
Abstract
Porous carbons with large surface areas, abundant mesopores and weak base sites are promising materials for the capture and conversion of CO2. However, it is still challenging to obtain such porous carbons in a facile and template-free way. Herein, nitrogen-decorated micro-mesoporous carbons were synthesized by direct carbonization of porous organic polymers, which were developed through alkylation-induced hyper-crosslink of rigid organic bases without the use of any templates. The synthesized carbons have ultrahigh surface areas of 2366–3580 m2/g, total pore volumes of 1.74–3.38 cm3/g and N contents of 1.50–3.24 wt%. As a consequence, the synthesized carbons enable highly efficient and selective adsorption of CO2 from CO2/N2 mixed gas, with the CO2 capacities of 1.50–2.03 mmol/g at 0 °C and 15 kPa, and IAST selectivities of 69–78 for CO2/N2 (0.15/0.85 vol) mixed gas at 0 °C and 100 kPa. After loaded with metal salts, the synthesized carbons also exhibit high activities for the catalytic conversion of CO2, with the TOFs of > 600 h−1 for cycloaddition of CO2 with propylene oxide at 100 °C and 1.0 MPa.
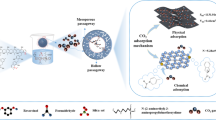
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Carbon dioxide (CO2) is a greenhouse gas and mainly emitted in the tail gas of thermal power plants, which consume large quantity of coals. The popularization of automobiles, which achieve energy through burning gasoline, also makes a great contribution to the emission of CO2. According to the report of Intergovernmental Panel on Climate Change (IPCC), the atmospheric concentration of CO2 has increased to a high level in comparison with a hundred years ago [1]. As a consequence, the global warming issue is more and more serious, thus inducing the continuous melting of glaciers and desertification of lands. Given the urgent situation, a consensus has been reached across the world that the emission of CO2 must be significantly reduced, so as to preserve the living environment of human beings. [2] Traditionally, the reducing of CO2 emission is realized by scrubbing with aqueous alkanolamine solutions [3, 4]. However, aqueous alkanolamine solutions are associated with some defects such as high volatility, large heat capacity and strong corrosion. Recently, ionic liquids (ILs) with extremely low volatility and structural designability were proposed as alternatives to aqueous alkanolamines for application in the reducing of CO2 emission [5,6,7]. Unfortunately, the high cost and large viscosity of ILs disfavor the practical use of ILs in the industry.
On the other hand, CO2 is a widely used C1 feedstock for the synthesis of value-added chemicals such as cyclic carbonates, carboxylic acids, urea-derivates, formamides, etc. [8,9,10,11,12]. Therefore, it is of great significance to capture CO2 from industrial tail gas and simultaneously convert it into value-added chemicals. Many advanced materials have been investigated for the capture and conversion of CO2, such as zeolites [13, 14], porous carbons [15,16,17], porous organic polymers (POPs) [18,19,20] and metal–organic frameworks (MOFs) [21,22,23]. Among these materials, porous carbons are particularly attractive, because they have intriguing features such as high thermal stability, tunable porous and chemical structure. The precursors for porous carbons are also widely available, including natural compounds [24,25,26,27], organic wastes [28,29,30] and synthetic polymers [31,32,33]. Ideal porous carbons for CO2 capture and conversion should have large surface areas, abundant mesopores and weak base sites. The large surface areas are favorable for the exposure of materials to reactants. The abundant mesopores are favorable for the diffusion of reactants in nanochannels. The weak base sites can not only selectively attract CO2 from gas phase through Lewis base–acid interaction, but also anchor metal ions that have catalytic activity for CO2 conversion.
Porous carbons with large surface areas are normally synthesized by chemical or physical activation of pre-carbonized solids [34,35,36,37,38,39,40]. The most frequently used activation agent is KOH, while others include NaNH2, K2CO3, ZnCl2, NH3, H2O and CO2. However, the activation process mainly results in micropores that are narrow in pore width. To construct mesopores, hard or soft templates should be employed in the preparation of carbon precursors [41,42,43,44,45]. The hard templates (e.g., SiO2) are removed by HF etching after carbonization process, while the soft templates (e.g., surfactants and block copolymers) are removed through thermal decomposition during carbonization process. The introduction of weak base sites can be realized by choosing precursors with weak base groups for carbonization [46, 47], or treating porous carbons with weak base compounds [48, 49]. Though with the progress, it is still challenging to synthesize porous carbons with large surface areas, abundant mesopores and weak base sites in a facile and template-free way.
The choice of precursors has great impact on the porosity and function of resultant carbons, thereby influencing their performance for CO2 capture and conversion. In this work, we developed a class of porous organic polymers (POPs) with abundant mesopores and weak base sites by alkylation-induced hyper-crosslink of rigid organic bases, without the use of any templates. It is then envisioned that the direct carbonization of such POPs may result in porous carbons with abundant mesopores and weak base sites: (1) the permanent mesopores formed by crosslinking structure are likely to be preserved during carbonization process; (2) the weak base sites can be in situ decorated into resultant porous carbons. To increase the surface areas of resultant porous carbons and simplify the synthetic process, POPs were mixed with KOH for direct carbonization. Scheme 1 illustrates the synthetic route for porous carbons with large surface areas, abundant mesopores and weak base sites. The porous carbons were systematically characterized for porous and chemical structure, and also examined for CO2 capture and conversion performance.

Synthetic route for porous carbons with large surface areas, abundant mesopores and weak base sites
Experimental
Chemicals
CO2 (99.99 vol%) and N2 (99.99 vol%) were supplied by Jiangxi Huasheng Co. Ltd., China. 1,4-Bis(chloromethyl)benzene (98 wt%) and p-phenylenediamine (99 wt%) were supplied by Shanghai Sigma Aldrich Co. Ltd., China. 1,2-Dichloroethane (99.5 wt%), SnCl4 (99 wt%), TiCl4 (99 wt%), 4,4′-bipyridine (98 wt%), hexamethylenetetramine (HMTA, 99 wt%), ethanol (99.7 wt%), iso-propylamine (99 wt%), KOH (90 wt%), CoCl2·6H2O (99 wt%), Zn(OAc)2·2H2O (99 wt%), N,N-dimethylformamide (DMF, 99 wt%), propylene oxide (99 wt%), tetrabutylammonium bromide (TBAB, 99 wt%) and methanol (99.9 wt%) were supplied by Shanghai Adamas Co. Ltd., China. All the chemicals were used as received.
Synthesis of POPs
POPs were synthesized by alkylation-induced hyper-crosslink of rigid organic bases according to the literature [50]. In brief, 1,4-bis(chloromethyl)benzene (2.0 g) was dissolved in 1,2-dichloroethane (30 mL), and then, SnCl4 or TiCl4 (6.0 mL) was added to the mixture under vigorous stirring at 0 °C. Subsequently, 4,4′-bipyridine, p-phenylenediamine or HMTA (1.6 g) was added to the mixture under static condition. The reaction was performed at 75 °C for 24 h under the protection of flowing N2. The formed solid was filtered out and washed with warm ethanol to remove residual reactants. The product was activated with iso-propylamine, followed by drying at 60 °C and 0.1 kPa for 24 h. The synthesized samples were denoted as POP-x, where x is the rigid organic base.
Synthesis of porous carbons
Porous carbons were synthesized by direct carbonization of POP-x samples. In brief, POP-x (1.0 g) was mixed with KOH (4.0 g) by manual grinding. The mixture was then loaded in a nickel crucible, transferred to a tube furnace, heated to 800 °C at a rate of 5 °C/min and kept at 800 °C for 2 h. The whole carbonization process was protected by flowing N2. After cooling down to the room temperature naturally, the product was washed with deionized water until the pH of water approaches 7. The synthesized samples were denoted as NPC-x, where NPC stands for nitrogen-decorated porous carbon and x is the rigid organic base.
Synthesis of metal-loaded catalysts
Metal-loaded catalysts were synthesized by impregnation of metal salts into the nanopores of porous carbons. In brief, CoCl2·6H2O (0.41 g) or Zn(OAc)2·2H2O (0.34 g) was dissolved in DMF (75 mL), and then, NPC-x (0.40 g) was added to the mixture. The mixture was vigorously stirred at 120 °C for 12 h under the protection of flowing N2. The solid product was filtered out, washed with warm DMF and finally dried at 60 °C and 0.1 kPa for 24 h. The synthesized samples were denoted as M@NPC-x, where M is the metal and x is the rigid organic base.
Characterizations
Before characterizations, each sample was degassed at 160 °C and 0.1 Pa for 6 h. N2 adsorption isotherms at − 196 °C were determined by a Micromeritics Gemini 2390a surface area analyzer to calculate the pore width distributions and porosity parameters. Pore width distributions were calculated using the Barrett–Joyner–Halenda (BJH) method. Surface areas were calculated using the Brunauer–Emmett–Teller (BET) equation in the relative pressure range of 0.02–0.15. Total pore volumes were calculated according to the amounts of N2 adsorbed at the relative pressure of 0.975. Micropore volumes were calculated using the t-plot method in the thickness range of 0.45–0.6 nm. Mesopore volumes were calculated by integrating the pore size distributions in the pore size range of 2–50 nm.
Thermogravimetric analysis (TGA) was performed on a Seiko 6300 TG/DTA system under the protection of flowing N2, with the heating rate set to 10 °C/min. Elemental analysis was performed on an Elementar Vario EL II system. Scanning electron microscope (SEM) images were taken on a Zeiss Auriga SEM/FIB Crossbeam system at an acceleration voltage of 5 kV. Transmission electron microscope (TEM) images were taken on a Zeiss Libra 200 FE system at an acceleration voltage of 200 kV. X-ray diffraction (XRD) patterns were collected on a PANalytical X’Pert Pro X-ray powder diffractometer with Cu-Kα radiation. The scanning range was set to 2θ = 15°–75°, and the step size was set to 0.02°. Raman spectra were collected on a Princeton Acton Trivista 555 spectrometer with λ = 532 nm laser excitation. X-ray photoelectron spectroscopy (XPS) spectra was collected on a Thermo Scientific ESCALAB250 X-ray photoelectron spectrometer with Al Kα radiation, and the binding energies were calibrated using the C 1 s peak at 284.9 eV. Inductively coupled plasma (ICP) analysis was performed on a Jarrel-AshJ-A1100 spectrometer.
Single gas adsorption
Before experiments, each sample was degassed at 160 °C and 0.1 Pa for 6 h. CO2 and N2 adsorption isotherms at 0 and 25 °C were determined by a Micromeritics Tristar II 3020 surface area analyzer.
Mixed gas adsorption
Mixed gas adsorption experiments were performed on a home-made fixed-bed adsorption system. In a typical run, a certain amount of NPC-x was loaded in a quartz tube, and then, the sample was stabilized at target temperature for 1 h under the protection of flowing Ar. Subsequently, the Ar flow was switched to a CO2/N2 mixed gas flow. The composition of inlet gas was adjusted by two Horiba Metron S500 mass flow controllers. The compositions of outlet gas were recorded online by an Agilent 7890B gas chromatograph.
Catalytic CO2 conversion
Catalytic CO2 conversion experiments were performed in a stainless steel autoclave with a Teflon tube (25 mL). The autoclave was first purged with CO2 to replace air and immersed in ice/water bath for 10 min. Then, specific amounts of M@NPC-x, propylene oxide and TBAB were loaded into the autoclave quickly. Subsequently, the autoclave was sealed and charged with CO2 to target pressure. After stirring at target temperature for a specific period of time, the excess CO2 was vented out and the autoclave was immersed in ice/water bath to halt the reaction. The liquid mixture was analyzed by an Agilent 7890A gas chromatography (GC) equipped with a flame ionization detector (FID), and the yields and selectivities were calculated using the internal standard method. Specifically, dodecane was used as the internal standard to calculate the correction factor of GC peaks. The amounts of reactant and product after reaction were then calculated by comparing the GC peak areas of reactant and product with that of dodecane. To examine the recyclability of catalysts, M@NPC-x was filtered out from reaction system, washed with methanol, dried at 60 °C and 0.1 kPa for 24 h and reused for catalytic CO2 conversion experiments.
Results and discussion
Structural characterizations
The carbon precursors (i.e., POP-x samples) developed by alkylation-induced hyper-crosslink of rigid organic bases were first characterized by N2 adsorption isotherms at − 196 °C to calculate the pore width distributions and porosity parameters (see Fig. 1, Table 1). It can be seen that the N2 adsorption isotherms display type II profiles, with considerable increase in N2 uptakes at the relative pressure of > 0.9. The pore width distributions cover a wide range of 0–50 nm, suggesting that both micropores and mesopores are present in POP-x samples. POP-x samples have moderate surface areas of 209–596 m2/g and total pore volumes of 0.21–0.73 cm3/g, following the sequence of POP-4,4′-bipyridine > POP-p-phenylenediamine > POP-HMTA. The mesopore volumes account for > 65% of the total pore volumes for POP-x samples, suggesting the predominantly mesoporous structure of POP-x samples. In addition, the elemental analysis results show that POP-x samples have the N contents of 3.25–5.82 wt%, with the highest value for POP-HMTA (see Table 1). This is within expectation since HMTA has higher N content than the other two organic bases. Furthermore, the TGA curves show that POP-x samples start to decompose at ~ 175 °C, and the loss of weight gradually slows down at > 250 °C (see Figure S1).

N2 adsorption isotherms at − 196 °C (a) and pore width distributions (b) of POP-x samples
The porous carbons (i.e., NPC-x samples) were then synthesized by direct carbonization of POP-x samples with the assistance of KOH at 800 °C. Actually, the effects of carbonization temperatures on the porosity parameters, N contents and CO2 uptakes of resultant NPC-4,4′-bipyridine were examined (see Figure S2, Table S1 and Figure S3). It is found that when the carbonization temperatures increase from 500 to 800 °C, the porous parameters of resultant porous carbons increase, while the N contents of resultant porous carbons decrease. However, further increasing the carbonization temperatures from 800 to 900 °C results in no product. The CO2 uptakes of NPC-4,4′-bipyridine synthesized at 800 °C are also higher than those of NPC-4,4′-bipyridine synthesized at other carbonization temperatures. Therefore, the carbonization temperature was set to 800 °C for all the carbon precursors in this work.
The synthesized NPC-x samples were systematically characterized for porous and chemical structure. Figure 2 shows the N2 adsorption isotherms at − 196 °C and pore width distributions of NPC-x samples, which display similar profiles as those of POP-x samples. Therefore, both micropores and mesopores are also present in NPC-x samples. The calculated porous parameters for NPC-x samples are summarized in Table 2. It is notably found that NPC-x samples have ultrahigh surface areas of 2366–3580 m2/g and total pore volumes of 1.74–3.38 cm3/g, following the sequence of NPC-4,4′-bipyridine > NPC-p-phenylenediamine > NPC-HMTA. This is consistent with the sequence of surface areas and total pore volumes for corresponding POP-x samples. Obviously, the surface areas and total pore volumes of NPC-x samples are much higher than those of POP-x samples, because KOH etching creates additional nanochannels in NPC-x samples during the carbonization process. The mesopore volumes account for > 50% of the total pore volumes for NPC-x samples, still suggesting the predominantly mesoporous structure of NPC-x samples. However, NPC-x samples have slightly higher percentages of micropore volumes than POP-x samples, since the nanochannels created by KOH etching are mainly microporous.

N2 adsorption isotherms at − 196 °C (a) and pore width distributions (b) of NPC-x samples
Figures 3 and 4 show the SEM and TEM images of NPC-x samples, respectively. In the SEM images, rough surfaces embedded with sponge-like mesopores can be clearly observed. In the TEM images, micropores and mesopores with disordered arrangements can be clearly observed. The observations from SEM and TEM images indicate that NPC-x samples are amorphous, and have micro-mesoporous structure. The micro-mesoporous structure is consistent with the pore size distributions and porosity parameters calculated from N2 adsorption isotherms. Figure 5 shows the XRD patterns and Raman spectra of NPC-x samples. In the XRD patterns, there are peaks arising from the (002) planes of graphitic carbons at ~ 24°. However, the peaks are very weak in intensities and broad in shapes, suggesting that the graphitic degrees of NPC-x samples are quite low. In the Raman spectra, there are peaks arising from the D bands of sp3 carbons at ~ 1350 cm−1, and the G bands of sp2 carbons at ~ 1590 cm−1. The peaks of D bands are weaker in intensities than those of G bands, implying that most carbon atoms are assembled within conjugated rings in NPC-x sample.

SEM images of NPC-4,4′-bipyridine (a, b), NPC-p-phenylenediamine (c, d) and NPC-HMTA (e, f)

TEM images of NPC-4,4′-bipyridine (a, b), NPC-p-phenylenediamine (c, d) and NPC-HMTA (e, f)

XRD patterns (a) and Raman spectra (b) of NPC-x samples
The N contents of NPC-x samples determined by elemental analysis are also summarized in Table 2. NPC-x samples have the N contents of 1.50–3.24 wt%, with the highest value for NPC-HMTA. This is consistent with the highest N content for corresponding POP-HMTA among POP-x samples. The N contents of NPC-x samples are lower than those of POP-x samples, because some N species are lost during carbonization process. However, the loss of N species for POP-4,4′-bipyridine (from 3.34 to 2.46 wt%) is not that significant as those for POP-p-phenylenediamine (from 3.25 to 1.50 wt%) and POP-HMTA (from 5.82 to 3.24 wt%). This can be explained by the fact that the N species in POP-4,4′-bipyridine are assembled within conjugated rings, while those in POP-p-phenylenediamine and POP-HMTA are not. The N species assembled within conjugated rings are more stable than those connected by single bonds [51]. As a result, the N species in POP-4,4′-bipyridine are more favorable to be preserved during carbonization process. Figure 6 shows the C 1 s and N 1 s spectra of NPC-x samples. The C 1 s spectra can be deconvoluted into peaks at ~ 284.6 and ~ 286.2 eV. The former ones are attributed to the signals of C−C bonds, while the latter ones are attributed to the signals of C−N bonds. The N 1 s spectra can be deconvoluted into peaks at ~ 398.6 and ~ 401.1 eV. The former ones are attributed to the signals of pyridinic N, while the latter ones are attributed to the signals of quaternary N. It is found that the percentage of pyridinic N in NPC-4,4′-bipyridine is higher than those in NPC-p-phenylenediamine and NPC-HMTA (see Table S2). This is understandable since the N species in POP-4,4′-bipyridine are pyridinic N, while those in POP-p-phenylenediamine and POP-HMTA are not. Therefore, more pyridinic N can be decorated into NPC-4,4′-bipyridine during carbonization process. Since there are lone pair electrons in pyridinic N, pyridinic N is more attractive to CO2 than quaternary N.

C 1 s (a) and N 1 s (b) spectra of NPC-x samples
Selective CO2 capture
Considering the ultrahigh surface areas, micro-mesoporous structure and N-decorated feature of NPC-x samples, they are believed to exhibit promising application in the capture and conversion of CO2. The selective CO2 capture performance of NPC-x samples was examined by both single and mixed gas adsorption experiments. Figure 7 shows the CO2 and N2 adsorption isotherms of NPC-x samples at 0 and 25 °C. The CO2 adsorption isotherms display non-ideal profiles, which is a result of the strong interaction between N species of NPC-x samples and CO2. The CO2 uptakes follow the sequence of NPC-4,4′-bipyridine > NPC-HMTA > NPC-p-phenylenediamine. NPC-4,4′-bipyridine has the highest CO2 uptakes among the three NPC-x samples because it has the highest surface area, micropore volume and percentage of pyridinic N. There is no significant difference in surface areas and micropore volumes for NPC-p-phenylenediamine and NPC-HMTA, but the N content of NPC-p-phenylenediamine is lower than that of NPC-HMTA. Therefore, NPC-HMTA has the second highest CO2 uptakes, while NPC-p-phenylenediamine has the lowest CO2 uptakes among the three NPC-x samples. The isosteric heats of CO2 adsorption on NPC-x samples were calculated by using the CO2 uptakes at different temperatures (see Figure S4). The CO2 adsorption heats are in the range of − 30 to − 34 kJ/mol, suggesting the moderately strong interaction between NPC-x samples and CO2. Therefore, the CO2 adsorbed by NPC-x samples may be easily stripped out with low energy penalty. The CO2 adsorption heats of NPC-4,4′-bipyridine are slightly more negative than those of NPC-p-phenylenediamine and NPC-HMTA, because NPC-4,4′-bipyridine has higher percentage of pyridinic N than the other two samples (see Table S2). In addition, the CO2 uptakes of NPC-x samples are significantly improved in comparison with those of POP-x samples (see Figure S5), which is mainly attributed to the much higher surface areas of NPC-x samples than POP-x samples.

CO2 (a, b) and N2 (c, d) adsorption isotherms of NPC-x samples at 0 °C (a, c) and 25 °C (b, d)
However, the N2 adsorption isotherms display nearly ideal profiles, which is a result of the weak interaction between NPC-x samples and N2. Due to the inert nature of N2, the N species in NPC-x samples have little contribution to the adsorption of N2. The N2 uptakes follow the sequence of NPC-4,4′-bipyridine > NPC-p-phenylenediamine ≈ NPC-HMTA. NPC-4,4′-bipyridine has the highest N2 uptakes among the three NPC-x samples because it has the highest surface area. The N2 uptakes of NPC-p-phenylenediamine and NPC-HMTA are similar, because there is no significant difference in surface areas for NPC-p-phenylenediamine and NPC-HMTA. The CO2 and N2 adsorption isotherms were then fitted by the dual-site Langmuir–Freundlich (DSLF) equation. The fitted parameters (see Tables S3–S6) were used to calculate the selectivities of CO2/N2 for NPC-x samples according to the ideal adsorption solution theory (IAST) [52], and results are shown in Fig. 8. The CO2/N2 selectivities of NPC-x samples follow the sequence of NPC-HMTA > NPC-4,4′-bipyridine > NPC-p-phenylenediamine. This is consistent with the sequence of N contents for NPC-x samples. In addition, the CO2/N2 selectivities of NPC-x samples are significantly improved in comparison with those of POP-x samples (see Figure S6), which is mainly attributed to the much higher CO2 uptakes of NPC-x samples than POP-x samples. The CO2 uptakes and CO2/N2 selectivities of NPC-x samples are summarized in Tables 3 and 4, respectively. Overall, these values are quite impressive and superior to most porous materials reported in the literature (see Tables S7 and S8).

IAST selectivities of CO2/N2 for NPC-x samples at 0 °C (a) and 25 °C (b)
Figure 9 shows the breakthrough curves for CO2/N2 mixed gas adsorption on NPC-HMTA at 25 °C. The breakthrough time for CO2 is ~ 10 min/g while that for N2 is almost instant, when the mixture of CO2/N2 (0.15/0.85 vol) was used for adsorption. It costs ~ 80 min/g for the concentrations of CO2 in outlet gas reaching the level of inlet gas. Therefore, NPC-x samples do enable highly efficient and selective adsorption of CO2 from CO2/N2 mixed gas. Given that there usually exists some moisture in the industrial stream, the breakthrough curves for humidified CO2/N2 mixed gas (RH = 20%) adsorption on NPC-HMTA at 25 °C were also determined (see Figure S7). They are similar to those for dry gas adsorption, indicating that the presence of minor moisture has no obvious effect on the selective adsorption of CO2 by NPC-x samples. The recyclability of NPC-x samples for CO2 adsorption is very important for the long-term use of these materials. Figure S8 shows the CO2 uptakes of NPC-HMTA at different adsorption–desorption cycles. The adsorption was performed at 25 °C and 100 kPa, and the desorption was performed at 80 °C and vacuum for 1 h. The loss of CO2 uptakes for NPC-HMTA is negligible after five adsorption–desorption cycles. Therefore, the adsorption of CO2 by NPC-x samples is highly reversible.

Breakthrough curves for CO2/N2 mixed gas adsorption on NPC-HMTA at 25 °C
Catalytic CO2 conversion
The synthesized M@NPC-x samples were first characterized by N2 adsorption, ICP and XPS to give the porosity parameters (see Figure S9 and Table S9), metal contents (see Table S9) and chemical nature of loaded metals (see Figure S10). The porosity parameters of M@NPC-x samples are decreased in comparison with those of NPC-x samples, which is within expectation because some of the nanopores in NPC-x samples are occupied by loaded metals. The M@NPC-x samples also have predominantly mesoporous structure, with the mesopore volumes accounting for > 50% of the total pore volumes. Especially for Co@NPC-HMTA, the mesopore volume of which accounts for 91% of the total pore volume. The metal contents of M@NPC-x samples are similar, with the values of 0.62–0.71 mmol/g. The Co 2p spectra of Co@NPC-4,4′-bipyridine show the Co 2p3/2 and 2p1/2 peaks at 780.6 and 795.8 eV, respectively, and two satellites peaks at 786.9 and 802.7 eV. This agrees with the Co 2p spectra of Co2+, suggesting that the loaded Co does not undergo oxidation. The Co 2p spectra of Co@NPC-4,4′-bipyridine show the Co 2p3/2 and 2p1/2 peaks at 780.6 and 795.8 eV, respectively, and two satellites peaks at 786.9 and 802.7 eV. This agrees with the Co 2p spectra of Co2+, suggesting that the loaded Co does not undergo oxidation. The Zn 2p spectra of Zn@NPC-4,4′-bipyridine show the Zn 2p3/2 and 2p1/2 peaks at 1022.4 eV and 1045.4 eV, respectively, also suggesting that the chemical state of loaded Zn is Zn2+.
The catalytic CO2 conversion performance of M@NPC-x samples was then examined by taking the cycloaddition of CO2 with propylene oxide as a model reaction. The yields, selectivities and TOFs of CO2 conversion reaction under different conditions are summarized in Table 5. It can be seen that both Co@NPC-x and Zn@NPC-x samples show excellent performance for catalytic CO2 conversion, giving the product yields of 97.6–99.5%, selectivities of 98.2–99.5% and TOFs of 614.1–716.8 h−1 at 100 °C and 1.0 MPa in 1.5 h (runs 1–4). The activities of M@NPC-x samples for catalytic conversion of CO2 are affected by many factors, such as the CO2 uptakes and porosity parameters. It is difficult to determine that which factor contributes more than others to the catalytic conversion of CO2. Herein, we can only make a qualitative analysis. Co@NPC-p-phenylenediamine has the lowest catalytic activity among the three Co@NPC-x samples, probably because NPC-p-phenylenediamine has the lowest CO2 uptakes among the three NPC-x samples. Although the CO2 uptakes of Co@NPC-HMTA are lower than those of NPC-4,4′-bipyridine, the percentage of mesopores in Co@NPC-HMTA is higher than that in NPC-4,4′-bipyridine (see Table S9). Since mesopores with large pore width are favorable for the diffusion of reactants, Co@NPC-HMTA has slightly higher catalytic activity than NPC-4,4′-bipyridine.

Co@NPC-4,4′-bipyridine still gives a product yield of 99.1% and TOF of 647.7 h−1 even after recycled for 5 times (run 5), which is comparable to the activity of fresh sample. However, the product yield is only 34.9% if no catalyst was used (run 6), and the product yield is only 21.5% if no TBAB was used (run 7). Therefore, the cooperation between M@NPC-x samples and TBAB is essential for the efficient conversion of CO2. It is noteworthy that Co@NPC-4,4′-bipyridine can give a product yield of 97.8% and TOF of 19.7 h−1 when the conversion reaction was performed at 30 °C and 0.3 MPa for 48 h (run 8). Therefore, M@NPC-x samples are also efficient for catalytic CO2 conversion under mild conditions. Overall, the activities of M@NPC-x samples are superior to most heterogenous catalysts reported in the literature (see Table S10). The superior activities of M@NPC-x samples are believed to be attributed to ultrahigh surface areas, micro-mesoporous structure and N-decorated feature of NPC-x samples.
Conclusions
In summary, nitrogen-decorated micro-mesoporous carbons were developed by direct carbonization of porous organic polymers, which were pre-synthesized through alkylation-induced hyper-crosslink of rigid organic bases without the use of any templates. The developed carbons were systematically characterized for porous and chemical structure and also examined for CO2 capture and conversion performance. It is found that the developed carbons have ultrahigh surface areas, abundant mesopores and weak base sites. As a result, the developed carbons enable highly efficient and selective adsorption of CO2 from CO2/N2 mixed gas. The CO2 capacities and CO2/N2 selectivities of developed carbons are superior to most porous materials reported in the literature. After loaded with metal salts, the developed carbons also exhibit high activities for the catalytic conversion of CO2. The activities of metal-loaded carbons for cycloaddition of CO2 with propylene oxide are also superior to most heterogenous catalysts reported in the literature. Based on the results obtained in this work, it is concluded that the developed carbons have promising application in the capture and conversion of CO2.
References
IPCC, AR5 Synthesis Report: Climate Change 2014, https://www.ipcc.ch/report/ar5/syr/.
Geden O (2016) The Paris Agreement and the inherent inconsistency of climate policymaking. Wires Clim Change 7:790–797
Rochelle GT (2009) Amine scrubbing for CO2 capture. Science 325:1652–1654
Rao AB, Rubin ES (2002) A technical, economic, and environmental assessment of amine-based CO2 capture technology for power plant greenhouse gas control. Environ Sci Technol 36:4467–4475
Chen FF, Huang K, Fan JP, Tao DJ (2018) Chemical solvent in chemical solvent: A class of hybrid materials for effective capture of CO2. AIChE J 64:632–639
Liu F, Huang K, Jiang L (2018) Promoted adsorption of CO2 on amine-impregnated adsorbents by functionalized ionic liquids. AIChE J 64:3671–3680
Zhang JB, Peng HL, Liu Y, Tao DJ, Wu PK, Fan JP, Huang K (2019) Highly efficient CO2 capture by polyethyleneimine plus 1-ethyl-3-methylimidazolium acetate mixed absorbents. ACS Sustain Chem Eng 7:9369–9377
Huang K, Zhang JY, Liu F, Dai S (2018) Synthesis of porous polymeric catalysts for the conversion of carbon dioxide. ACS Catal 8:9079–9102
Niu D, Wu Z, Zhang L, Du R, Xu H, Zhang X (2016) Synthesis of cyclic carbonates from epoxides and CO2 in acetonitrile via the synergistic action of BMIMBr and electrogenerated magnesium. Chin J Catal 37:1076–1080
Zhang Z, Xie Y, Li W, Hu S, Song J, Jiang T, Han B (2008) Hydrogenation of carbon dioxide is promoted by a task-specific ionic liquid. Angew Chem Int Ed 47:1127–1129
Shi F, Deng Y, SiMa T, Peng J, Gu Y, Qiao B (2003) Alternatives to phosgene and carbon monoxide: synthesis of symmetric urea derivatives with carbon dioxide in ionic liquids. Angew Chem Int Ed 42:3257–3260
Bi QY, Lin JD, Liu YM, Xie SH, He HY, Cao Y (2014) Partially reduced iridium oxide clusters dispersed on titania as efficient catalysts for facile synthesis of dimethylformamide from CO2, H2 and dimethylamine. Chem Commun 50:9138–9140
Srivastava R, Srinivas D, Ratnasamy P (2005) Zeolite-based organic–inorganic hybrid catalysts for phosgene-free and solvent-free synthesis of cyclic carbonates and carbamates at mild conditions utilizing CO2. Appl Catal A Gen 289:128–134
Zhu A, Jiang T, Han B, Zhang J, Xie Y, Ma X (2007) Supported choline chloride/urea as a heterogeneous catalyst for chemical fixation of carbon dioxide to cyclic carbonates. Green Chem 9:169–172
Ma X, Zou B, Cao M, Chen SL, Hu C (2014) Nitrogen-doped porous carbon monolith as a highly efficient catalyst for CO2 conversion. J Mater Chem A 2:18360–18366
Molla RA, Iqubal A, Ghosh K, Islam M (2016) Nitrogen-doped mesoporous carbon material (N-GMC) as a highly efficient catalyst for carbon dioxide fixation reaction with epoxides under metal-free condition. ChemistrySelect 1:3100–3107
Huang K, Liu F, Fan JP, Dai S (2018) Open and hierarchical carbon framework with ultralarge pore volume for efficient capture of carbon dioxide. ACS Appl Mater Interfaces 10:36961–36968
Liu F, Huang K, Wu Q, Dai S (2017) Solvent-free self-assembly to the synthesis of nitrogen-doped ordered mesoporous polymers for highly selective capture and conversion of CO2. Adv Mater 29:1700445
Wu Q, Huang K, Liu F, Zhang P, Jiang L (2017) Pyridine-functionalized and metallized meso-macroporous polymers for highly selective capture and catalytic conversion of CO2 into cyclic carbonates. Ind Eng Chem Res 56:15008–15016
Huang K, Liu F, Jiang L, Dai S (2017) Aqueous and template-free synthesis of meso-macroporous polymers for highly selective capture and conversion of carbon dioxide. Chemsuschem 10:4144–4149
Beyzavi MH, Klet RC, Tussupbayev S, Borycz J, Vermeulen NA, Cramer CJ, Stoddart JF, Hupp JT, Farha OK (2014) A hafnium-based metal-organic framework as an efficient and multifunctional catalyst for facile CO2 fixation and regioselective and enantioretentive epoxide activation. J Am Chem Soc 136:15861–15864
Li PZ, Wang XJ, Liu J, Lim JS, Zou R, Zhao Y (2016) A triazole-containing metal-organic framework as a highly effective and substrate size-dependent catalyst for CO2 conversion. J Am Chem Soc 138:2142–2145
Gao WY, Wu H, Leng K, Sun Y, Ma S (2016) Inserting CO2 into aryl C–H bonds of metal-organic frameworks: CO2 utilization for direct heterogeneous C–H activation. Angew Chem Int Ed 55:5472–5476
Peng HL, Zhang JB, Zhang JY, Zhong FY, Wu PK, Huang K, Fan JP, Liu F (2019) Chitosan-derived mesoporous carbon with ultrahigh pore volume for amine impregnation and highly efficient CO2 capture. Chem Eng J 359:1159–1165
Huang K, Li ZL, Zhang JY, Tao DJ, Liu F, Dai S (2019) Simultaneous activation and N-doping of hydrothermal carbons by NaNH2: An effective approach to CO2 adsorbents. J CO2 Util 33:405–412
Zhan Y, Han Q, Pan S, Kan X, Mi J, Liu F, Cao Y, Au C, Jiang L (2019) Biomass-derived hierarchically porous carbons abundantly decorated with nitrogen sites for efficient CO2 catalytic utilization. Ind Eng Chem Res 58:7980–7988
Zhang JY, Zhang JB, Li M, Wu Z, Dai S, Huang K (2020) Solvent-free and one-pot synthesis of ultramicroporous carbons with ultrahigh nitrogen contents for sulfur dioxide capture. Chem Eng J 391:123579
Biswal M, Banerjee A, Deo M, Ogale S (2013) From dead leaves to high energy density supercapacitors. Energy Environ Sci 6:1249–1259
Sun L, Tian C, Li M, Meng X, Wang L, Wang R, Yin J, Fu H (2013) From coconut shell to porous graphene-like nanosheets for high-power supercapacitors. J Mater Chem A 1:6462–6470
Gao S, Geng K, Liu H, Wei X, Zhang M, Wang P, Wang J (2015) Transforming organic-rich amaranthus waste into nitrogen-doped carbon with superior performance of the oxygen reduction reaction. Energy Environ Sci 8:221–229
Lee JM, Briggs ME, Hasell T, Cooper AI (2016) Hyperporous carbons from hypercrosslinked polymers. Adv Mater 28:9804–9810
Zhang C, Kong R, Wang X, Xu Y, Wang F, Ren W, Wang Y, Su F, Jiang JX (2017) Porous carbons derived from hypercrosslinked porous polymers for gas adsorption and energy storage. Carbon 114:608–618
Saeed AM, Rewatkar PM, Majedi Far H, Taghvaee T, Donthula S, Mandal C, Sotiriou-Leventis C, Leventis N (2017) Selective CO2sequestration with monolithic bimodal micro/macroporous carbon aerogels derived from stepwise pyrolytic decomposition of polyamide-polyimide-polyurea random copolymers. ACS Appl Mater Interfaces 9:13520–13536
Wang J, Kaskel S (2012) KOH activation of carbon-based materials for energy storage. J Mater Chem 22:23710–23725
Huang K, Chai SH, Mayes RT, Veith GM, Browning KL, Sakwa-Novak MA, Potter ME, Jones CW, Wu YT, Dai S (2015) An efficient low-temperature route to nitrogen-doping and activation of mesoporous carbons for CO2 capture. Chem Commun 51:17261–17264
Yue L, Xia Q, Wang L, Wang L, DaCosta H, Yang J, Hu X (2018) CO2 adsorption at nitrogen-doped carbons prepared by K2CO3 activation of urea-modified coconut shell. J Colloid Interfaces Sci 511:259–267
He X, Ling P, Yu M, Wang X, Zhang X, Zheng M (2013) Rice husk-derived porous carbons with high capacitance by ZnCl2 activation for supercapacitors. Electrochim Acta 105:635–641
Wang X, Liu CG, Neff D, Fulvio PF, Mayes RT, Zhamu A, Fang Q, Chen G, Meyer HM, Jang BZ, Dai S (2013) Nitrogen-enriched ordered mesoporous carbons through direct pyrolysis in ammonia with enhanced capacitive performance. J Mater Chem A 1:7920–7926
Sui ZY, Meng QH, Li JT, Zhu JH, Cui Y, Han BH (2014) High surface area porous carbons produced by steam activation of graphene aerogels. J Mater Chem A 2:9891–9898
Nandi M, Okada K, Dutta A, Bhaumik A, Maruyama J, Derks D, Uyama H (2012) Unprecedented CO2 uptake over highly porous N-doped activated carbon monoliths prepared by physical activation. Chem Commun 48:10257–10356
Han SJ, Hyeon T (1999) Simple silica-particle template synthesis of mesoporous carbons. Chem Commun 19:1955–1956
Kim TW, Ryoo R, Gierszal KP, Jaroniec M, Solovyov LA, Sakamoto Y, Terasaki O (2005) Characterization of mesoporous carbons synthesized with SBA-16 silica template. J Mater Chem 15:1560–1571
Meng Y, Gu D, Zhang F, Shi Y, Yang H, Li Z, Yu C, Tu B, Zhao D (2005) Ordered mesoporous polymers and homologous carbon frameworks: amphiphilic surfactant templating and direct transformation. Angew Chem Int Ed 44:7053–7059
Zhao J, Shu Y, Zhang P (2019) Solid-state cTAB-assisted synthesis of mesoporous Fe3O4 and Au@Fe3O4 by mechanochemistry. Chin J Catal 40:1078–1084
Kan X, Chen X, Chen W, Mi J, Zhang JY, Liu F, Zheng A, Huang K, Shen L, Au C, Jiang L (2019) Nitrogen-decorated, ordered mesoporous carbon spheres as high-efficient catalysts for selective capture and oxidation of H2S. ACS Sustain Chem Eng 7:7609–7618
Ferrero GA, Fuertes AB, Sevilla M (2015) N-doped porous carbon capsules with tunable porosity for high-performance supercapacitors. J Mater Chem A 3:2914–2923
Zhu D, Cheng K, Wang Y, Sun D, Gan L, Chen T, Jiang J, Liu M (2017) Nitrogen-doped porous carbons with nanofiber-like structure derived from poly (aniline-co-p-phenylenediamine) for supercapacitors. Electrochim Acta 224:17–24
Wang X, Lee JS, Tsouris C, DePaoli DW, Dai S (2010) Preparation of activated mesoporous carbons for electrosorption of ions from aqueous solutions. J Mater Chem 20:4602–4608
Watanabe H, Asano S, Fujita SI, Yoshida H, Arai M (2015) Nitrogen-doped, metal-free activated carbon catalysts for aerobic oxidation of alcohols. ACS Catal 5:2886–2894
Mi J, Liu F, Chen W, Chen X, Shen L, Cao Y, Au C, Huang K, Zheng A, Jiang L (2019) Design of efficient, hierarchical porous polymers endowed with tunable structural base sites for direct catalytic elimination of COS and H2S. ACS Appl Mater Interfaces 11:29950–29959
Arrigo R, Havecker M, Schlogl R, Su DS (2008) Dynamic surface rearrangement and thermal stability of nitrogen functional groups on carbon nanotubes. Chem Commun 44:4891–4893
Myers AL, Prausnitz JM (1965) Thermodynamics of mixed-gas adsorption. AIChE J 11:121–127
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21978052 and 22008033), Natural Science Foundation of Jiangxi Province (20192ACB21016) and Natural Science Foundation of Jiangsu Higher Education Institution (19KJB150041).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Maude Jimenez.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the supplementary information.
Rights and permissions
About this article

Cite this article
Wu, X., Guan, R., Zheng, WT. et al. Developing porous organic polymers as precursors of nitrogen-decorated micro-mesoporous carbons for efficient capture and conversion of carbon dioxide. J Mater Sci 56, 9315–9329 (2021). https://doi.org/10.1007/s10853-021-05835-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-05835-z