
Abstract
A series of Pt-VOx/γ-Al2O3 catalysts with vanadium loading corresponding to 5 and 20 wt% V2O5 was prepared by impregnation method. The catalytic performance of the obtained materials was investigated in propene deep oxidation reaction. The catalysts were characterized by N2 physisorption, chemical analysis, X-ray diffraction (XRD), temperature-programmed reduction (TPR), electron paramagnetic resonance (EPR) spectroscopy, and UV–Vis diffuse reflectance spectroscopy. Alternating current (AC) electrical conductivity measurements (differential steps technique) were performed in situ during propene oxidation, using an own design reactivity cell. The variation in the measured electrical conductance brings information about the relative oxidation state of the catalyst's surface during reaction. It was found by TPR, EPR, and UV–Vis that the presence of platinum facilitates the reduction of V5+ to V4+ species. The catalytic performance of the investigated catalysts was discussed in relation to their structure, reducibility, and conductance.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Catalytic combustion is largely used for controlling the emission of toxic, volatile organic compounds (VOCs) due to its flexibility and energy saving in comparison with the classical thermal combustion processes [1]. It offers the advantage that VOC’s can be removed from effluents up to very low levels at relatively low temperatures. Noble metals (such as Pt or Pd) dispersed on high surface area metal oxides (like alumina [2, 3] or silica [4]) or on activated carbon [5] are typical combustion catalysts. These types of catalysts are the most active for combustion, but they are rather unstable and expensive [6]. Alternatively, vanadium, tungsten, molybdenum, uranium, chromium, cobalt, titanium, and cerium oxides are frequently used as the oxidation catalysts. Such catalysts can be regarded as electronic promoters owing to their redox activity, as they are all oxides of polyvalent metals. Due to this variability of electronic configuration, these catalysts are extremely active in oxidation reactions [7, 8].
Vanadium oxides are used in industrial applications such as gas sensors, electrochemical and optical switching devices, reversible cathode materials for Li batteries, and as catalysts [7]. The catalysts containing vanadia have been extensively used in oxidation reactions like (1) selective oxidation or oxidative dehydrogenation (ODH) of hydrocarbons [9,10,11,12,13] (for instance, V2O5/Al2O3 is a good catalyst for oxidative dehydrogenation of short chain alkanes and (2) total oxidation of air pollutants including hydrocarbons, aromatics, chlorinated compounds, furans, and dioxins [14,15,16].
The oxidation of hydrocarbons over vanadia or supported vanadia catalysts occurs via the Mars van Krevelen (redox) mechanism [17], in which the hydrocarbon reacts with lattice oxygen resulting in the appearance of anion vacancies. The re-oxidation of the catalyst occurs in a separate step with gas-phase oxygen. Based on this mechanism, the redox properties of the catalyst are important factors since they determine its activity and selectivity. For instance, an easily reducible catalyst has (at least initially) a high activity (its long-term action is dependent also on the facile re-oxidation). In agreement with these considerations, Vassileva et al. [18] found that Pd/V2O5/Al2O3 catalysts were more active than V2O5/Al2O3 and Pd/Al2O3 ones in benzene oxidation, whereas Garcia et al. [16] showed that vanadium-promoted Pt/Al2O3 catalysts were very active in total combustion of propane, more reactive than V-free Pt/Al2O3 catalyst. These results were attributed to the promotion of reduction of vanadium oxide by metal addition.
Surface conductivity of a catalyst is sensitive to a redox process induced by the presence of the chemisorbed species but also to many other changes (e.g., phase composition and structure) occurring during operation in catalytic conditions [19]. Thus, conductivity measurements are especially useful for studying redox-active materials under catalytic reaction conditions. It has been demonstrated that information concerning the changes of the topography of the catalyst surface, when exposed to certain oxidizing, reducing, or inert atmospheres, can be obtained by electrical properties' measurements [20, 21]. More extended information about the nature of the interaction between catalyst's surface and reactants is obtained when these measurements, performed in gas flow and under the influence of the reactants/temperature (in situ conditions), are associated with other physicochemical characterization techniques (microscopic/spectroscopic).
The aim of this paper is to study the catalytic properties of a series of Pt-VOx/γ-Al2O3 catalysts in propene oxidation, by using information concerning vanadia reducibility obtained by means of electrical conductivity measurements (performed in situ), EPR spectroscopy, diffuse reflectance UV–Vis spectroscopy, and TPR. The catalysts have been also characterized by XRD, BET surface area, and ICP-AES techniques. For better understanding of the redox processes, the equivalent materials without Pt addition have also been examined.
Experimental
Materials and synthesis
The 5 wt% V2O5/γ-Al2O3 and 20 wt% V2O5/γ-Al2O3 samples, denoted as V5Al and V20Al, respectively, were prepared by a classical wet impregnation method using a commercial alumina support (Degussa, oxide C, SBET = 108 m2/g) and NH4VO3 from Fluka, as described elsewhere [22]. The amount of the active oxide required for the formation of a theoretical monolayer was estimated as corresponding to 0.145 wt% V2O5 per one m2 of the support [23]. Therefore, in the case of γ-Al2O3 support used in this work, the monolayer coverage would correspond to 15.7 wt% V2O5/Al2O3. As for the supported vanadium oxide catalysts, the nature of the support and the vanadium loading are parameters playing a key role in their catalytic performance [24, 25]. Thus, the loading values of vanadia (5 and 20 wt%) were, respectively, chosen here to correspond to sub- and over-monolayer formation. The corresponding concentration of vanadium atoms is 2.8 and 11.2%, respectively.
To obtain (0.4 wt%)Pt-(5 wt%)V2O5/γ-Al2O3 and (0.4 wt%)Pt-(20 wt%)V2O5/γ-Al2O3 catalysts, denoted as PtV5Al and PtV20Al, respectively, the corresponding V2O5/γ-Al2O3 samples were impregnated with a solution of hexachloroplatinic acid (H4PtCl6•6H2O). In a typical experiment, the required quantity of a precursor necessary to obtain a 0.4 wt% Pt was dissolved in a minimum volume of distilled water containing the immersed V2O5/γ-Al2O3 powder. The mixture was stirred at room temperature for 4 h. After drying overnight at 120 °C, the samples were calcined in air flow at 500 °C for 5 h and then cooled down under the same gas flow.
Characterization
The samples were primarily characterized using BET, ICP-AES, and XRD techniques. BET specific surface areas of the samples were determined from N2 adsorption at − 196 °C. Prior to surface area determination, the powders were out-gassed at 400 °C. The concentrations of the supported vanadium and platinum were determined by chemical analysis using inductively coupled plasma atomic emission spectroscopy (ICP-AES). For Pt analysis, the catalysts were dissolved in a mixture of HCl + HNO3 and heated up to 250–300 °C. Similarly, prior to vanadium analysis, the samples were dissolved in a mixture of inorganic acids (H2SO4 + HNO3) and heated up to 250–300 °C. X-ray diffraction of the investigated catalysts was performed using a Phillips PW 1710 diffractometer under the CuKα incident beam, within 2θ range of 10°–70°. The measurements were taken with a 0.02° step and 1-s interval between steps.
The EPR spectra of the samples, before and after their use in the electrical conductivity experiments, were recorded at liquid nitrogen temperature with a Bruker ELEXSYS E580 spectrometer operating at X-band microwave frequency domain with a 100 kHz field modulation. Computer simulations of the EPR spectra were carried out with EPRsim32 software [26]. The intensity of EPR signals was obtained as double integral of the spectrum. Temperature-programmed oxidation of the samples (10 mg) in air was carried out in EPR tubes in static conditions. The samples were heated progressively in tubular furnace with 10 °C/min heating rate, and the spectra were recorded after quenching at liquid nitrogen.
Diffuse reflectance UV–Vis spectra were recorded by using a PerkinElmer Lambda 35 spectrophotometer, equipped with an integrating sphere. The measurements were carried out in the spectral range of 1100–200 nm, using spectralon as a reference. The reflectance measurements were converted to absorption spectra using the Kubelka–Munk function F(R).
The TPR experiments were performed using a ChemBET-3000 Quantachrome equipped with a TCD sensor. The samples (0.05–0.1 g) were heated from ambient temperature up to 850 °C with a temperature ramp of 10 °C/min, and the overall flow of a 5 vol.% H2 in Ar gas mixture was measured as 70 cm3/min. To maintain system stability/sensibility and also to avoid water vapor influence, a silica trap was added to the experimental TPR setup.
Electrical conductivity measurements and catalytic tests
The variations in the AC electrical conductance of the catalyst powders as a function of temperature and composition of the gaseous atmosphere were measured in situ, using the differential steps technique (DST) and a specially designed reaction cell fully described elsewhere [27, 28]. The experiments were intended to simulate as good as possible the real conditions of testing the catalysts [27, 28, 29]. Briefly, this cell consists of two coaxial tantalum cylinders as electrodes (CE) supported on a frit, embedded in a Pyrex glass tube and connected by tungsten wires to a precision RLC bridge (TESLA BM484), which allows simultaneous measurements of the AC parallel electrical conductance, Gp, at a fixed frequency (1592 Hz) and real-time monitoring of the composition of the inlet/outlet gas by online gas chromatography (Fig. 1). The annular space between the electrodes was filled with the catalyst powder (1.5 cm3, grain fraction between 0.25 and 0.5 mm). Gases can enter and exit from the cell connected online with a GC-TCD which monitored in continuous the qualitative and quantitative gas composition.

Schematic diagram of simultaneous electrical–catalytic measurements, with: 1-tungsten wires, 2-coaxial tantalum electrodes, 3-ceramic frit
The same batch of catalyst was used for measurements under specific cycles of heating–cooling in various atmospheres in gas flow, by changing the gases according with a specific protocol. The gases used were of research grade (99.99% purity). In the present study, the protocol of sample activation and changing the gases was as follows:
The surface stabilization by evacuation was intentionally avoided; instead, three heating–cooling cycles in dry argon (DAr) flow were used to 'clean' the surface of atmospheric contaminants (DAr1 DAr2 DAr3). This treatment was followed by heating–cooling cycle in dry oxygen (DO), and the surface stability was checked by measuring the surface conductance on re-heating in dry argon (DAr4). Finally, the conductance was also obtained when the sample was heated in a propene-air mixture of 1:22 molar ratio, namely CT run. Before each heating run (between 30 and 400 °C with a ramp of 2 °C/min), the sample was flushed for 30 min with the corresponding gas at room temperature. Cooling was done in the same atmosphere as heating, then the cell was closed, and the sample was kept overnight in this atmosphere.
The catalytic performances in propene oxidation (selected as a model reaction for VOC’s abatement) were evaluated during the CT run of the electrical conductivity protocol, by periodically sampling the effluent into the gas chromatograph (Fig. 1). A flow of a C3H6-air mixture of 1:22, at a contact time of 1.1 s, was used. Gas analysis was performed online with a GC (TCD detector) equipped with two parallel columns (Porapak Q and molecular sieves 5 Å) with helium as a carrier gas.
C3H6 conversion (C) was calculated from propene consumption, based on the formula:
where [C3H6]out and [C3H6]in represent the C3H6 concentration (vol%) at the reactor outlet and inlet, respectively.
The selectivity to a product (SP) was defined as the concentration ratios of the propene converted into a specified component, P, to the total transformed propene and calculated accordingly to the following equation:
where [C3H6]P is the concentration of propene transformed to product P (vol%). These transient values obtained, even if not absolute as in the case of stationary state data, are informative in terms of the trend of propene conversion.
In the following, a used sample denotes the sample removed from the conductivity cell after completion of the protocol described above.
Results and discussion
Catalyst characterization
Basic characteristics of the investigated Pt-VOx/γ-Al2O3 catalysts, including BET surface areas and the weight percent of vanadium deposited on γ-Al2O3 as determined by chemical analysis, are listed in Table 1. Based on the vanadium content and specific surface area, the vanadium surface coverage (V atoms per nm2) was calculated for each catalyst. For comparison, the results obtained for the reference materials without Pt (VOx/γ-Al2O3) are also included in Table 1.
The obtained surface areas of the supported vanadia catalysts were slightly lower than that characteristic of alumina support (108 m2/g) and decreased with the increasing vanadium oxide loading. The small decrease is due to the added mass and indicates that the overall morphology of the alumina support is preserved [30]. Either the addition of platinum did not essentially modify morphology (the BET surface areas of Pt-containing catalysts are almost the same as for their precursors).
The actual amount of vanadium deposited on γ-alumina for PtV20Al sample was found to be the same to the desired value, 11.2 wt% nominal, while for the low loading catalyst slightly lower amount of vanadium (2.60 wt% respect to 2.80 wt% nominal) was found in the final powders. Similar results were obtained for the samples without Pt. The content of Pt found by chemical analysis remained in agreement with the expected loading (0.4 wt% Pt).
The vanadia dispersed on alumina support were extensively studied. Literature data [7, 31] mention that at low loading, vanadia appear dispersed as monovanadates consisting mainly of VO4 units, showing a V=O double bond and three M–O–V bonds on the support. At moderately higher vanadia loadings, polyvanadate species forming a two-dimensional network on the support exist. If vanadia loading exceeds the monolayer coverage, V2O5 crystallites appear. A theoretical monolayer for two-dimensional isolated monovanadates/polyvanadates coverage corresponds to a surface density of 2.9 V atoms/nm2 or 8.8 V atoms/nm2, respectively [32, 33]. Crystalline V2O5 species appears only at much higher coverage, i.e., 14.2 V/nm2, although it could start to form at coverage of 4.4 V/nm2 [34].
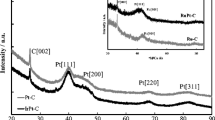
The studied samples with low loading vanadia show a surface density value of ~ 3 atoms/nm2 (Table 1) which is near the theoretical monolayer coverage of monovanadate surface species, as it was found in the literature and mentioned above [32, 33]. So, it could be supposed that vanadia in V5Al and PtV5Al mainly exist as monovanadates and some polyvanadates species if a good dispersion of vanadia was obtained. The VOx surface densities calculated for V20Al and PtV20Al catalysts are higher than the characteristic value for theoretical monolayer coverage of polyvanadates, so the formation of crystalline V2O5 surface species was expected. Indeed, for the low-vanadia loading samples only γ-alumina diffraction lines can be observed in the XRD pattern (Fig. 2). This can be explained considering that either the vanadium oxide is uniformly distributed on the alumina surface or that vanadium oxide does not form a crystalline phase.

XRD patterns of PtV5Al and PtV20Al catalysts
However, for PtV20Al distinct weak diffraction peaks at 15°, 20°, 26°, and 31° were present (the value of 2θ are characteristic to vanadium pentoxide according to JCPDS 9–387). It was also observed that the average peak intensity of alumina decreased for this sample. No diffraction lines attributed to platinum were observed in XRD patterns.
Reducibility of Pt-VOx/γ-Al2O3 catalysts
Insight into the reducibility of surface VOx species was achieved by means of the temperature-programmed reduction experiments. The reduction with H2 was performed in the temperature range of 100–800 °C. Figure 3 shows typical TPR profiles for the investigated.

TPR profiles for investigated VOx/γ-Al2O3 catalysts with or without Pt. Experimental conditions: 5 vol% H2 in Ar, total flow rate of 70 cm3/min, from RT to 850 °C, heating rate 10 °C/min)
Pt-VOx/-Al2O3 catalysts. For the sake of comparison, the H2 consumption profiles registered for the samples without Pt are presented as well. Comparing these reduction profiles could provide indirect information about the interaction between the active components of the catalysts, namely vanadium oxides and Pt.
It was shown in the literature that the TPR patterns strongly depend on VOx surface coverage. At low vanadium loading, tetrahedral vanadium species (VO4) were formed preferentially by the reaction of the precursor with surface hydroxyl groups of the support. With the increase in the vanadium loading, the small clusters could polymerize into large structures. Eventually, higher vanadium loading led to the formation of V2O5 crystallites, as proved by XRD, which would be more difficult to be reduced. Indeed, it was shown that bulk V2O5 exhibited four major reduction peaks centered at 632, 672, 725, and 830 °C, corresponding to the succeeding consecutive reduction steps: V2O5 → V6O13 → V2O4 → V2O3 [35 and references therein].
In case of the VOx/γ-Al2O3 catalysts, TPR profiles showed a broad reduction peak with a maximum at 510 °C for V5Al and at 525 °C for V20Al. These peaks should be assigned to the presence of well-dispersed monomeric and polymeric surface VO4 on alumina support, as it was reported previously [35]. The temperature of the maximum H2 consumption increases with increasing of vanadia loading. This is in line with other studies [36] and is because less reducible square-pyramidal polymeric structures, i.e., polyvanadates, become predominant with increasing the V loading. For V20Al sample, two supplementary reduction peaks at 628 °C and 670 °C were found. These peaks are related to the reduction of even less reducible crystalline V2O5, which appears for this set of samples, as evidenced by XRD and UV–Vis data.
The reduction with H2 was also performed for Pt/Al2O3 sample in order to verify if the presence of platinum affects the redox behavior of alumina (not shown here). The only peak that appears, at 280 °C, was assigned to the presence of platinum oxide based on the hydrogen consumption, which was very small.
For Pt-containing catalysts, the H2 consumption corresponding to the reduction of PtOx could not be detected due to the small Pt loading (0.4 wt%), in agreement with the previous literature [16]. By comparing the reduction profiles of VAl with PtVAl catalysts, it is clear that the presence of Pt dramatically shifts the reduction toward lower temperatures. This effect has been associated with H2 spillover promoted by Pt over VOx species [16, 37]. It can be stated that the first peak (at ~ 250 °C) corresponds to the reduction of mono- and poly-vanadates species. The second reduction peak (at 275 °C) obtained only for PtV20Al sample is probably associated with the reduction of V2O5 crystallites, whose presence was highlighted by XRD and UV–VIS.
Identification of reduced centers—EPR spectra
To observe the changes in the amount of reduced species, data obtained from the recording of EPR spectra of the studied catalysts were used. The 3d5 configuration of vanadium makes possible for the existence of few reduced states, among which V4+ (3d1 configuration) can be easily detected by EPR spectroscopy, being paramagnetic [38,39,40,41,42]. The shape of a signal strongly depends on the number of oxo ligands attached to V4+ core [41], but the most common surface vanadium(IV) species are monoxo vanadyls VO2+. Usually, the EPR signals show nearly axial symmetry (ge gxx gyy gzz) [39] with well-resolved hyperfine structure (A tensor) due to the interaction between electron and nuclear (51 V, 99.75%, I = 7/2) spin magnetic moments.
The representative EPR spectra of VAl and PtVAl fresh and used catalysts are shown in Fig. 4, while the simulated spin Hamiltonian parameters are listed in Table 2. Computer simulations of experimental spectra revealed the presence of four types of V4+ species varying in their abundance, which depended on vanadium content. Type I and type II (Table 2) represent well-known isolated surface vanadyl VO2+ sites with the type II exhibiting slightly bigger magnetic anisotropy and Azz parameter than type I (probably due to a more distorted surroundings). Type I vanadyls are present for all the investigated samples, and their concentration is the highest for low loading VOx samples as well as for V20Al one. Type II sites are present solely for PtV20Al. According to Yoshida et al. [43], vanadium oxide is stabilized by interaction with γ-alumina as square pyramids while Van Reijen and Cossee [44] reported that on γ-alumina support V4+ appears in vanadyl configuration (VO2+). This reconciles with the type I and II signals observed in this study.

X-band EPR spectra of a PtV20Al-used, b V20Al-used, c V5Al-used, and d V5Al samples. Experimental spectra (black line) and simulated spectra (blue line) are shown
For the highest V loading, additional signal appears, namely type III. Type III represents clustered surface sites composed of polynuclear mixed valence vanadyl surface complexes containing V–O–V bridges, which segregate into nanocrystalline V2O5 and spread over alumina surface. Such magnetically coupled species give rise to a single, unresolved axial EPR signal due to inhomogeneous broadening caused by intracrystalline electron–electron dipolar interactions [38, 42]. Apart from isolated and nanocrystalline species, an additional signal (type IV) contributes to the experimental spectra of all investigated samples being more intense for higher vanadium loading. Type IV is an isotropic signal due to magnetically coupled vanadium(IV) surface sites, subject to long-range dipole–dipole interactions, giving rise to a broad background of relatively low intensity.
Due to the high sensitivity of EPR method, the spectra reveal the presence of even a small amount of V4+ ions in the fresh samples (Fig. 5). Comparison of the integral EPR intensities shows a clear increase with increasing V loading from 5 to 20 wt%. The fresh samples containing Pt showed more intense signals than those obtained for the samples without Pt. Vanadium(IV) signals are much more intense for the used samples (in the catalytic tests coupled with conductivity measurements), and the change of intensity with respect to the fresh sample (V5Al) is higher for Pt-containing catalysts. This means that the concentration of paramagnetic V4+ species increases during the catalytic reaction and the sample becomes reduced. Such reduction process is clearly facilitated by the presence of platinum.

Comparison of EPR integral intensity of investigated VAl and PtVAl fresh and used samples
Changes in EPR intensity are shown in Fig. 6. Both sample types (with and without Pt) exhibit a maximum of reduction at the same temperature of c.a. 200 °C. At higher temperature, re-oxidation of vanadium (IV) leads to reduction in its EPR signal. However, Pt-containing sample is characterized by higher number of reduced sites and it seems to be more sensitive to the reduction/oxidation conditions.

Relative changes of EPR integral intensity of V5Al and PtV5Al samples subject to thermal treatment in air at progressively increasing temperatures
UV–Vis spectra
The UV–Vis light absorption of supported metal oxides depends on the structure of the surface species formed and the oxidation state of metallic ion [45]. Thus, by using diffuse reflectance UV–Vis (DR UV–Vis) spectroscopy it is possible to gain more insight into the evolution of VOx species as a function of vanadia loading and the oxidation state of vanadium ions as well.
The DR UV–Vis spectra of fresh and used samples are presented in Fig. 7. For all the studied catalysts, a similar spectral pattern is observed. It is featured with a broad band that spreads from 200 to 500 nm for V5 samples, and from 200 to 550 nm for V20 samples. The absorption bands located around 240 nm, which are present for all samples, can be assigned to the presence of the isolated, tetrahedrally coordinated V5+ species, while the absorption near 300–500 nm can be related to the polymeric VOx, in 2D-square pyramidal coordination. For V20Al and PtV20Al samples (Fig. 7b), an extended absorption toward longer wavelengths (higher than 500 nm) is present, indicating the formation of crystalline V2O5, in accordance with XRD data [16, 34, 46].

Diffuse reflectance UV–Vis spectra for a V5Al and PtV5Al, b V20Al and Pt V20Al catalyst recorded before and after catalytic test (used)
The broad band observed in the spectral region of 600–900 nm is due to d-d transitions characteristic for V4+ and/or V3+ species [45]. This band is present for all samples, so reduced vanadium ions are present even in fresh samples as also shown with EPR measurements. However, the d-d band is more intense for the samples examined after the catalytic test indicating a deeper reduction during the reaction, in agreement with EPR results. Among the fresh samples, the PtV20Al is initially reduced to the greater degree (the band corresponding to V4+ d-d transitions is more intense) than the V5Al one. These observations correlate well with the reducibility of the investigated catalysts envisaged from TPR data.
Catalytic performances in propene oxidation
VOx/Al2O3 was generally studied in selective oxidation of hydrocarbons [9,10,11,12,13] and poorly studied for total oxidation of hydrocarbon [15, 16]. For example, in propene oxidation, Zhao et al. [47] used different loading of vanadia deposited on alumina support as catalysts and found a good selectivity for acrolein formation, while Li et al. [48] found that a 10%V2O5/TiO2 catalyst showed a pronounced selectivity to acetone in the presence of water.
In this study, the catalytic performances of the VOx/-Al2O3 and Pt-VOx/-Al2O3 catalysts for propene oxidation was evaluated as a function of reaction temperature and only carbon oxides were obtained. The main oxidation product was CO2, but CO was also detected. The measured conversion of C3H6 reached ~ 100% at 400 °C for V20Al and PtV5Al catalysts as can be seen in Fig. 8. By the addition of Pt, the activity is significantly improved and the propene conversion gradually increases starting at 200 °C.

Changes of propene conversion with reaction temperature (C3H6:air = 1:22 molar ratio) over V5Al, V20Al, PtV5Al, and PtV20Al catalysts
The calculated values of conversion in deep oxidation of C3H6 and its selectivity to CO2 at 215 °C and 400 °C are listed in Table 3. The established activity of the investigated catalysts (measured up to 350 °C) generally follows an order of: V5Al < V20Al < PtV5Al < PtV20Al. At ~ 200 °C, the C3H6 conversion has a significant increase when Pt is added, with the major product being CO2. So, it can be stated that a synergy between Pt and vanadia species exists. At the same time, PtV20Al catalyst has the highest conversion. However, above 350ºC the order of activity was changed, namely V5Al < PtV20Al < PtV5Al ~ V20Al.
From the inspection of Table 3, it can be concluded that the oxidation of propene took place with rather high selectivity to CO over the samples with high loading of vanadia. The highest selectivity to CO2 (SCO2) was obtained for V5Al and PtV5Al, while the lowest one was obtained for V20Al sample. On the other hand, SCO2 for high loading vanadia samples decreases with increasing the temperature.
The performance of the Pt-based catalysts for total oxidation of hydrocarbons is usually governed by several factors such as (1) the oxidation state of the Pt species and dimension of the metallic particles and (2) redox properties of the catalytic materials. In the literature [16], it was found that on Pt-V/Al2O3 catalyst the enhanced performance was attributed to the enhanced reducibility of these catalysts by Pt-VOx interaction. This conjecture is fully supported by our TPR and EPR data shown above.
For better understanding of the role of the redox properties of Pt-VOx/Al2O3 catalysts in propene oxidation, the variation in conductivity measured during the catalytic reaction runs was performed.
In situ electrical conductivity measurements
The changes in the relative oxidation level of the catalyst’s surfaces under reaction conditions can be investigated by following the evolution of the electrical properties of the solid in relation to a specific composition of a gas phase. Exposure to oxygen of a n-type oxide leads to a decrease in its conductivity with respect to that in an inert gas, as a result of oxygen adsorption and consecutive oxidation. On the contrary, flushing with inert gas (e.g., argon) facilitates desorption of pre-adsorbed oxygen ions or even formation of anion vacancies acting as electron donors. At the same time, interaction with a reducing gas, as propene, results in either formation of anion vacancies VO (when reaction follows a Mars van Krevelen mechanism) acting as electron donors or in consumption of the oxygen ad-ions. Both variants result in the increase in electron carrier concentration and thus in the increase in conductivity [20].
The increase/decrease in the electrical conductivity of a solid in the presence of various gases depends on the type of semiconductor. Thus, it is important to firstly determine the semiconductor type (n or p) of the investigated catalysts.
In Fig. 9 Arrhenius plots of the variation in conductance (G) with temperature when flushing the Pt-V catalysts with inert gas or oxygen are presented. For comparison, we also showed conductance curves for alumina support and VAl catalysts. As a result of thermal cycling of alumina in an inert atmosphere (DAr3) and in oxidizing atmosphere (DO), changes of conductance with temperature generate an unique U-shape, reaching a maximum in the lower temperature region (~ 50–80 °C) followed by a decrease in conductance. This characteristic of alumina is due to the loss of various adsorbed species of water [49] and was not observed in the case of the supported catalysts, indicating that the adsorption/migration of the water from the support onto the catalyst’s surface is reduced by vanadium oxide deposition. This indicates that the dynamic of vanadium oxide surface is less influenced by the adsorption of water and also that the oxide phase deposition is homogeneous, as resulted also from XRD and UV–Vis.

Arrhenius plots of the variation in conductance (G), measured in S, with the temperature in inert atmosphere (DAr) and oxygen (DO) for: a γ-Al2O3, V5Al and PtV5Al and b V20Al and PtV20Al samples
For temperatures higher than ~ 100 °C, the adsorbed water was practically desorbed and the electronic conductivity prevails to the detriment of the ionic one, due to a much smaller size of the carriers in first case, which implies a much greater mobility of them. Consequently, the electrical conductance results are discussed in terms of electron transfer. As shown in Fig. 9, all VAl samples behave as n-type semiconductors (higher conductance in argon, lower conductance in oxygen), and the same was noticed for their platinum counterparts. The n-type behavior is by far more obvious for the highly loaded samples, which show also much higher conductivity. Practically, the plots in DO and DAr are differentiated starting from 130 °C, indicating this temperature as representative for the onset of oxygen adsorption/surface oxidation (associated with electronic carrier consumption).
One can further study the behavior of the catalysts in the presence of the reaction mixture C3H6: air 1:22, based on the differential steps technique, i.e., to determine the role of the redox properties of the catalysts’ surface in the reaction evolution.
The changes of electric conductance with temperature when heating the samples in the catalytic test gas mixture are shown in Fig. 10. For comparison, the variation in conductance measured in DO atmosphere is shown, these values being characteristic for oxidized samples. It was observed that the reaction mixture induces a higher increase in conductance, in spite of the presence of oxygen in the feed. Considering the n-type behavior of the samples, this increase in conductance indicates that the dominant process above 130 °C is the surface reduction in the samples by propene. The surface reduction could be due either to the consumption of adsorbed oxygen or to the reaction of propene with surface lattice oxygen. The increase in conductance of all samples in the presence of catalytic mixture is higher than in inert atmosphere (when the increase is mainly due to desorption of adsorbed oxygen). This means that the reduction of catalyst in CT run is not only due to the consumption of adsorbed oxygen, but it is also associated with consuming of lattice oxygen by propene, via a Mars van Krevelen mechanism (i.e., the propene oxidation uses lattice oxygen, while the gaseous oxygen replaces the removed oxygen species in a separate, subsequent step). Overall, as also resulted from EPR and UV–Vis investigations of the samples before and after the contact with C3H6:air mixture, in the conditions of these experiments, the surface reduction (by propene) is faster than its re-oxidation, since the samples appear as being more reduced after the catalytic test, in spite of the presence of oxygen in the feed.

Comparison between temperature dependence of electric conductance (G) in DO and C3H6:air 1:22 runs for: a 5wt% and b 20wt% V2O5 loading samples
This trend for variation in conductance was more obvious for V20Al and PtV20Al (with stronger tendency for the Pt-containing catalyst), as seen in Fig. 9b. Furthermore, in the temperature range from 130 °C to 320 °C, the increase in the conductance for PtV20Al sample in CT run (relative to the values of the oxidized sample) is more pronounced than for V20Al. At 320 °C, the shift of the conductance ΔG = GCT – GO, defined as the difference between the conductance of the samples in the presence of catalytic mixture (GCT) and the conductance of the oxidized sample (GO), is 17.5 nS for PtV20Al higher than 12.4 nS for V20Al. This supports the assertion that at low temperature, Pt favors the reduction of V5+, as deduced also from TPR and from the spectroscopic characterization of the catalysts.
In this temperature range (130 to 320 °C), the conversion of C3H6 is higher for PtV dual catalysts. Therefore, the in situ electrical measurements complemented with spectroscopic characterization induced the conclusion that enhancement in the alkene conversion is due to the presence of highly reducible vanadium species. As resulted from both electrical and TPR measurements, the reducibility depends on the vanadia loadings and more important on the presence of Pt. This result remains in agreement with literature data [15, 16]. In this temperature range, the order of activity follows the same order with the reducibility of samples, namely V5Al < V20Al < PtV5Al < PtV20Al.
The decrease in conductance above 320 °C for PtV20Al sample in CT run indicates that the re-oxidation of the catalyst’s surface is faster than its reduction. Since this effect was noticed particularly for Pt-containing samples (PtV5Al presents lower conductance in catalytic mixture between 285 and 385 °C, comparing with the values obtained in DO atmosphere), it is possible that the presence of platinum favors the increase in the re-oxidation rate at high temperature. After passing 320 °C, the order of catalyst’s performances is also changed and the conversion of C3H6 over PtV20Al becomes limited at ~ 85%. This is probably due to the oxygen dissociation on the Pt surface at higher temperatures (resulting in the decrease in conductance), the rate determining step becoming in this case the activation of the C-H bond, as was also reported in the literature for propane oxidation [15, 50].
On the other hand, V20Al is the only sample that shows a continuous increase in conductance in CT. V20Al catalyst presents a high conversion (100% at 400 °C), but it has also the lowest CO2 selectivity (~ 54% at 400 °C). It is obvious from variation in the electrical conductance that in the case of this sample, the predominant step is the reduction in the surface by propene. The re-oxidation and, thus, the replacement of the lattice oxygen lost during the reduction step are lower, and the catalyst’s surface became excessively reduced to carry the reaction toward full oxidation product CO2. The over-reduction of vanadia seems to be responsible for the increased concentration of CO in detrimental of CO2.
The electrical measurements indicate that the ability of reduction/re-oxidation of the samples is crucial factors in determining the activity and selectivity of the reaction. The overall reducibility depends also on the VOx loading (thus, on the dominant vanadia structure) and also on the presence of Pt. It is not clear for the moment which is the surface species responsible for the CO formation. But, coupling all the data obtained, it can be affirmed that the presence of isolated/polymerized V5+ tetrahedrally coordinated species favors the total oxidation of propene.
Conclusions
The surface modifications of Pt-VOx/γ-Al2O3 catalysts were investigated as a function of vanadia amount and Pt addition in propene deep oxidation. The results are consistent with the presence of vanadium species in various degrees of polymerization and different oxidation states, mainly V5+ (but reduced vanadium is present even in fresh samples), as shown by EPR, UV–Vis, and the in situ electrical conductivity measurements.
The AC electrical measurements performed in situ, when adequately used, are a useful tool to investigate gas–solid and solid–solid electronic interactions and thus can become a method of choice for catalytic studies. Therefore, it was shown by the electrical properties measurements that the presence of Pt facilitates the reduction of the vanadia phase at low temperature, but also its re-oxidation at high temperature. A higher reducibility of vanadium oxide represents in the present case a higher conversion of propene. But the reducibility is not the only factor which determines the catalytic performances for deep propene oxidation; the fast surface re-oxidation also controls the selectivity of the reaction (CO2/CO ratio).
References
Liotta LF (2010) Catalytic oxidation of volatile organic compounds on supported noble metals. Appl Catal B Environ 100:403–412. https://doi.org/10.1016/j.apcatb.2010.08.023
Pina MP, Irusta S, Menéndez M et al (1997) Combustion of volatile organic compounds over platinum-based catalytic membranes. Ind Eng Chem Res 36:4557–4566. https://doi.org/10.1021/ie9700876
Garcia T, Solsona B, Murphy DM et al (2005) Deep oxidation of light alkanes over titania-supported palladium/vanadium catalysts. J Catal 229:1–11. https://doi.org/10.1016/j.jcat.2004.09.018
Okumura K, Kobayashi T, Tanaka H, Niwa M (2003) Toluene combustion over palladium supported on various metal oxide supports. Appl Catal B Environ 44:325–331. https://doi.org/10.1016/S0926-3373(03)00101-2
Wu JC-S, Lin Z-A, Tsai F-M, Pan J-W (2000) Low-temperature complete oxidation of BTX on Pt/activated carbon catalysts. Catal Today 63:419–426. https://doi.org/10.1016/S0920-5861(00)00487-9
Zang M, Zhao C, Wang Y, Chen S (2019) A review of recent advances in catalytic combustion of VOCs on perovskite-type catalysts. J Saudi Chem Soc 23:645–654. https://doi.org/10.1016/j.jscs.2019.01.004
Guerrero-Pérez MO (2017) Supported, bulk and bulk-supported vanadium oxide catalysts: a short review with an historical perspective. Catal Today 285:226–233. https://doi.org/10.1016/j.cattod.2017.01.037
Bratan V, Chesler P, Vasile A et al (2011) Surface properties and catalytic oxidation on V2O5-CeO2 catalysts. Rev Roum Chim 56(10–11):1055–1065
Wachs IE, Weckhuysen BM (1997) Structure and reactivity of surface vanadium oxide species on oxide supports. Appl Catal A Gen 157:67–90. https://doi.org/10.1016/S0926-860X(97)00021-5
Concepción P, Navarro MT, Blasco T et al (2004) Vanadium oxide supported on mesoporous Al2O3: Preparation, characterization and reactivity. Catal Today 96:179–186. https://doi.org/10.1016/j.cattod.2004.06.151
Le BJ, Auroux A, Forissier M, Vedrine JC (1996) Active sites of V2O5/γ-Al2O3 catalysts in the oxidative dehydrogenation of ethane. J Catal 162:250–259. https://doi.org/10.1006/jcat.1996.0282
Liu J, Mohamed F, Sauer J (2014) Selective oxidation of propene by vanadium oxide monomers supported on silica. J Catal 317:75–82. https://doi.org/10.1016/j.jcat.2014.06.003
Held A, Kowalska-Kuś J, Nowińska K, Góra-Marek K (2017) Potassium-modified silica-supported vanadium oxide catalysts applied for propene epoxidation. J Catal 347:21–35. https://doi.org/10.1016/j.jcat.2016.12.001
Debecker DP, Bouchmella K, Delaigle R et al (2010) One-step non-hydrolytic sol–gel preparation of efficient V2O5-TiO2 catalysts for VOC total oxidation. Appl Catal B Environ 94:38–45. https://doi.org/10.1016/j.apcatb.2009.10.018
Liao W-M, Liu Y-R, Zhao P-P et al (2020) Total oxidation of propane over Pt-V/SiO2 catalysts: Remarkable enhancement of activity by vanadium promotion. Appl Catal A Gen 590:117337. https://doi.org/10.1016/j.apcata.2019.117337
Garcia T, Agouram S, Taylor SH et al (2015) Total oxidation of propane in vanadia-promoted platinum-alumina catalysts: Influence of the order of impregnation. Catal Today 254:12–20. https://doi.org/10.1016/j.cattod.2015.01.038
Heynderickx PM, Thybaut JW, Poelman H et al (2009) Kinetic modeling of the total oxidation of propane over anatase and vanadia sputter deposited catalysts. Appl Catal B Environ 90:295–306. https://doi.org/10.1016/j.apcatb.2009.03.020
Vassileva M, Andreev A, Dancheva S, Kotsev N (1989) Complete Catalytic oxidation of benzene over supported vanadium oxides modified by palladium. Appl Catal 49:125–141. https://doi.org/10.1016/S0166-9834(00)81428-1
Ovenston A, Walls JR (1987) D.C. conductivity used as a sensor for the high-temperature H2 chemisorption on commercial supported nickel catalysts. Sensors Actuat 12:159–178. https://doi.org/10.1016/0250-6874(87)85014-X
Bratan V, Munteanu C, Hornoiu C et al (2017) CO oxidation over Pd supported catalysts—In situ study of the electric and catalytic properties. Appl Catal B Environ 207:166–173. https://doi.org/10.1016/j.apcatb.2017.02.017
Vasile A, Bratan V, Hornoiu C et al (2013) Electrical and catalytic properties of cerium-tin mixed oxides in CO depollution reaction. Appl Catal B Environ 140–141:25–31. https://doi.org/10.1016/j.apcatb.2013.03.042
Scurtu M, Caldararu M, Bratan V et al (2012) In situ study of the electrical/dielectric properties of alumina supported vanadia used as combustion catalysts. Env Eng Manag J 1:185–191
Bond GC, Tahir SF (1991) Vanadium oxide monolayer catalysts Preparation, characterization and catalytic activity. Appl Catal 71:1–31. https://doi.org/10.1016/0166-9834(91)85002-D
Corma A, García H (2002) Lewis acids as catalysts in oxidation reactions: from homogeneous to heterogeneous systems. Chem Rev 102:3837–3892. https://doi.org/10.1021/cr010333u
Blasco T, Nieto JML (1997) Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl Catal A Gen 157:117–142. https://doi.org/10.1016/S0926-860X(97)00029-X
Spałek T, Pietrzyk P, Sojka Z (2005) Application of the genetic algorithm joint with the Powell method to nonlinear least-squares fitting of powder EPR spectra. J Chem Inf Model 45:18–29. https://doi.org/10.1021/ci049863s
Cǎldǎraru M, Sprînceana D, Popa VT, Ionescu NI (1996) Surface dynamics in tin dioxide-containing catalysts II. Competition between water and oxygen adsorption on polycrystalline tin dioxide. Sensors Actuators B Chem 30:35–41. https://doi.org/10.1016/0925-4005(95)01746-I
Chesler P, Hornoiu C, Bratan V et al (2015) Carbon monoxide sensing properties of TiO2. Rev Roum Chim 60:227–232
Caldararu M, Munteanu C, Chesler P et al (2007) Supported oxides as combustion catalysts and as humidity sensors. Tuning the surface behavior by inter-phase charge transfer. Microporous Mesoporous Mater 99:126–131. https://doi.org/10.1016/j.micromeso.2006.07.039
Baldychev I, Gorte RJ, Vohs JM (2010) The impact of redox properties on the reactivity of V2O5/Al2O3 catalysts. J Catal 269:397–403. https://doi.org/10.1016/j.jcat.2009.11.022
Klose F, Wolff T, Lorenz H et al (2007) Active species on γ-alumina-supported vanadia catalysts: nature and reducibility. J Catal 247:176–193. https://doi.org/10.1016/j.jcat.2007.01.013
Weckhuysen BM, Keller DE (2003) Chemistry, spectroscopy and the role of supported vanadium oxides in heterogeneous catalysis. Catal Today 78:25–46. https://doi.org/10.1016/S0920-5861(02)00323-1
Al-Ghamdi SA, de Lasa HI (2014) Propylene production via propane oxidative dehydrogenation over VOx/γ-Al2O3 catalyst. Fuel 128:120–140. https://doi.org/10.1016/j.fuel.2014.02.033
Kokumai TM, Cantane DA, Melo GT et al (2017) VOx-Pt/Al2O3 catalysts for hydrogen production. Catal Today 289:249–257. https://doi.org/10.1016/j.cattod.2016.09.021
Schwarz O, Habel D, Ovsitser O et al (2008) Impact of preparation method on physico-chemical and catalytic properties of VOx/γ-Al2O3 materials. J Mol Catal A Chem 293:45–52. https://doi.org/10.1016/j.molcata.2008.07.009
Mitran G, Ahmed R, Iro E et al (2018) Propane oxidative dehydrogenation over VOx/SBA-15 catalysts. Catal Today 306:260–267. https://doi.org/10.1016/j.cattod.2016.12.014
Ferreira RSG, de Oliveira PGP, Noronha FB (2004) Characterization and catalytic activity of Pd/V2O5/Al2O3 catalysts on benzene total oxidation. Appl Catal B Environ 50:243–249. https://doi.org/10.1016/j.apcatb.2004.01.006
Serwicka EM (1989) ESR study of supported H3PMo12O40/K3PMo12O40 catalysts for the oxidation of acrolein. Zeitschrift für Physikalische Chemie 165:S95–101. https://doi.org/10.1524/zpch.1989.165.Part_1.095
Pietrzyk P, Góra-Marek K (2016) Paramagnetic dioxovanadium(IV) molecules inside the channels of zeolite BEA–EPR screening of VO2 reactivity toward small gas-phase molecules. Phys Chem Chem Phys 18:9490–9496. https://doi.org/10.1039/C6CP01046F
Rossetti I, Fabbrini L, Ballarini N et al (2008) V2O5–SiO2 systems prepared by flame pyrolysis as catalysts for the oxidative dehydrogenation of propane. J Catal 256:45–61. https://doi.org/10.1016/j.jcat.2008.02.028
Pietrzyk P, Sojka Z, Dzwigaj S, Che M (2007) Generation, identification, and reactivity of paramagnetic VO2 centers in zeolite BEA for model studies of processes involving spin pairing, electron transfer, and oxygen transfer. J Am Chem Soc 129:14174–14175. https://doi.org/10.1021/ja076689q
Brückner A, Kondratenko E (2006) Simultaneous operando EPR/UV–vis/laser–Raman spectroscopy: a powerful tool for monitoring transition metal oxide catalysts during reaction. Catal Today 113:16–24. https://doi.org/10.1016/j.cattod.2005.11.006
Yoshida S, Iguchi T, Ishida S, Tarama K (1972) Some physico-chemical properties of vanadium oxide supported on silica or γ-alumina. Bull Chem Soc Jpn 45:376–380. https://doi.org/10.1246/bcsj.45.376
van Reijen LL, Cossee P (1966) Electron spin resonance study of rearrangements in the co-ordination of Cr5+ and V4+ complexes due to chemisorption. Discuss Faraday Soc 41:277–289. https://doi.org/10.1039/DF9664100277
Kondratenko EV, Baerns M (2001) Catalytic oxidative dehydrogenation of propane in the presence of O2 and N2O—the role of vanadia distribution and oxidant activation. Appl Catal A Gen 222:133–143. https://doi.org/10.1016/S0926-860X(01)00836-5
Wu Z, Kim H-S, Stair PC et al (2005) On the structure of vanadium oxide supported on aluminas: UV and visible Raman spectroscopy, UV−Visible diffuse reflectance spectroscopy, and temperature-programmed reduction studies. J Phys Chem B 109:2793–2800. https://doi.org/10.1021/jp046011m
Zhao C, Wachs IE (2008) Selective oxidation of propylene over model supported V2O5 catalysts: Influence of surface vanadia coverage and oxide support. J Catal 257:181–189. https://doi.org/10.1016/j.jcat.2008.04.022
Li M, Shen J (2001) Selective oxidation of propylene to acetone over supported vanadia catalysts. React Kinet Catal Lett 72:263–267. https://doi.org/10.1023/A:1010586727662
Caldararu M, Postole G, Hornoiu C et al (2001) Electrical conductivity of γ-Al2O3 at atmospheric pressure under dehydrating/hydrating conditions. Appl Surf Sci 181:255–326. https://doi.org/10.1016/S0169-4332(01)00393-2
García-Diéguez M, Chin Y-H, Iglesia E (2012) Catalytic reactions of dioxygen with ethane and methane on platinum clusters: mechanistic connections, site requirements, and consequences of chemisorbed oxygen. J Catal 285:260–272. https://doi.org/10.1016/j.jcat.2011.09.036
Acknowledgements
The authors are thankful to the scientific services of IRCELYON. The research carried out in Poland was done with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00–12-023/08).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest statement
The authors declare no conflicts of interest.
Additional information
This article is dedicated to the loving memory of Dr. Monica Caldararu, who developed the differential steps technique, a method for in situ analysis of catalysts based on the changes of their electrical conductivity in reaction conditions.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bratan, V., Chesler, P., Hornoiu, C. et al. In situ electrical conductivity study of Pt-impregnated VOx/γ-Al2O3 catalysts in propene deep oxidation. J Mater Sci 55, 10466–10481 (2020). https://doi.org/10.1007/s10853-020-04779-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-04779-0