless semiempirical constant, \(V\) is the atomic volume, and \(\gamma_{A}^{S}\) is the surface energy of the pure subscripted component. The coefficient captures the change in coordination at the surface; when the segregant B atom is at the surface rather than in the bulk, it has gone from being surrounded by A atoms to being only two-thirds in contact. The coefficient 0.237 accounts for the surface relaxation due to surface electron density distribution and surface geometry, which reduces the exposed surface area. \(\Delta E_{el}\) accounts for the elastic strain effects that contribute to segregation [31].
where K is the bulk modulus, G is the shear modulus, \(r\) is the atomic radius.
The terms \(\Delta G_{c}^{mix}\) and \(\Delta G_{gb}^{mix}\) can be postulated to describe the extremes in the thermodynamic state of a nanocrystalline system in which either only the bulk crystal or grain boundary exists. In a sharp grain boundary limit, i.e., the thickness of the grain boundary region is infinitesimally small, \(d \to 0\), only a single crystal exists, which reduces Eq. (3) to a classical regular solution
where \(k\) is the Boltzmann constant and \(T\) is the temperature. On the other extreme end when \(d=t\), a hypothetical system with only the grain boundary exists which again simplifies Eq. (3) to
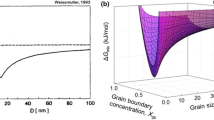
Considering the coexistence of both the bulk crystal and grain boundary, the global thermodynamic state can then be postulated as an interpolation of Equations. (9) and (10) in addition to the interaction parameters accounting for the transition region which yields Eq. (3). This equation yields a 3D free energy surface in grain boundary composition–grain size space as shown in Fig. 1. This free energy surface describes how the material system’s energy changes as a function of its grain boundary solute content (\(X_{gb} )\), which is an approximation for the system’s degree of segregation, and as a function of the final microstructure’s average grain size (\(d\)). Therefore, this surface shows the most prominent and influential variable quantities that affect the degree of stability that the nanocrystalline structure is capable of attaining. A nanocrystalline minimum in this surface denotes a state of stability at which the benefit to material properties of a nanocrystalline microstructure are retained. By using Eq. (1) in (3), the nanocrystalline stability can be evaluated as a function of the global dopant content.

Schematic of a Gibbs free energy surface with a minimum in the grain boundary region. The energy minimizing grain boundary dopant content and the preferred grain size correspond the minimum in the free energy surface
An important feature of this approach is that it will not only identify the dopant compositions that lead to a reduction in the grain boundary energy but will also identify the stable grain size. These quantities for the stable state of the material system can be directly read from the corresponding axes of the generated free energy surface at which a minimum occurs, as shown in Fig. 1. This effect can be clearly seen in the 2D plot in Fig. 2, where the blue line is the free energy curve for the nanocrystalline state of the material, and the red line is the free energy of the bulk unsegregated large-grained material. If the nanocrystalline state’s curve is concave in shape, as determined by sign of its second derivative, and any point on the curve exhibited a free energy value lower than that of the material system’s unsegregated bulk solution, a stable nanocrystalline state exists. Otherwise, the most energetically favorable state for the material system is that of the bulk solution at a large grain size, attainable by the occurrence of grain growth.

Gibbs free energy plot showing the energy minimizing grain boundary solute content corresponding to the minimum in the energy surface. Blue line denotes nanocrystalline state, red line denotes bulk large-grained state
Results and discussion
Analysis of grain boundary segregation
Through the use of the regular nanocrystalline solution model (Eq. (3)), NC Al has been evaluated for thermodynamic stability through grain boundary segregation using fifty-one potential dopants. The input parameters used in the model were obtained from the literature. The two thermodynamic parameters, which together contain all of the most relevant physics of the problem: enthalpies of segregation and mixing were obtained for all of the fifty-one systems from Ref. [29]. All other material parameters, including pure substance grain boundary energies and atomic volumes, were obtained from [24, 26, 27, 32, 33]. To form stability surfaces based on Eq. (3) throughout the entire design space for each binary Al-dopant system, the grain size was varied from 10 to 100 nm and grain boundary solute content was varied throughout its whole range of 0 to 100 at%. This variation was performed in steps of 1%, in order to provide sufficient resolution to accurately determine the stable state of the system.
Using the fifty-one dopant elements, free energy surfaces were constructed and utilized to create a set of stability predictions for binary NC Al-dopant systems. This stability was achieved through free energy minimization at the grain boundary regions. 3D Gibbs free energy surfaces were constructed for each NC Al-dopant combination to assess thermodynamically stable nanocrystalline states and global compositions. From these studies three candidate dopants: magnesium (Mg), lanthanum (La), and silicon (Si) were selected as exhibiting the necessary characteristics to segregate to the grain boundary and provide a stable nanocrystalline state. This selection was done by examining the free energy surfaces created for each material and identifying surfaces exhibiting minima in the grain boundary dopant content-grain size regime. A unique characteristic of each of the three identified dopants is that they possess positive enthalpy of segregation; due to this fact, these three exhibit a tendency for segregation toward the grain boundaries. However, of the three, only Mg possesses a positive enthalpy of mixing [29]. Being that this thermodynamic characteristic describes the solute’s ability to form a solid solution with the Al solvent, Mg may be identified as the most promising dopant for binary Al systems. From a thermodynamics perspective, a positive enthalpy of mixing implies that the solvent and dopant atoms do not have an affinity for each other thus essentially separate out leading to the dopants segregating to the disordered grain boundaries. Additionally, since the regular nanocrystalline regular solution empirical stability criterion (see Section Empirical analysis of nanostructure stability) is only valid for positive enthalpy of mixing systems, Mg is the only dopant for which the model confidently predicts stability against phase precipitation. Figure 3 shows the necessary minima in the Gibbs free energy surfaces for NC Al–Mg, Al-La, Al-Si, and Al-Ag systems, respectively, where (a)-(c) show stable nanocrystalline states whereas (d) shows an unstable nanocrystalline state. This distinction can be seen by the concavity of the surface, as determined by its second derivative. Concave surfaces result in stable nanocrystalline minima, while convex surfaces denote instability of the microstructure with respect to grain growth. As such, in Al–Mg, Al-La, Al-Si nanocrystalline systems, the grain boundary segregated state is energetically favorable to that of the large-grained bulk solution. On the other hand, Al-Ag in Fig. 3d with a convex free energy surface will always have an unstable nanocrystalline structure thus will be susceptible to grain coarsening. By evaluating the grain boundary solute content \({(X}_{gb})\) and grain size \((d)\) at which the minima in the free energy occur, the degree of segregation and the resultant microstructure’s grain size can be predicted. For a global dopant content of \(X=10\%,\) a maximum achievable grain boundary dopant content \(X_{gb,Mg} = 25\%\) was predicted for Al–Mg, \(X_{gb,La} = 35\%\) for Al-La, and \(X_{gb,Si} = 34\%\) for Al-Si. The dopant content at which the nanocrystalline minimum occurs is a function of the degree of segregation that a material system exhibits. Therefore, it is dependent on the enthalpy of segregation and is different for each solvent–solute pair. In the case of binary aluminum systems, a global Mg content of 13% was found to promote the highest degree of stability for the Al–Mg nanocrystalline system. 10% La or Si content was necessary for stability, with higher contents corresponding to smaller final crystallize sizes without affecting the existence of a stable state.

Gibbs free energy surfaces as function of the grain boundary dopant content, \(X_{gb}\) and grain size, \(d\) for a NC Al–Mg, b Al-La, c Al-Si and d Al-Ag systems, for a global dopant content of \(X = 10\%\)
Additionally, the three stable nanocrystalline systems show a minimum achievable grain size of 10 nm. However, the actual grain size that can be achieved during manufacturing is ultimately dependent on the initial grain size of the raw aluminum powder that is alloyed to create the segregated system as well as the processing methods and conditions. Smaller initial grain sizes typically result in smaller grain sizes in stabilized nanocrystalline systems. Figure 4 shows the variation in Gibbs free energy, as a function of grain size, for both the case of an unstable nanocrystalline state and a stable nanocrystalline state. It can be seen that, for an unstable nanocrystalline material, the free energy of the system decreases with an increase in grain size. Being that the energy minimized state is thermodynamically stable, this shows that the stable microstructure in this case is that of a bulk large-grained material. In contrast to this, for the case of a stable nanocrystalline state, it can be seen that the energy minimum occurs at a small nanoscale microstructure, with the free energy of the large-grained microstructure being greater than that of our stable nanocrystalline state. The resulting plots of Gibbs free energy vs grain size can in essence be used as a quick tool for identifying material systems for which a stable nanocrystalline state exists.

Evolution of Gibbs free energy with grain size for a unstable and b stable nanocrystalline state
Stability against second phase precipitation
It is likely that the dopant element interacting with the solvent element could lead to the formation of second phase precipitates once the maximum solubility limit within the nanocrystalline metal is reached. Binary nanocrystalline systems tend to exhibit rampant grain growth following second phase precipitation if the precipitate phase is energetically favorable to one in which the material’s alloying element is segregated to the material’s grain boundaries. This effect is due to the precipitate phase consuming dopant atoms from the grain, which reduces its stabilizing effect on the microstructure. By this effect, precipitation of secondary phases due to the presence of dopants can disrupt the necessary segregation required for grain stability. This comparison can be seen visually in Fig. 5 where the blue curve represents the nanocrystalline state, and the red curve represents the lowest energy state of the bulk large-grained microstructure and any precipitating phases. In the case that a precipitating phase is of a lower energy state than the nanocrystalline regime, the stabilizing solute in the material’s grain boundary is utilized in the precipitation of the second phase, effectively reducing its presence in the grain boundary and thus its stabilization effect. As can be seen in Fig. 5, for each chosen material system the free energy of the nanocrystalline state is lower than that of the bulk solution and any precipitating phases; this means that Al–Mg, Al-La, and Al-Si are stable against phase precipitation under dopant contents of 20%.

Gibbs free energy contours for Al–Mg, Al-La, Al-Si systems for a global solute content of \(X = 0.2\%\)
Influence of global dopant content on grain stabilization
The effect of varying global dopant content \(X\) on the free energy of a material system for the range of dopant contents most commonly used to synthesize binary nanocrystalline systems is shown in Fig. 6(a); this range is chosen to promote stability without inciting secondary phase precipitation. It can be seen that a higher Mg dopant content, under the limit set by the onset of phase precipitation (\(X \le 13\% ),\) yields a higher degree of thermodynamic stability against grain growth. To this end, the optimal range of dopant content for each material system can be established. Within this optimal range, as shown by Fig. 6a, as global solute content is increased, so is the degree of segregation seen in the material, as read by the grain boundary solute content value at which the minimum in the curve exists. It is intuitive to say the stability of the nanocrystalline structure will increase with an increase in the global dopant content. However, this apparently is not true as can be seen in Fig. 6b; although an increase in the global dopant increases the grain boundary content, it also leads to an increase in the free energy which trends the nanocrystalline system toward instability. When the concentration of the added dopant is in a supersaturated state in the nanocrystalline matrix, such thermodynamic conditions will promote second phase precipitates to nucleate and grow in the grain boundaries and grain interiors. Thus in NC Al–Mg, a global dopant content above 13% will be accompanied by the precipitation secondary phases in the grain boundary region in order to reduce the free energy. Precipitation of secondary intermetallic phases can disrupt the necessary segregation required for grain stability.

Influence of a variation in global Mg dopant content \(X\) on grain boundary stabilization in NC Al a Mg dopant content in the dilute limit and b high Mg dopant content
Additionally, a material system’s preference for the size of the solute atoms can be seen in Fig. 6b. From the results of the regular nanocrystalline solute model, a variation in solute atomic volume showed a two-regime effect; at grain boundary solute contents below 19% a large solute atomic volume resulted in a reduction in grain boundary energy presumably due to the large atoms’ ability to reduce the grain boundary’s free volume more effectively than small solute atoms. At grain boundary solute contents above 19%, small solute atomic volumes are preferred due to their lesser atomic misfit in the grain boundaries, and therefore lesser lattice strain effects. These lattice strain effects directly influence the material system’s enthalpies of mixing and segregation as described in Eqs. (6) and (7), respectively. This concept suggests that comparing the atomic volumes of the solvent and solute elements can indicate the propensity for the system in question to form a stable nanocrystalline state.
The thermodynamic effect of an addition of dopant excess \(\left( {\Gamma } \right)\) on grain boundary energy is described by the Gibbs adsorption isotherm
where \(\gamma\) is the reduced grain boundary energy as a result of solute segregation, \(\gamma_{0}\) is the average grain boundary energy of the pure solvent material, \(\Delta H^{seg}\) is the system’s enthalpy of segregation, and \(X\) is the global dopant content [27]. The Gibbs adsorption isotherm predicts that as the degree of dopant segregation is increased, the resulting microstructure’s final grain size becomes smaller. By varying the global dopant content in the nanocrystalline metal it can be shown that the model’s free energy surfaces predict the effects of the Gibbs Adsorption Isotherm, Eq. (13), correctly; that is, as dopant content is increased, so is the content in the grain boundary, and therefore the final nanostructure’s stable grain size is reduced as a function of solute excess. This relation provides a means for controlling the resultant microstructure of a manufacturing process in which a binary nanocrystalline system is created. However, this control is limited in that no final nanostructure can be created with a final grain size smaller than that of the raw aluminum powder that is initially used in the process. The grain size is known to have a considerable influence of the material properties. The influence is efficiently described by the Hall–Petch relationship which follows
The Hall–Petch relationship predicts that the material’s strength is inversely proportional to its grain size. This, in turn, by the commutative property, means that as the global dopant content is increased, so does the Hall–Petch effect’s contribution toward the strength of the material. Additionally, as grain boundary segregation of dopant increases the excess free volume in the grain boundary is decreased. Therefore, the grain boundary’s excess free energy is also decreased. By this mechanism, an increase in global solute content also increases the degree of stability of the nanocrystalline material, up to a limit set by the onset of phase precipitation at which point the precipitation of phases consumes dopants in the grain boundary reducing its stabilizing effect. In order to assess these effects, the free energy curves corresponding to an increase in global solute content were systematically created from the regular nanocrystalline regular solution model. While grain boundary solute content was varied throughout its full range, zero content to full saturation, the grain size was restricted by the breakdown of the Hall–Petch regime. Being that the regular nanocrystalline regular solution model is based on the assumptions of Hall–Petch, the model is only valid while those assumptions hold. According to Trelewicz [32], the Hall–Petch regime begins to break down into a description of amorphous solids at a grain size of approximately 10 nm; as such, the model’s grain size parameter was varied through the remainder of the nanocrystalline regime, 10 to 1000 nm. This breakdown is described by the Hall–Petch relationships of Eqs. (12) and (13), where \(\sigma_{0}\) is the intercept value of stress from a material’s empirically derived Hall–Petch plot, and \(k_{y}\) is the slope of the same curve.
Empirical analysis of nanostructure stability
In order to analytically assess the stability of NC Al against both grain growth and phase precipitation, an empirical stability criterion derived from the results of the regular nanocrystalline regular solution model was employed,
The parameters \(\Delta H_{mix}\) and \(\Delta H_{seg}\) which capture the thermodynamics of the grain interior and grain boundary environment were estimated for each NC Al-dopant system using Eqs. (6) and (7), respectively. The criterion was calibrated using regression analysis of the original regular nanocrystalline regular solution model’s stability predictions utilizing a large dataset of binary nanocrystalline alloys which yielded the fit coefficients a \(= 0.567\) and \(c = 4.425\) [33]. Since the dataset which was used was not restricted to aluminum solvents, the empirical stability criterion’s predictions may exhibit systematic errors for aluminum systems due to uncertainties in the fit coefficients. The output of stability criterion Eq. (14) can be visualized in the form of a stability map that is characterized by the stable, metastable, and unstable regions separated by curves corresponding to an equal ratio between the enthalpy of segregation and the model’s adjusted enthalpy of mixing.
The empirical stability criterion Eq. (14) was applied to each NC Al-dopant system to provide an additional source of data with which to reaffirm the predicted thermodynamically nanocrystalline states. The analysis was limited to NC Al-dopant systems with a positive \(\Delta H_{mix}\), which have a higher propensity to segregate to grain boundaries. According to the regular nanocrystalline regular solution criterion, solvent-dopant systems with sufficiently large enthalpies of segregation in relation to their model-adjusted enthalpy of mixing are predicted to be stable against both grain growth and phase precipitation. Figure 7 shows a stability map for the NC Al-dopant systems which exhibit positive \(\Delta H_{mix}\). Three regions are present: the Bulk Stable Region where grain boundary segregation does not result in a stabilized nanocrystalline structure due to its free energy being greater than that of the bulk solution, the Metastable Region in which macroscopic phase separation would be preferential (despite the presence of a nanocrystalline state stable against grain growth), and the Stable Nanocrystalline Region for which the nanocrystalline state is stable against both grain growth and phase separation being that it is the lowest free energy state available. In order for a material to be chosen as a viable dopant for the purposes of this research, the resulting binary alloy must reside in the stable nanocrystalline region. As can be seen by Fig. 7, the only binary system predicted to exhibit this characteristic with a positive enthalpy of mixing is Al–Mg. As such, Mg has been identified as the most promising dopant to successfully stabilize nanocrystalline aluminum’s microstructure. The empirical stability criterion’s predictions are therefore consistent with observations from the free energy surfaces.

Empirical stability map for binary aluminum systems with positive enthalpy of mixing
Additionally, by examining the material system’s enthalpy of segregation and grain boundary interaction parameters it can be shown that there is a necessary minimum value in order for segregation to occur sufficiently to reduce the grain boundary energy to a level at which a stable nanocrystalline state is possible. This observation can be used as an additional tool to screen potential dopant elements and reaffirm the stability map’s predictions; systems with increasingly negative values of the grain boundary interaction parameter exhibit the tendencies necessary to provide grain boundary stability.
Conclusion
The work described herein has been done in order to predict the dopant elements and their respective compositions for which nanocrystalline aluminum’s grain size can be stabilized. The regular nanocrystalline solution model applied for binary nanocrystalline aluminum alloys predicts thermodynamic stability against grain growth and phase precipitation for Mg, La, and Si dopants. These dopants, when added to the material system above the required global dopant content for stability, promote segregation to the grain boundaries resulting in a reduction in grain boundary energy. A global Mg content of 13% was found to promote the highest degree of stability in the binary Al–Mg nanocrystalline system. 10% La or Si content was necessary for stability, with higher contents corresponding to smaller final crystallize sizes without affecting the existence of a stable state. Additional dopant addition beyond this critical minimum global solute content results in a characteristic reduction in average microstructure grain. These predictions can be used to inform which experimental materials shall best be created to facilitate investigations with the goal of establishing manufacturing techniques for bulk nanocrystalline aluminum alloys to be used in industrial applications such as power generation and transportation.
While the results derived from the modified Regular Nanocrystalline Solution model presented in this work are relatively reasonable, it must be noted that both the chemical and mechanical interaction terms of the model represent simplified dynamics corresponding to interactions based on bond energies and elastic effects only. As such, dynamic interactions such as atomic hysteresis effects have been neglected. Additionally, the enthalpies of mixing and segregation used in this work are subject to the limitations of Miedema’s model. While these effects are expected to be small in terms of the results presented herein, in some cases deviation between experimental and modeled results is to be expected.
References
Gutkin Y (2011) Nanostructured metals and alloys processing, mechanical properties and applications. Woodhead Publishing Series, New Delhi, pp 329–374
Ovid’ko I (2018) Review on superior strength and enhanced ductility of metallic nanomaterials. Prog Mater Sci 94:462–540
Han B (2005) Improvement of toughness and ductility of cryomilled Al-Mg alloy via microstructural modification. Met Mat Trans A 36(8):2081–2091
Tellkamp VL, Lavernia EJ (1999) Processing and mechanical properties of nanocrystalline 5083 Al alloy. Nanostructured Mater 12(1–4):249–252
Youssef KM, Scattergood RO, Murty KL, Koch CC (2006) Nanocrystalline Al-Mg alloy with ultrahigh strength and good ductility. Scr Mater 54(2):251–256
Pun S (2017) Nanocrystalline Al-Mg with extreme strength due to grain boundary doping. Mater Sci Eng 696:400–406
Mishnaevsky L, Levashov E (2015) Micromechanical modelling of nanocrystalline and ultrafine grained metals: A short overview. Comput Mater Sci 96:365–373
Gleiter H (2000) Nanostructured materials: basic concepts and microstructure. Acta Mater 48(1):1–29
Tucker GJ, McDowell DL (2011) Non-equilibrium grain boundary structure and inelastic deformation using atomistic simulations. Int J Plast 27(6):841–857
Divinski S, Rosner H, Wilde G (2009) Functional Nanostructured Materials - Microstructure, Thermodynamic Stability and Atomic Mobility. Front Nanosci 1:1–50
Tamaki J, Fujimori K, Akiyama M, Harada N, Miura N, Yamazoe N (1994) Grain-size effects in tungsten oxide-based sensor for nitrogen oxides. J Electrochem Soc 141(8):2207
Westbrook J, Wood D (1961) Embrittlement of grain boundaries by equilibrium segregation. Nature 192(4809):1280–1281
Luo J, Gupta V, Yoon D, Meyer H (2005) Segregation-induced grain boundary pre-melting in nickel-doped tungsten. Appl Phys 87(23):231902
Devaraj A et al (2019) Grain boundary segregation and intermetallic precipitation in coarsening resistant nanocrystalline aluminum alloys. Acta Mater 165:698–708
Peng S, Wei Y, Gao H (2020) Nanoscale precipitates as sustainable dislocation sources for enhanced ductility and high strength. PNAS 117(10):5204–5209
Joshi A, Shastry CR, Levy M (1981) Effect of heat treatment on solute concentration at grain boundaries in 7075 aluminum alloy. Metall Trans A 12(6):1081–1088
Viswanadham RK, Sun TS, Green JAS (1980) Grain boundary segregation in Al-Zn-Mg alloys-implications to stress corrosion cracking. Metall Mater Trans A 11(1):85–89
Vasudevan AK, Doherty RD (1987) Grain boundary ductile fracture in precipitation hardened aluminum alloys. Acta Metall 35(6):1193–1219
Moran MJ, Shapiro HN, Boettner DD, Bailey MB (2010) Fundamentals of engineering thermodynamics. Wiley, pp 836–838
Gong M, Liu F, Chen Y (2017) Thermal stability of nanocrystalline materials: thermodynamics and kinetics. Int Mater Rev 62(6):303–333
Weissmuller J (1993) Alloy effects in nanostructures. Nanostruct Mater 3(1–6):261–272
Birringer R (1992) Metastable phase formation in the solid state, Klost. Irsee, pp 1–346
Liu F, Kirchheim R (2004) Nanoscale grain gowth inhibited by reducing grain boundary energy through solute segregation. J Cryst Growth 264(1–3):385–391
Trelewicz J, Schuh C (2009) Grain boundary segregation and thermodynamically stable binary nanocrystalline alloys. MIT Phys Rev B 79(9):094112
Saber M, Kotan H, Koch CC (2013) Thermodynamic stabilization of nanocrystalline binary alloys. J Appl Phys 113(6):063515
Chookajorn T, Murdoch H, Schuh C (2012) Design of stable nanocrystalline alloys. Science 337(60970):951–954
Murdoch H, Schuh C (2013) Stability of binary nanocrystalline alloys against grain growth and phase separation. Acta Mater 61(6):2121–2132
Saber M, Kotan H, Koch CC (2013) A predictive model for thermodynamic stability of grain size in nanocrystalline ternary alloys. J Appl Phys 114(10):103510
Murdoch H, Schuh C (2013) Estimation of grain boundary segregation enthalpy and its role in stable nanocrystalline alloy design. J Mater Res 28(16):2154–2163
Miedema AR, Boom R, De Boer FR (1975) On the heat of formation of solid Alloys. J Common Met 41(2):283–298
McLean D (1957) Grain boundaries in metals. Oxord Clarendon Press, London, pp 263–274
Trelewicz J, Schuh C (2007) The Hall-Petch breakdown in nanocrystalline metals: a crossover to glass-like deformation. Acta Mater 55(17):5948–5958
Murdoch H (2013) Design of a stable nanocrystalline alloy. MIT Thesis, pp 1–200
Acknowledgements
Authors gratefully acknowledged financial support by the Department of Energy (DOE) within project number DE-EE0009116 through the Advanced Manufacturing Office. PK also thankfully acknowledges the startup grant by the University of New Mexico.
Author information
Authors and Affiliations
Contributions
Jake Hohl did Conceptualization, Writing—original draft, Investigation, Visualization; Pankaj Kumar did Funding acquisition, Writing—review & editing; Mano Misra did Funding acquisition, Writing-review & editing; Leslie Mushongera did Funding acquisition, Conceptualization, Writing–original draft, Supervision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Handling Editor: Avinash Dongare.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hohl, J., Kumar, P., Misra, M. et al. Thermodynamic stabilization of nanocrystalline aluminum. J Mater Sci 56, 14611–14623 (2021). https://doi.org/10.1007/s10853-021-06224-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06224-2