
Abstract
Morphological effects on the electrochemical behaviors of Tin Dioxide (SnO2) have recently been explored in several studies. Nevertheless, the preparations of SnO2 in those works are mostly based on wet-chemistry methods, while the usage of dry-chemistry methods is still lack of investigation. In this work, for the first time, we report the successful fabrication of SnO2 particles attached on carbon fibers via a simple two-step Chemical Vapor Deposition (CVD) approach. CVD growth parameters (temperature, growth time and gas inlet flow rate) were systematically varied to observe the changes in morphologies (in terms of particle density and size distribution) as well as the formation of core–shell Sn/SnO2 structures; and the electrochemical properties of the as-synthesized samples were studied. Our experiments indicate strong correlations among the synthesis conditions, morphologies and electrochemical behaviors. Overall, our findings provide new insights into CVD as a morphological tailoring approach for electrochemical applications—in addition to its simple apparatus and short synthesizing duration, the process is easily extendable for the preparation of other metal and metal oxide particles.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metal oxides (RuO2 [1, 2], MnO2 [3,4,5], Co3O4 [6,7,8,9], V2O5 [10, 11], SnO2 [12,13,14,15,16,17,18,19,20,21,22]), owing to their excellent electro-activities, are presenting themselves as an important class of materials for electrochemical applications (supercapacitors and Li-ion batteries). Among those metal oxides, the earth-abundant Tin Dioxide (SnO2) has considerably attracted research attention due to its relatively acceptable theoretical capacity (782 mA h g−1), low cost, and non-toxicity [12, 13]. It is well-accepted that the electrochemical properties of SnO2 are significantly affected by size effects and geometrical factors. Hence, during the past decades, there have been dedicated works focusing on the control of synthesis processes to achieve the optimized morphology. Liu et al. prepared hierarchical SnO2 nanostructures by hydrothermal method, in which the sizes and morphologies of the samples could be tuned by varying the precursors’ concentrations (SnCl2·2H2O and C6H5Na3O7·2H2O). Thus, the easy access of electrolyte anions was guaranteed by the thin thickness and hierarchical nature of intermingled nanosheets, leading to the enhanced electrochemical performance [14]. Likewise, using the similar method, Yang and co-workers assembled nanocrystals SnO2 into highly ordered hollow microspheres by varying the amount of SnCl4·5H2O and HCl. The high capacitance value was attributed to the multilayered and mesoporous structures [15]. Laterally, SnO2 quantum dots (~ 2.4 nm) were successfully fabricated by Bonu et al. by modifying the soft chemical method, which had previously been used to synthesize SnO2 nanoparticles (~ 25 nm). In this work, the phenomenon of diffusion layer formation changed the cyclic voltammetry profile of SnO2 quantum dots and therefore made them superior in terms of Columbic efficiency [16].
It could be seen that these aforementioned SnO2 morphological-control processes are all based on wet-chemistry methods (hydrothermal method [14, 15], soft chemical method [16]). The common disadvantage of these methods is the involvement of many chemicals with stepwise and complicated synthesis techniques, which often take more than half a day to complete [14,15,16]. Dry-chemistry methods, on the other hand, are still lack of exploration. In this work, we focus on chemical vapor deposition (CVD) as a new prospective approach to SnO2 preparation and morphological control. The main advantages of CVD are short synthesizing duration, affordable cost, high deposition rate, high versatility, and adaptability to large-scale processing [23]. This approach has shown promising results in the fabrication of SnO2 nanostructures, including SnO2 nanowires (NWs) [24], nanoporous SnO2 thin film [25], and SnO2 nanorods (NRs) [26]. In addition, another attractive feature offered by CVD is the morphological tailoring ability, enabled by the systematic tuning of several CVD operational parameters. In their study on the fabrication of multi-walled carbon nanotubes (MWCNTs), Venkatesan and co-workers revealed that the reaction temperature and the precursor flow rate were directly proportional to the mean diameter of MWCNTs, while the process time showed an inversely proportional relationship [27]. Similarly, the CVD-synthesized MoS2 monolayer was optimized by Kang et al. in which the coverage of MoS2 flakes was influenced by the carrier gas amount and deposition time; while their size and shape were affected by the growth temperature [28]. On the other hand, the deposition rate of C coating on carbon steel prepared by Chowdhury et al. increased with the increase in CVD reactant gas (CH4) flow rate [29]. Lu and co-workers observed longer and thicker ZnO nanorods (NRs) as the synthesis duration increased [30]. In brief, the CVD operational parameters (temperature, growth time, carrier gas/reactant gas flow rate) play a significant role in the formation of different material structures (particles, thin film, nanowires, nanorods, etc.). However, the involvement of CVD method in material synthesis and morphological tailoring for electrochemical applications is still lack of investigation at this moment.
In this study, we report the fabrication of SnO2 particles attached on carbon fibers via the CVD route, in which the effects of aforementioned CVD variables (temperature, growth time, and gas inlet flow rate) are the main focuses. Our findings indicate some correlations between these three parameters and morphological features of the as-prepared particles. Electrochemical characterizations were furthermore performed on some chosen samples to evaluate the effects of morphological changes. Herein, carbon fibers as substrate can offer several advantages, including the cost-effectiveness, disposability, flexibility, lightweight, and high conductivity [31, 32]. Good electrical contact between SnO2 and carbon fibers has previously been reported [33]; besides, the network structure of carbon fibers implies the high level of active sites, which is beneficial for electrochemical processes [34]. Moreover, it is also interesting that by employing carbon fibers as substrate for particles’ deposition – unlike structures such as thin film, particles can easily dispersed and inserted into the macrolayers of fibers, which maximizes the deposition capability. Finally, we obtain SnO2 particles of various sizes and compositions, some of which might have core–shell Sn/SnO2 structure.
Experimental procedure
Chemicals and materials
Tin (II) chloride anhydrous powder (99.99% trace metals basis) was purchased from Sigma-Aldrich and used as the precursor for the CVD growth of SnO2 without further purification. Commercially available Carbon Fiber Paper (CFP) was purchased from Fuel Cell Store (Toray Carbon Paper TGP H090, 280 µm thickness). A piece of CFP (1.5 cm × 1.5 cm), which consists of layers of conductive and macroporous 3D carbon fibers, was used as substrate for the process. Heat treatment (800 °C, 240 min) was performed on the CFP for PTFE (polytetraflouroethylene) removal before the experiments.
Synthesis of SnO2 particles
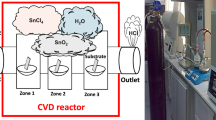
The SnO2 particles were synthesized by a two-step CVD approach (Fig. 1). In the first step, Sn particles were deposited on carbon fiber paper (CFP) by the CVD process. Anhydrous SnCl2 powder (2.0 g) was placed in a quartz boat centered in a tube furnace with a CFP locating at a downstream position as a supporter of Sn particles. Temperature (T), growth time (t) and flow rate of Ar/H2 (95:5 V%) (v) were systematically varied to observe the effects of those CVD parameters on the morphologies of as-synthesized samples (Table 1). In the second step, the samples were annealed under atmosphere at 300 °C for 60 min to oxidize Sn into SnO2.

Schematic diagram of CVD process for SnO2 particles growth
Materials characterizations
The morphological features (size distribution and particle density) of the prepared samples were examined by scanning electron microscopy (SEM, Zeiss SUPRA40, Germany). The samples were then characterized by X-ray diffraction (XRD, Bruker D8 Advanced Thin Film) and transmission electron microscopy (TEM, JEM2010F) to acquire the crystallographic and fine structures information, respectively. In addition, the near-surface chemical composition was studied by X-ray photoelectron spectroscopy (XPS, AXIS Ultra) with an Al Kα X-ray source (1486.7 eV) using CasaXPS software. Due to the charging effect during measurement, the adventitious carbon C 1 s at 284.8 eV was referred for peak shifts calibration.
Electrochemical characterizations
The electrochemical evaluations were performed in the typical three-electrode system under 2 M KOH electrolyte, in which the fabricated samples, Pt rod, and Ag/AgCl electrode served as the working, counter and reference electrode, respectively. The Cyclic Voltammetry (CV) was recorded within a potential range from − 0.9 to 0.6 V at scan rates of 5, 10, 20, 50, 100 mV s−1. Galvanic Discharge was carried out at current density of 1 A/g, with a cut off voltage of − 0.8 to 0.5 V.
Results and discussion
In CVD process, the volatile SnCl2 sublimed (above 600 °C) and reacted with H2 to form Sn and gaseous HCl; while carrier gas Ar played the role of transporting and depositing Sn onto the carbon fibers. The formation of Sn particles consists of two sequential steps. The first step is the nucleation of Sn particles from gaseous precursors, followed by the growth of particles. Sn particles are then oxidized to become SnO2. The chemical reactions involved in the process are listed as follows:
Structural and morphological analysis
Effects of temperature
To observe the effects of temperature, the growth time and gas inlet (Ar/H2) flow rate were kept constant at 60 min and 100 sccm, respectively. The temperature was varied from 700 to 900 °C in steps of 100 °C. The samples were characterized under Scanning Electron Microscopy (SEM), as illustrated by Fig. 2a–c. An increase in the particle diameter is observed as the temperature increases (0.5–7 µm at 700 °C, 7–10 µm at 800 °C and 8–20 µm at 900 °C). Two competing aspects are considered to explain the temperature effects on the particle growth process. Firstly, at higher temperature, the sintering rate becomes high, which significantly enhances the coagulation of small particles to form larger particles [35, 36]. Secondly, due to volume expansion, the residence time of reactants decreases as the temperature increases, implying the less chance for the generated nuclei to grow into larger particles by sintering and coagulation [37]. Since the particle size increases with the CVD temperature, the particle growth mechanism in this experiment should be dominated by the first aspect. It is worth mentioning that at 800 °C, we witness narrower particle size distribution. Based on this observation, the temperature-dependent coagulation process is proposed. As the temperature rises from 700 to 800 °C, small particles with size of 0.5–1 µm coagulate into medium size (7–10 µm). At 900 °C, medium-size particles (8–10 µm) are mixed with large-size particles (16–20 µm), implying the continuity of those medium size particles’ coagulation process.

The SEM images (magnification 500×) of SnO2 particles at different temperatures: a 700 °C (with zoom-in inset of magnification 5000×), b 800 °C, c 900 °C, respectively; d Schematic illustration of temperature-dependent particle-growth process
Effects of growth time
To observe the effects of growth time, the temperature and gas inlet (Ar/H2) flow rate were maintained at 700 °C and 100 sccm, respectively. The growth time was varied from 30 to 90 min in steps of 30 min. Morphological differences, in terms of particle density and size distribution, are clearly observed within the microsphere structures (Fig. 3). Firstly, broader particle size distribution is observed as the deposition time increases (0.3–5 µm after 30 min, 0.5–7 µm after 60 min and 1.5–20 µm after 90 min). We notice that there is merely a slight increase in the particle size as the growth time rises from 30 to 60 min; however, after 60 min, this trend becomes much more significant. On the other hand, as the growth time is extended from 30 to 60 min, there is a substantial increase in the particle density; nonetheless, the density does not show much difference as the time is prolonged to 90 min. In brief, as the deposition time rises from 30 to 60 min, we observe considerably higher particle density; while from 60 to 90 min, the particle-growth process is dominated by the change of size distribution. There is insignificant morphological change after 90 min due to the depletion of SnCl2 precursor. Based on this observation, a time-dependent mechanism of particle-growth is proposed. From 30 to 60 min, the as-produced Sn is dispersed in different positions on carbon fibers, implying an increase in the number of nucleation sites. After 60 min, the disperse process slow down, whereas the existing Sn particles continue growing their size. At 90 min, several particles have sufficient time to grow to relatively large size (more than 20 µm). Until now, the effect of growth time on CVD-synthesized particles is still lack of study; however, there have been research on other CVD-synthesized structures. Kang et al. showed that the coverage of MoS2 triangular flakes and the number of layers increased as the deposition time increased [28]. Likewise, Ivanova et al. also reported an increase in Co thin films’ thickness [38]; while Lu and co-workers described thicker and longer ZnO nanorods as they extended the CVD synthesis duration [30]. Overall, it can be concluded that during the CVD process, the amount of deposited materials increases with the growth time, which is in agreement with our observation.

The SEM images (magnification 500×) of SnO2 particles at different growth times: a 30 min (with zoom-in inset of magnification 5000×), b 60 min (with zoom-in inset of magnification 5000×), c 90 min, respectively; d Schematic illustration of time-dependent particle-growth process
Effects of gas inlet (Ar/H2) flow rate
To observe the effects of Ar/H2 flow rate, the temperature and growth time were respectively kept constant at 700 °C and 60 min, while the flow rate were set at 25, 50, 100, 150 and 175 sccm. At different flow rates of Ar/H2, we observe significant changes in the density and size distribution of the as-synthesized particles on carbon fibers. Two competing factors are considered to explain the gas flow’s effects. The first factor is the diffusion capability of carrier gas (Ar). At low flow rate, the Sn precursors tends to land on the existing nuclei continuously due to the incapability of efficient diffusion; while at higher flow rate, the carrier gas has more power to disperse the Sn in different positions, forming particles with smaller size and higher density. Similar observations showing this correlation were described by Jang et al. [35], Wegner et al. [39], and Youn et al. [40]. The second factor is the amount of reactant gas (H2) passed through the CVD tube. At higher flow rate, there will be higher amount of reactant gas within the same duration, implying the higher deposition rate of Sn. A similar effect was reported by Chowdhury and co-workers, in which the higher flow rate of CH4 as reactant gas led to the higher deposition rate of C coating on Carbon Steel [29]. Based on the morphological analysis under SEM, it could be deduced that at low Ar/H2 flow rate (25–100 sccm), the particle-growth process is dominated by the first factor. Low-density and large-size spherical particles (about 80 µm) are observed at 25 sccm. Besides, it can be noticed that there are some tiny and fine particles (below 1 µm) unevenly and sparsely scattered on the carbon fibers surface. Due to the limited diffusion, the Sn tends to land on the existing nuclei in the overall microscale; however, in the nanoscale it might be stopped and “dropped” on the carbon fibers during the process. When the flow rate becomes higher (Fig. 4b and c), these tiny particles disappear since nanoscopically, the carrier gas can efficiently transport the Sn. In the microscale, as the Ar/H2 flow rate rises to 100 sccm, the particle density becomes higher and the particle size becomes smaller due the enhanced diffusion capability of carrier gas (2–30 µm at 50 sccm and 0.5–7 µm at 100 sccm, respectively). On the other hand, at higher flow rate, i.e., 150 sccm and 175 sccm, the trend becomes different. At 150 sccm, there are dispersed small particles (below 10 µm) mixed with moderately large particles (15–45 µm). Under higher SEM magnification, we furthermore observe spherical particles with size below 1 µm are distributed on the surface of those moderately large particles (15–45 µm) and carbon fibers (Fig. 4d). At 175 sccm, similar observation is noticed as there are dispersed small particles (diameter less than 10 µm) mixed with larger-size particles (15–35 µm). However, instead of spherical particles, there are irregular-shape nanoparticles (size below 100 nm) with very high density explicitly observed on the surface of carbon fibers (Fig. 4f). It is worth mentioning that this phenomenon could not be observed at lower gas flow rates. Overall, an increase in particle density with increasing gas flow rate suggests the diffusion effect of carrier gas (Ar); while the existence of those large-size particles implies the higher deposition rate of Sn, i.e., higher amount of reactant gas (H2) transferred within the same duration. In brief, both aforementioned factors contribute to the particle-growth process as the gas inlet flow rate goes beyond 100 sccm.

The SEM images (magnification 500×) of SnO2 particles at different flow rates: a 25 sccm, b 50 sccm, c 100 sccm, d 150 sccm (with zoom-in inset of magnification 5000×), e 175 sccm, respectively; f the 175 sccm sample at higher magnification of 5000×; g Schematic illustration of gas-flow-dependent particle-growth process
Furthermore, it is interesting to notice the effects of gas flow rate on the surface roughness of as-synthesized particles. The particles’ surface is completely smooth at the Ar/H2 flow rate of 100 sccm under a variety of temperatures and growth times (Fig. 5d–f). Nonetheless, the smoothness is not maintained as the gas flow rate varies, suggesting that an optimum value of gas flow rate is essential to synthesize particles with completely smooth surface. The particles’ surfaces are not completely smooth (25 sccm, Fig. 4a), or experience some arbitrary cracks (50 sccm and 150 sccm, Fig. 5a and b), or witnessed the “organized” breakage of spherical particles to form 2D hexagonal micro-flakes—herein, the signals of micro-flakes formation could be anticipated from the angular patterns on the surface of spherical particles (175 sccm, Fig. 5c). The electrochemical impacts of these morphological features have been reported in several studies. Xu et al. proposed crack propagation as a possible mechanism for capacity fade in electrodes [41]. On the other hand, structures like micro/nano-flowers or micro/nano-flakes are believed to possess large spaces for mitigating the volume change during charge/discharge process of electrochemical devices [42]. We suspect these aforementioned features can be attributed to the local gas pressure difference on the particles’ surfaces at different gas flow rates. Bernoulli’s principle shows that there are correlations between the fluid (gas or liquid) flow speed (v) and the local pressure (p) [43,44,45]. As the gas flow is varied, the local pressure therefore undergoes deviations, leading to stress [46, 47] and subsequently, has some effects on the particles’ surfaces. A thorough investigation on synthesis study and surface science may give a detailed understanding of this phenomenon, which is beyond the scope of our focus. To the best of our knowledge, such effects of CVD gas flow rate variation on the surface roughness of particles have never been reported in any previous studies so far. These observations may therefore open new potential for further investigation in future.

Cracks observed on particles’ surface at a 50 sccm, b 150 sccm; c 2D hexagonal micro-flakes formation observed at 175 sccm; d–f Some representatives of “smooth” surface of particles synthesized at 100 sccm under different temperatures and growth times: d 700 °C, 60 min, e 900 °C, 60 min, f 700 °C, 90 min
Crystallographic and elemental analysis
As mentioned in the previous part, the SnO2 particles were prepared by a two-step CVD route—Sn particles were first deposited on carbon fibers, followed by the oxidation process to become SnO2. The crystalline structures of the samples before and after the oxidation process are further revealed by XRD. In Fig. 6a, the strong characteristics peaks at 30.5°, 32°, 43.8° and 44.9° are attributed to the crystallographic planes of Sn (200), (101), (220) and (211), respectively, indicating the successful deposition of Sn particles (JCPDS No. 04-0673) [48]. After the thermal treatment, the peaks at ~ 33.8°, 37.9°, 51.8° and 54.5°, corresponding to the crystallographic planes SnO2 (101), (200), (211) and (220) respectively, demonstrate the successful conversion of Sn into SnO2 (JCPDS No. 41-1445) [13]. All the other peaks labeled by “Δ” are ascribed to carbon fibers [49, 50]. It is important to notice that for samples with large particle’s size (for example, the sample prepared at 700 °C, 60 min under Ar/H2 flow rate of 25 sccm), the huge size of particles hinders the oxidation process, i.e., Sn is not fully converted to SnO2. As a result, XRD patterns of both Sn and SnO2 can be observed (Fig. S3). This suggests that SN25 might have a core–shell structure, where the inner core consists of Sn metal, and the outer layer consists of SnO2 from Sn metal oxidation.

a XRD analysis before and after oxidation; b XPS overall spectra, c Sn 3d pattern, d C 1 s pattern
The XPS survey spectra of SnO2 are demonstrated in Fig. 6b–d, in which only Sn, O, and, C elements are identified. The narrow scan of Sn 3d are indicated in Fig. 6c—the binding energies of Sn 3d5/2 and Sn 3d3/2 are at around 487 and 496 eV, respectively; with a 9 eV peak-to-peak separation, which further verifies the formation of SnO2 particles on the carbon fibers [51]. The C 1 s spectrum is shown in Fig. 6d, which can be decomposed into 4 components associated with carbon in different oxidized levels: C–C (non-oxidized), C–O, C=O and O=C–O. It suggests the oxidation of carbon fibers during the annealing process.
The samples were further characterized by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). The micro-size particles have a spherical shape (Fig. 7a); while several irregular shapes, in addition to the spherical shape, were found within the nano-size particles (Fig. 7b–c). Overall, the TEM images are in agreement with our previous SEM. The HRTEM result indicates that the samples are crystalline, with the inter-planar spacing d(110) is about 0.31 nm, which is very close to the value of d(110) in SnO2 (0.33 nm) [52].

TEM and HRTEM analysis: a Microsphere, b Nanosphere, c Nanoparticles; d HRTEM image
Electrochemical characterizations
To evaluate the effects of morphological changes on the electrochemical behaviors of the as-prepared particles, Cyclic Voltammetry (CV) and Galvanic Discharge were conducted by using the three-electrode system in 2 M KOH solution. Our experiment focused on the three samples synthesized at 700 °C, 60 min under different Ar/H2 flow rates—175 sccm, 100 sccm, and 25 sccm, which were respectively assigned as SN175, SN100, and SN25. The samples’ electrochemical behaviors, as affected by their substantial morphological differences (in terms of particle density and size distribution), are expected to exhibit significant differences. There have recently been dedicated works exploring the electrochemical properties of SnO2 in alkaline electrolyte (KOH) [16,17,18,19,20]; and in general, there are two charge storage mechanisms. The first one involves the surface adsorption/desorption of electrolyte cations (K+) on SnO2 at the electrode/electrolyte interface, signified by rectangular-shape CV curves:
while the second mechanism involves the intercalation/de-intercalation of electrolyte cations (K+) into the SnO2 crystalline matrix, signified by Faradaic redox peaks:
Figure 8a–d illustrates the CV curves of these three samples beside the CV curve of the substrate (pure carbon fibers) at different scan rates. From Fig. 8b–d, the observed CV curves of SN100 and SN25 are simply the enlargements of the pure carbon fibers’ CV curve. The resulting shapes are believed to originate from the combination of the pure carbon fibers’ CV curve and the rectangular CV curve, suggesting that there is only the first mechanism involved in the charge storage processes of these two samples. At 5 mV s−1, the specific capacity of SN100 is 10.2 C g−1, which is slightly higher than SN25 (4.7 C g−1). On the other hand, SN175 outperforms the other two samples with a specific capacity of 97.9 C g−1 at the same scan rate. The most prominent feature detected in the CV curve of SN175 is a discernible couple of redox reactions’ peaks (the oxidation and reduction peaks are at 0.5 V and 0.2 V, respectively). Similar redox peaks were also experienced by a few previous works [16,17,18], and they were attributed to the second mechanism, i.e., the intercalation/de-intercalation of electrolyte cations into the bulk of SnO2. Compared to the background, this couple of peaks become more prominent as the scan rate decreases. The reason is that at low scan rates, electrolytes ions have sufficient time to access the interior reaction sites of SnO2, leading to the more intense redox peaks and also, the higher capacity; whereas at higher scan rate, their interaction is restricted by the insufficient residence time [53]. Furthermore, with high scan rate, the oxidation and reduction peaks are respectively shifted toward positive and negative potentials, which can be ascribed to the limitation of ionic diffusion to keep pace with the electronic neutralization in redox reaction [54, 55]. Owing to the contribution of both aforementioned mechanisms, the charge storage in SN175 is not limited to the surface (or near-surface) reactions, implying the enhanced specific capacity compared to SN100 and SN25.

CV curves at different scan rates of samples: a SN175, b SN100, c SN25; d Pure carbon fibers in KOH electrolyte at scan rate of 50 mV s−1; e CV curves of 3 analyzed samples and pure carbon fibers at scan rate of 50 mV s−1; f Rate capabilities
The Galvanic Discharge curves of the three analyzed samples at a current density of 1 A g−1 are illustrated in Fig. 9. The non-linear discharging curves are overall in agreement with the Cyclic Voltammetry results. For SN175, a plateau observed at the potential of 0.2 V confirms the reduction reaction, i.e., the intercalation of electrolyte cations K+ into the SnO2 crystalline structure. As a result, Fig. 9 indicates the discharging time of SN175 is dramatically enhanced (277.5 s) compared to SN100 (26.7 s) and SN25 (9.3 s). The specific capacity can also be calculated from the Galvanic Discharge curve, which is directly proportional to the discharging time if the current densities are equal for every sample [56]. Accordingly, SN175 has the calculated specific capacity of 277.5 C g−1, which is substantially higher than those values of SN100 and SN25 (26.7 C g−1 and 9.3 C g−1, respectively).

Discharge curves of 3 analyzed samples at a current density of 1 A/g
Overall, it can be seen that SN100 has slightly higher specific capacity than SN25, while SN175 outperforms both SN100 and SN25. By comparing among these samples, we can correlate their electrochemical behaviors to the morphological features in terms of particles’ size and density. Firstly, the existence of high-density irregular-shape nanoparticles in SN175 means the larger specific surface area, i.e., the higher level of contact between electrolyte ions (K+) and electrode’s active material (SnO2 particles). This feature enables more surface adsorption / desorption and more probability for the intercalation process of K+ ions, therefore enhances both surface and Faradaic charge storage. Secondly, the nanoscale morphology allows shorter conducting pathway for electron transfer from carbon fibers to the active material / electrolyte interface, leading to the better electric conductivity. On the other hand, the microscale morphologies of SN100 and SN25 imply their lower specific capacities compared to SN175. Herein SN100 still shows slightly higher specific capacity, which can be ascribed to the stronger surface adsorption/desorption supported by the larger specific surface area. Furthermore, apart from the aforementioned factors, the lowest performance of SN25 can also be attributed to its large particles’ size, which hinders the oxidation process of Sn and subsequently, inhibits the electro-activity of the active material. In this case, the variation of Ar/H2 flow rate in CVD process enables significant morphological changes among the as-synthesized samples and hence, shows the effects on their electrochemical behaviors.
Conclusion
In summary, SnO2 particles attached on carbon fibers were successfully fabricated via a two-step CVD approach. We have studied the effects of CVD growth parameters (temperature, growth time and gas inlet flow rate) on the as-prepared samples’ morphologies. The temperature mainly affects the particles’ size; while the growth time and gas flow rate have impacts on both particle density and size distribution. Possible growth mechanisms were proposed based on those observations. Among the aforementioned parameters, gas flow rate shows the most significant influence and therefore, samples synthesized under different flow rates were electrochemically characterized to evaluate the dependence of their electrochemical behaviors on the morphologies. We found that the sample synthesized under high Ar/H2 flow rate (175 sccm) outperforms the other two analyzed samples in terms of specific capacity. This better performance can be ascribed to the larger specific surface area and the shorter electron conducting pathway supported by the nanoscale morphology. Overall, our study provides new insights into CVD as an alternative to the traditional wet-chemistry approach for electrochemical applications – in addition to the morphological tailoring potential, the process offers simple apparatus, short synthesizing duration and extendibility for the synthesis of other metal and metal oxide particles.
References
Ryu I, Yang M, Kwon H, Park HK, Do YR, Lee SB, Yim S (2014) Coaxial RuO2–ITO nanopillars for transparent supercapacitor application. Langmuir 30(6):1704–1709. https://doi.org/10.1021/la4044599
Bi R-R, Wu X-L, Cao F-F, Jiang L-Y, Guo Y-G, Wan L-J (2010) Highly dispersed RuO2 nanoparticles on carbon nanotubes: facile synthesis and enhanced supercapacitance performance. J Phys Chem C 114(6):2448–2451. https://doi.org/10.1021/jp9116563
He Y, Chen W, Li X, Zhang Z, Fu J, Zhao C, Xie E (2012) Freestanding three-dimensional graphene/MnO2 composite networks as ultralight and flexible supercapacitor electrodes. ACS Nano 7(1):174–182. https://doi.org/10.1021/nn304833s
Chen K, Noh YD, Li K, Komarneni S, Xue D (2013) Microwave-hydrothermal crystallization of polymorphic MnO2 for electrochemical energy storage. J Phys Chem C 117(20):10770–10779. https://doi.org/10.1021/jp4018025
Kim JS, Shin SS, Han HS, Oh LS, Kim DH, Kim J-H, Kim JY (2013) 1-D structured flexible supercapacitor electrodes with prominent electronic/ionic transport capabilities. ACS Appl Mater Interfaces 6(1):268–274. https://doi.org/10.1021/am404132j
Rakhi RB, Chen W, Cha D, Alshareef HN (2012) Substrate dependent self-organization of mesoporous cobalt oxide nanowires with remarkable pseudocapacitance. Nano Lett 12(5):2559–2567. https://doi.org/10.1021/nl300779a
Meher SK, Rao GR (2011) Ultralayered Co3O4 for high-performance supercapacitor applications. J Phys Chem C 115(31):15646–15654. https://doi.org/10.1021/jp201200e
Deori K, Ujjain SK, Sharma RK, Deka S (2013) Morphology controlled synthesis of nanoporous Co3O4 nanostructures and their charge storage characteristics in supercapacitors. ACS Appl Mater Interfaces 5(21):10665–10672. https://doi.org/10.1021/am4027482
Guan Q, Cheng J, Wang B, Ni W, Gu G, Li X, Nie F (2014) Needle-like Co3O4 anchored on the graphene with enhanced electrochemical performance for aqueous supercapacitors. ACS Appl Mater Interfaces 6(10):7626–7632. https://doi.org/10.1021/am5009369
Zhu J, Cao L, Wu Y, Gong Y, Liu Z, Hoster HE, Vajtai R (2013) Building 3D structures of vanadium pentoxide nanosheets and application as electrodes in supercapacitors. Nano Lett 13(11):5408–5413. https://doi.org/10.1021/nl402969r
Sathiya M, Prakash AS, Ramesha K, Tarascon JM, Shukla AK (2011) V2O5-anchored carbon nanotubes for enhanced electrochemical energy storage. J Am Chem Soc 133(40):16291–16299. https://doi.org/10.1021/ja207285b
Wang Y, Huang ZX, Shi Y, Wong JI, Ding M, Yang HY (2015) Designed hybrid nanostructure with catalytic effect: beyond the theoretical capacity of SnO2 anode material for lithium ion batteries. Sci Rep. https://doi.org/10.1038/srep09164
Hu Z, Xie Y, Wang Y, Mo L, Yang Y, Zhang Z (2009) Polyaniline/SnO2 nanocomposite for supercapacitor applications. Mater Chem Phys 114(2–3):990–995. https://doi.org/10.1016/j.matchemphys.2008.11.005
Liu Y, Jiao Y, Zhang Z, Qu F, Umar A, Wu X (2014) Hierarchical SnO2 nanostructures made of intermingled ultrathin nanosheets for environmental remediation, smart gas sensor, and supercapacitor applications. ACS Appl Mater Interfaces 6(3):2174–2184. https://doi.org/10.1021/am405301v
Yang Y, Ren S, Ma S, Hao C, Ji M (2015) Hollow tin dioxide microspheres with multilayered nanocrystalline shells for pseudocapacitor. Electrochim Acta 155:437–446. https://doi.org/10.1016/j.electacta.2014.12.166
Bonu V, Gupta B, Chandra S, Das A, Dhara S, Tyagi A (2016) Electrochemical supercapacitor performance of SnO2 quantum dots. Electrochim Acta 203:230–237. https://doi.org/10.1016/j.electacta.2016.03.153
Cui H, Liu Y, Ren W, Wang M, Zhao Y (2013) Large scale synthesis of highly crystallized SnO2 quantum dots at room temperature and their high electrochemical performance. Nanotechnology 24(34):345602. https://doi.org/10.1088/0957-4484/24/34/345602
Thorat GM, Jadhav HS, Chung W-J, Seo JG (2018) Collective use of deep eutectic solvent for one-pot synthesis of ternary Sn/SnO2@C electrode for supercapacitor. J Alloy Compd 732:694–704. https://doi.org/10.1016/j.jallcom.2017.10.176
He C, Xiao Y, Dong H, Liu Y, Zheng M, Xiao K, Lei B (2014) Mosaic-structured SnO2@C porous microspheres for high-performance supercapacitor electrode materials. Electrochim Acta 142:157–166. https://doi.org/10.1016/j.electacta.2014.07.077
Chen M, Wang H, Li L, Zhang Z, Wang C, Liu Y, Gao J (2014) Novel and facile method, dynamic self-assemble, to prepare SnO2/rGO droplet aerogel with complex morphologies and their application in supercapacitors. ACS Appl Mater Interfaces 6(16):14327–14337. https://doi.org/10.1021/am5036169
Wu M, Zhang L, Wang D, Xiao C, Zhang S (2008) Cathodic deposition and characterization of tin oxide coatings on graphite for electrochemical supercapacitors. J Power Sour 175(1):669–674. https://doi.org/10.1016/j.jpowsour.2007.09.062
Sephra PJ, Baraneedharan P, Sivakumar M, Thangadurai TD, Nehru K (2018) Size controlled synthesis of SnO2 and its electrostatic self- assembly over reduced graphene oxide for photocatalyst and supercapacitor application. Mater Res Bull 106:103–112. https://doi.org/10.1016/j.materresbull.2018.05.038
Bekermann D, Barreca D, Gasparotto A, Maccato C (2012) Multi-component oxide nanosystems by chemical vapor deposition and related routes: challenges and perspectives. Cryst Eng Comm 14(20):6347. https://doi.org/10.1039/c2ce25624j
Zervos M, Othonos A (2012) A systematic study of the nitridation of SnO2 nanowires grown by the vapor liquid solid mechanism. J Cryst Growth 340(1):28–33. https://doi.org/10.1016/j.jcrysgro.2011.11.063
Liu Y, Koep E, Liu M (2005) A highly sensitive and fast-responding SnO2 sensor fabricated by combustion chemical vapor deposition. Chem Mater 17(15):3997–4000. https://doi.org/10.1021/cm050451o
Pan J, Zhang J, Shen H, Xiong Q, Mathur S (2012) Correction to “switchable wettability in SnO2 nanowires and SnO2@SnO2 heterostructures”. J Phys Chem C 116(25):13835–13836. https://doi.org/10.1021/jp305167t
Venkatesan S, Visvalingam B, Mannathusamy G et al (2018) Effect of chemical vapor deposition parameters on the diameter of multi-walled carbon nanotubes. Int Nano Lett 8:297–308. https://doi.org/10.1007/s40089-018-0252-4
Kang SK, Lee HS (2019) Study on Growth parameters for monolayer MoS2 synthesized by CVD using solution-based metal precursors. Appl Sci Converg Technol 28(5):159–163. https://doi.org/10.5757/asct.2019.28.5.159
Chowdhury MA, Nuruzzaman DM, Rahaman ML (2011) The effect of gas flow rate on the thin film deposition rate on carbon steel using thermal CVD. Int J Chem Reactor Eng. https://doi.org/10.2202/1542-6580.2673
Lu W, Jiang C, Caudle D, Tang C, Sun Q, Xu J, Song J (2013) Controllable growth of laterally aligned zinc oxide nanorod arrays on a selected surface of the silicon substrate by a catalyst-free vapor solid process—a technique for growing nanocircuits. Phys Chem Chem Phys 15(32):13532. https://doi.org/10.1039/c3cp51558c
Yu M, Zhai T, Lu X, Chen X, Xie S, Li W, Tong Y (2013) Manganese dioxide nanorod arrays on carbon fabric for flexible solid-state supercapacitors. J Power Sour 239:64–71. https://doi.org/10.1016/j.jpowsour.2013.03.083
Luo Y, Jiang J, Zhou W, Yang H, Luo J, Qi X, Yu T (2012) Self-assembly of well-ordered whisker-like manganese oxide arrays on carbon fiber paper and its application as electrode material for supercapacitors. J Mater Chem 22(17):8634. https://doi.org/10.1039/c2jm16419a
Saha MS, Li R, Cai M, Sun X (2007) High electrocatalytic activity of platinum nanoparticles on SnO2 nanowire-based electrodes. Electrochem Solid State Lett 10(8):B130. https://doi.org/10.1149/1.2745632
Zhang Y, Hu Z, Liang Y, Yang Y, An N, Li Z, Wu H (2015) Growth of 3D SnO2 nanosheets on carbon cloth as a binder-free electrode for supercapacitors. J Mater Chem A 3(29):15057–15067. https://doi.org/10.1039/c5ta02479j
Jang HD, Hwang DW, Kim DP, Kim HC, Lee BY, Jeong IB (2004) Preparation of cobalt nanoparticles by hydrogen reduction of cobalt chloride in the gas phase. Mater Res Bull 39(1):63–70. https://doi.org/10.1016/j.materresbull.2003.09.023
Okuyama K, Kousaka Y, Tohge N, Yamamoto S, Wu JJ, Flagan RC, Seinfeld JH (1986) Production of ultrafine metal oxide aerosol particles by thermal decomposition of metal alkoxide vapors. AIChE J 32(12):2010–2019. https://doi.org/10.1002/aic.690321211
Jang HD, Jeong J (1995) The effects of temperature on particle size in the gas-phase production of TiO2. Aerosol Sci Technol 23(4):553–560. https://doi.org/10.1080/02786829508965337
Ivanova AR (1999) The effects of processing parameters in the chemical vapor deposition of cobalt from cobalt tricarbonyl nitrosyl. J Electrochem Soc 146(6):2139. https://doi.org/10.1149/1.1391904
Wegner K, Walker B, Tsantilis S, Pratsinis SE (2002) Design of metal nanoparticle synthesis by vapor flow condensation. Chem Eng Sci 57(10):1753–1762. https://doi.org/10.1016/s0009-2509(02)00064-7
Youn W, Kim C, Hwang N (2013) Effect of the carrier gas flow rate on the microstructure evolution and the generation of the charged nanoparticles during silicon chemical vapor deposition. J Nanosci Nanotechnol 13(10):7127–7130. https://doi.org/10.1166/jnn.2013.7669
Xu J, Deshpande RD, Pan J, Cheng YT, Battaglia VS (2015) Electrode side reactions, capacity loss and mechanical degradation in lithium ion batteries. J Electrochem Soc. https://doi.org/10.1149/2.0291510jes
Meng X, Zhou M, Li X, Yao J, Liu F, He H, Zhang Y (2013) Synthesis of SnO2 nanoflowers and electrochemical properties of Ni/SnO2 nanoflowers in supercapacitor. Electrochim Acta 109:20–26. https://doi.org/10.1016/j.electacta.2013.07.052
Lindsay GA (1952) Pressure energy and Bernoulli's principle. Am J Phys 20(2):86–88. https://doi.org/10.1119/1.1933123
Qin R, Duan C (2017) The principle and applications of Bernoulli equation. J Phys Conf Ser 916:012038. https://doi.org/10.1088/1742-6596/916/1/012038
Arakeri JH (2000) Bernoulli’s equation. Resonance 5(8):54–71. https://doi.org/10.1007/bf02837937
Nagaev EL (1991) Surface forces and chemical potential of small particles. Phys Status Solidi (b) 167(2):381–404. https://doi.org/10.1002/pssb.2221670202
Sanfeld A, Steinchen A (2000) Surface energy, stress, capillary-elastic pressure and chemical equilibrium constant in nanoparticles. Surf Sci 463(3):157–173. https://doi.org/10.1016/s0039-6028(00)00644-0
Arora N, Jagirdar BR (2014) From (Au5Sn·AuSn) physical mixture to phase pure AuSn and Au5Sn intermetallic nanocrystals with tailored morphology: digestive ripening assisted approach. Phys Chem Chem Phys 16(23):11381–11389. https://doi.org/10.1039/c4cp00249k
Ungár T, Gubicza J, Ribárik G, Pantea C, Zerda T (2002) Microstructure of carbon blacks determined by X-ray diffraction profile analysis. Carbon 40(6):929–937. https://doi.org/10.1016/s0008-6223(01)00224-x
Si Q, Matsui M, Horiba T, Yamamoto O, Takeda Y, Seki N, Imanishi N (2013) Carbon paper substrate for silicon–carbon composite anodes in lithium–ion batteries. J Power Sour 241:744–750. https://doi.org/10.1016/j.jpowsour.2013.05.090
Li F, Song J, Yang H, Gan S, Zhang Q, Han D, Ivaska A, Niu L (2009) One step synthesis of graphene/SnO2 nanocomposites and its application in electrochemical supercapacitors. Nanotechnology 20(45):455602. https://doi.org/10.1088/0957-4484/20/45/455602
Rani R, Sharma S (2016) Preparation and Characterization of SnO2 Nanofibers via Electrospinning. Adv Nanopart 05(01):53–59. https://doi.org/10.4236/anp.2016.51006
Yang Y, Ren S, Song X, Guo Y, Si D, Jing H, Ji M (2016) Sn@SnO2 attached on carbon spheres as additive-free electrode for high-performance pseudocapacitor. Electrochim Acta 209:350–359. https://doi.org/10.1016/j.electacta.2016.05.105
Dar F, Moonooswamy K, Es-Souni M (2013) Morphology and property control of NiO nanostructures for supercapacitor applications. Nanoscale Res Lett 8(1):363. https://doi.org/10.1186/1556-276x-8-363
Li X, Xiong S, Li J, Bai J, Qian Y (2012) Mesoporous NiO ultrathin nanowire networks topotactically transformed from α-Ni(OH)2 hierarchical microspheres and their superior electrochemical capacitance properties and excellent capability for water treatment. J Mater Chem 22(28):14276. https://doi.org/10.1039/c2jm32559d
Du D, Lan R, Humphreys J, Xu W, Xie K, Wang H, Tao S (2017) Synthesis of NiMoS4 for high-performance hybrid supercapacitors. J Electrochem Soc. https://doi.org/10.1149/2.0071713jes
Acknowledgements
The authors acknowledge the support from the Ministry of Education and the National University of Singapore (Grant Nos: R284-000-142-112, R284-000-150-112). S.H.Y. and J.X.Y. would like to acknowledge NUS Research Scholarship funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Joshua Tong.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nguyen, T.K., Yu, S.H., Yan, J. et al. SnO2-anchored carbon fibers chemical vapor deposition (CVD) synthesis: effects of growth parameters on morphologies and electrochemical behaviors. J Mater Sci 55, 15588–15601 (2020). https://doi.org/10.1007/s10853-020-05121-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-05121-4