
Abstract
Diopside-based ceramics and glass–ceramics have been studied because of their applications in electronics and biomedicine. However, since diopside glass presents poor internal nucleation ability, sintering combined with surface crystallization of powdered glasses has been reported to obtain diopside glass–ceramics. On the other hand, in this work, we explore the effect of an efficient nucleating agent (Fe2O3) to induce copious internal nucleation in this glass, which enabled the production of single-phase diopside glass–ceramics by the traditional route. The crystallization kinetics of a diopside glass (25CaO·25MgO·50SiO2) containing 8.26 mol% of Fe2O3 was investigated under isothermal conditions by differential thermal analysis (DTA) and was modeled by the Johnson-Mehl-Avrami-Kolmogorov-Erofeev (JMAKE) equation. The crystals formed were iron-diopside—the X-ray diffraction pattern was indexed to the ferric-diopside card (Ca0.991(Mg0.641Fe0.342)(Si1.6Fe0.417)O6). Through a systematic DTA study, we successfully determined the mechanism and kinetics of crystallization of this material, which provided relevant information to guide the development of this novel type of internally crystallized glass–ceramic.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Diopside (25CaO·25MgO·50SiO2, hereafter denoted as CMS2) is an important mineral of the pyroxene group, which presents a monoclinic structure of the C2/c4 space group [1, 2]. Partially or totally crystallized CMS2-based materials have gained increased interest because of their potential applications as artificial bones, dental roots and crowns [3], as well as sealants for solid oxide fuel cells [4,5,6]. Such relevance can be associated with an adequate combination of high chemical durability, significant microwave dielectric properties and very high flexural strength (400 MPa) [7, 8]. However, diopside-based glass–ceramics (GC) containing different additives, such as La2O3, ZnO, B2O3, SrO and Cr2O3, present poor internal nucleation ability. For this reason, they have been mostly obtained by sintering with concurrent (surface) crystallization of glass powders, which often leads to residual porosity and other problems that are intrinsic to the sintering processes.
In principle, glass–ceramics can also be obtained from controlled internal crystallization of certain glasses. The process consists of forming a glass piece (e.g., by conventional melt-quenching method) and then submitting it to a heat treatment to induce internal nucleation and crystal growth [9,10,11]. The addition of nucleating agents, such as TiO2, ZrO2, P2O5, Cr2O3, Fe2O3, WO3 and noble metals, is essential to foster the nucleation process in the interior of most glasses. Nevertheless, they are rather specific: Each glass composition demands a different nucleating agent [9,10,11], which is empirically found.
Only a few studies on the role of such catalyzers in diopside glass–ceramics have been published [3, 12, 13]. For example, Nonami and Tsutsumi [3] obtained CaO–MgO–2SiO2–0.375TiO2–0.007Ag2O glass–ceramic samples containing a small amount of diopside—between 15 and 25% (vol%)—as the only microcrystalline phase distributed in the glass. According to the authors, the brown color is a result of silver colloids inside the glass. Heat treatment at 870 °C induced the growth of such silver colloids and the subsequent precipitation of 1–2-μm diopside crystals, causing a whitish color. Despite the small amount of diopside, the bending strength of this glass–ceramic was surprisingly very high (400 MPa), making the material suitable for use as artificial bones. This result motivated us to study further and develop diopside glass–ceramics.
In the previous work, one of us [12] discovered the effect of Fe2O3 as a potential nucleating agent for diopside glass, although it was only superficially explored. In such a study, the authors analyzed the influence of Fe2O3 on the nucleation kinetics of 25CaO·25MgO·50SiO2.xFe2O3 glassy samples, with x equal to 3.9 and 7.9 mol%. In samples containing 7.9 mol% of Fe2O3, heat-treated at 720 °C for nucleation and subsequently at 940 °C for crystal growth, transmission electron microscope images showed the presence of nano-sized crystalline aggregates uniformly distributed throughout the specimen volume. These crystals could not be detected by X-ray diffraction because of their small sizes and low volume fraction. The authors suggested that they could be magnetite (Fe3O4). After a subsequent heat treatment at 800 °C for 2 h, they observed the presence of diopside crystals.
Recently, Zhang et al. [13] studied the effect of Cr2O3 on the crystallization kinetics of a diopside glass. They reported that the maximum achievable solubility of Cr2O3 in diopside was 3 wt%, and that a crystalline spinel phase was formed by the addition of 3 wt% Cr2O3–MgCr2O4—which acted as a nucleating agent for the diopside crystals.
In these previous studies, the crystallization kinetics of diopside glass containing a nucleating agent was not characterized. In addition, the properties of the resulting glass–ceramics were only cursorily described. However, the determination of the crystallization mechanism and kinetics may provide key information necessary for producing novel, optimized glass–ceramic materials via controlled internal crystallization. One method to do so is through isothermal treatments via DTA. Although a few studies have focused on this method to investigate the crystallization mechanism for inorganic glasses—by far, most authors often use non-isothermal DTA techniques [14, 15]. Thus, this study aims to determine the crystallization mechanism and kinetics, in a range of temperatures, of a diopside glass containing an efficient nucleating agent, Fe2O3.
Experimental details
Different amounts of Fe2O3 were tested; 6.54 and 8.26 mol% were enough to induce copious internal nucleation. However, in this article, we will focus on the 8.26 mol% composition for which we carried out a comprehensive study. To obtain the desired glass composition, 8.26 mol% of Fe2O3 was added exceeding 100 mol% of the stoichiometric 25CaO·25MgO·50SiO2 (CMS2) diopside composition, which led to a 25CaO·25MgO·50SiO2.8.26Fe2O3 mol% composition, hereafter denoted as CMS2-Fe.
To obtain vitreous samples of the CMS2 and CMS2-Fe, appropriate proportions of CaCO3 (Alfa Aesar > 99.5%), MgO (Alfa Aesar, 99.95%), SiO2 (Alfa Products, 99.9%) and Fe2O3 (Alfa Aesar 99.99%) were thoroughly mixed in a rotary mixer for 3 h. A 60-gram batch was melted in a platinum crucible at 1500 °C for 2 h, and then, plate samples were obtained by pouring the molten material in a thin metallic mold at room temperature. The melting process was followed by fast cooling between two metallic plates, and the procedure was repeated four times to promote homogenization. To relax the thermal stresses arising from the non-uniform cooling process, the vitreous plates were submitted to an annealing treatment at 60 °C for 1 h below the respective glass transition temperatures.
The glass transition temperatures (Tg), the onset crystallization temperatures (Tc) and the peak crystallization temperature (Tp) of CMS2 and CMS2-Fe samples were determined through differential thermal analysis (DTA) measurements performed in air at a heating rate of 10 °C/min in a temperature range of 20 °C to 1100 °C using a 2910 Differential Thermal Analyzer from TA Instruments.
To verify the formation of crystals in the CMS2-Fe glass volume, the sample underwent two subsequent heat treatments: first at 705 °C for 15 min and subsequently at 780 °C for 15 min. One side of the sample was polished by removing approximately 200 μm from its surface to reveal its internal crystals. The polished surface was then subjected to chemical etching in a 0.1% HCl–0.3% HF vol% for 15 s. Scanning electron micrographs and energy-dispersive X-ray spectra (EDX) of the samples were obtained in a Zeiss microscope model Sigma equipped with electron gun by field emission (SEM-FEG).
Determination of the temperature of maximum nucleation rate (TNmax) of the CMS2-Fe sample
The temperature of maximum nucleation rate (TNmax.) of the CMS2-Fe sample was determined by the DTA method proposed by Marotta et al. [16] using 30-mg pieces. The small monolithic pieces of vitreous samples were previously submitted to isothermal nucleation treatments in an electrical furnace at 645 °C, 655 °C, 665 °C, 675 °C, 685 °C, 695 °C and 705 °C for 2 h. These pre-nucleated samples were then subjected to DTA analysis using a heating rate of 10 °C/min to obtain the trace corresponding to each nucleation temperature. The temperature of maximum crystal nucleation rate (TNmax) corresponding to the maximum difference between Tp’ (DTA crystallization peak of the sample without pre-nucleation isothermal treatment) and Tp (DTA crystallization peak of a pre-nucleated vitreous sample) was then determined.
Isothermal crystallization mechanism and kinetics by DTA of the CMS2-Fe sample
The isothermal crystallization method through DTA was used to determine the mass crystallized fraction (α) as a function of the treatment time and the Avrami coefficient (n), later described, of the CMS2-Fe glass. The DTA measurements were performed using monolithic pieces of around 30 mg and a heating rate of 10 °C/min up to the isothermal treatment temperature. The time at the isothermal condition varied according to the temperature of study (760, 770, 780 and 790 °C). The experiments were interrupted after complete crystallization of each sample, that is, after the complete formation of the exothermic peak in the DTA trace as inferred by the return of the curve to its baseline.
Results and discussion
Figure 1 shows the DTA traces of a monolithic piece and a powdered sample (< 100 μm) of the CMS2 and CMS2-Fe glasses. From this figure, we determined the characteristic temperatures of each sample, which are presented in Table 1. As shown in that table, the addition of Fe2O3 led to a decrease in the Tg, Tc and Tp and, consequently, of the value of ΔT in relation to the CMS2 glass.

DTA traces of CMS2 and CMS2-Fe glassy samples in bulk and powder forms at a heating rate of 10 °C/min
It is well established that the approximation of the crystallization peak temperatures of a powdered sample and the same material in monolithic form (ΔΤp) is an indication that the nucleation process happens predominantly in the sample interior [17]. Thus, a simple way of assessing whether internal nucleation is occurring is to verify the value of the ΔΤp. Whereas for the pure CMS2 glass sample, which presents only surface nucleation, ΔΤp ≈ 100 °C, for the material containing Fe, ΔΤp ≈ 18 °C (Fig. 1). This result indicates that this amount of iron oxide added in the sample acted as an effective catalyzer for internal nucleation in diopside glass.
To prove that crystal nucleation really occurred in the glass interior, the sample was subjected to a heat treatment at 705 °C for 15 min (for nucleation) followed by another at 780 °C for 15 min to have its crystals revealed. As shown in Fig. 2, after the surface layer removal, multiple crystals could be observed in the sample cross section, indicating that the formation of crystals indeed occurred in the sample volume. To confirm that iron atoms were present in solid solution in the crystalline phase, an EDX analysis was made inside the crystal (square) and in the glass region (circle) (Fig. 2b). As shown in Fig. 2c, the EDX spectrum collected inside the crystal and in the glass region is quite similar, showing that Fe is indeed incorporated in the crystalline phase.

Electron micrograph of the cross section of a CMS2–9Fe glass sample heat-treated at 705 °C/15 min for nucleation and subsequently at 780 °C/15 min for crystal development. a Small (< 5 μm) crystals are uniformly distributed in the sample interior. b SEM micrograph showing details where the EDX analysis was performed. c EDX spectra obtained inside the crystal (square) and in the glass region (circle)
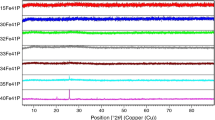
Figure 3 shows the X-ray diffraction pattern of a powdered glassy CMS2 and CMS2-Fe samples that underwent a crystallization treatment at the peak crystallization temperature (Tp) to induce (almost) full crystallization. We compared the XRD patterns of pure diopside (CMS2) and iron-containing diopside glass (CMS2-Fe) samples. From this comparison, we verified that the XRD patterns present significant differences, mainly in the 26–38 two-theta range. The diffraction pattern of the CMS2 crystallized sample was indexed as diopside (25CaO·25MgO·50SiO2, ICSD 30522 card). The XRD pattern of CMS2-Fe sample that could only be fully indexed to the ferric-diopside phase (Ca0.991(Mg0.641Fe0.342)(Si1.6Fe0.417)O6, ICSD 85691 card) was considered. In this phase, Fe2+ ions can substitute Mg2+, whereas Fe3+ can substitute Si4+ [1]. Mossbauer measurements (not shown) indicate that iron can be present as Fe+2 and Fe+3 in the original glass with a content of 16 and 84%, respectively.

X-ray diffraction patterns of the crystallized CMS2 and CMS2-Fe glassy samples: a CMS2 X-ray diffraction pattern indexed to the ICSD File 30522 (25CaO·25MgO·50SiO2 phase) and CMS2-Fe X-ray diffraction pattern indexed to the 85691 ICSD File (Ca0.991(Mg0.641Fe0.342)(Si1.6Fe0.417)O6 phase). b Comparison of the XRD patterns of CMS2 and CMS2-Fe samples showing the 2θ range where the differences are more significant)
Figure 4 shows the nucleation curve as a function of the temperature of nucleation treatment obtained by the method developed by Marotta [16] based on DTA data. The value of the temperature of maximum nucleation (TNmax.) is approximately 10 °C lower than Tg (682 °C). Although being rare to achieve a TNmax lower than Tg, this fact was also observed in a cordierite glass, which only exhibits internal nucleation as a result of the addition of a large amount of TiO2 [18].

Plot of Tp’–Tpversus temperature of nucleation treatment. Tp corresponds to the peak crystallization temperature in the DTA trace of monolithic pieces previously submitted to nucleation treatments at different temperatures for 2 h. Tp’ is the crystallization peak in the DTA curve of the as-obtained glassy sample
Crystallization kinetics of the CMS2-Fe glass by the isothermal DTA method
The kinetics of isothermal crystallization can be described based on a theory developed by Johnson and Mehl, Avrami, Kolmogorov and Erofeev, known as JMAKE phase transformation theory [19]. They proposed the following equation for general use:
where αv(t) is the volume fraction transformed; n is the Avrami coefficient, a numerical factor related to the nucleation mechanism and growth morphology of crystals; and the coefficient K is related to the crystal shape, nucleation and growth rates.
Equation (1) can be linearized, resulting in the following logarithmic form:
The isothermal DTA method consists of heating a glass sample at a constant rate in the DTA equipment up to the desired temperature of isothermal treatment and maintaining it at that temperature during the exothermic crystallization transition, i.e., from the beginning to the end of crystallization peak. Figure 5a shows the normalized DTA crystallization peaks as a function of time between 760 and 790 °C for the CMS2-Fe glass. The results indicate a single, smooth process of isothermal crystallization with the formation of ferric-diopside. The exothermic crystallization peaks of the ferric-diopside phase shift to lower temperatures and become narrower and more intense, confirming the expected progressive increase in the crystallization rate as the temperature of the isothermal treatment increases.

a Time dependence of the DTA signal during isothermal annealing of CMS2-Fe glassy samples at 760 °C, 770 °C, 780 °C and 790 °C. b Time dependence of the crystallized mass fraction (αm) and crystallized volume fraction (αv) during isothermal annealing of CMS2-Fe glassy samples at 760 °C, 770 °C, 780 °C and 790 °C
The typical experimental points of the sigmoidal curves represent a variation in the crystallized mass fraction, αm, as a function of time, t, for the different isothermal temperatures. They were inferred from the crystallization peak areas. The crystallized mass fraction can be calculated as follows [20, 21]:
where (At) is the partial area of the peak at the time t and (A) is the total area of the peak, as indicated in the inset of Fig. 5a.
The data obtained by isothermal heating in the DTA equipment refer to the crystallized mass fraction (αm). However, to interpret these data through JMAKE equation, it is necessary to convert them into a volumetric crystallized fraction (αv), according to the following Eq. (4):
where αv and αm are the crystallized volume and mass fractions, respectively, being ρg = 2.87 g/cm3 and ρc = 3.27 g/cm3, the reported values for the diopside glass and crystal densities, respectively.
The experimental points in Fig. 5b formed sigmoidal curves of crystallized mass fraction (αm) and crystallized volume fraction (αv) as a function of time, t, for different temperatures. The density difference (13%) causes differences between the two data sets. As expected, the time necessary for full crystallization decreases as the temperature increases from 760 °C to 790 °C.
Once the time dependence of crystallized volume fraction during isothermal annealing experiments is obtained, the Avrami coefficient, n, can be determined by the slope of the graph ln(−ln (1 − αv)) versus ln t (Eq. 2). According to the literature, n can vary from 0.5 to 4.0 depending on the crystallization mechanism and crystal morphology [22].
To determine the Avrami coefficient, the experimental αv curves were used. Graphs of ln[−ln (1 − αv)] versus ln (t) resulting from the crystallization isotherms at 790, 780, 770 and 760 °C are shown in Fig. 6. According to the previous studies, for 0 < α < 0.1, the ln[−ln (1 − αv)] values are strongly affected by small variations in αv, since it is difficult to determine an exact measure of the area under the crystallization peak at the beginning, which may consequently lead to incorrect values. In addition, the final stages of crystallization (αv~ 0.8) can also cause measurement difficulties and curvature in the linearized graphs [23, 24]. To minimize such possible problems, only values corresponding to the crystallized volume fraction range (0.3 < αv < 0.7) were used to estimate the value of n [23, 24]. The results shown in Fig. 6 were determined by least squares fitting of the experimental data in this interval and correspond to the line slopes.

Ln[(− ln(1 − αv)] versus ln t (logarithm of time) plots for the isothermal crystallization of the CMS2-Fe glass at four temperatures well above the TNmax. The continuous lines resulted from linear fittings of the experimental data at each temperature. The numbers above the lines refer to the Avrami coefficients
This procedure resulted in an average value of n = 3.2 ± 0.3. Considering that the median value of n is very close to 3, the mechanism of crystal growth for the CMS2-Fe glass can be interpreted as interface-controlled crystal growth from a fixed number of nuclei during the isothermal crystallization process in the DTA [22]. This value of n is approximately temperature independent. Consequently, the CMS2-Fe glass undergoes internal nucleation, which occurs during the heating path in the DTA, when the sample temperature crosses the nucleation peak at 670–680 °C, independent of the chosen (final) crystallization temperature.
This value of n ~ 3 clearly indicates the growth of three-dimensional crystals from a constant number of nucleation sites in the sample interior. We have not discovered the nature of the nucleation sites yet. However, the crystal nuclei were clearly formed on the heating path, crossing the nucleation curve maximum at 672 °C and reaching temperatures from 760 to 790 °C (Fig. 6).
As seen in the above results, through specific thermal treatments it is possible to produce microstructured diopside glass–ceramics. This study could be extended into four fronts: (1) to determine the chemical mechanism of nucleation; (2) to characterize the properties of the current glass–ceramics; (3) to add a little more Fe2O3 to further boost the nucleation rates; or (4) to increase the treatment time at the temperature of maximum nucleation to develop nano-structured glass–ceramics (nano-GC). Such nano-GC could perhaps show interesting, unusual properties.
Conclusions
We induced copious internal nucleation in a diopside glass by adding enough Fe2O3, which enabled the production of microstructured diopside glass–ceramics. We then studied the crystallization mechanism and kinetics of such promising material. The value of the Avrami exponent n ~ 3 indicates the growth of tridimensionally shaped crystals from a fixed number of internal nucleation sites. Taken in toto, these results are relevant and could guide the development of novel diopside glass–ceramics.
References
Redhammer GJ (1998) Mössbauer spectroscopy and rietveld refinement on synthetic ferri-Tschermak’s molecule CaFe3+(Fe3+Si)O6 substituted diopside. Eur J Mineral 10:439–452
Morimoto N (1988) Nomenclature of pyroxenes. Miner Pet 39:55–76
Nonami T, Tsutsumi S (2000) Press-formable CaO-MgO-SiO2-TiO2-Ag2O glass as a biomaterial. J Biomed Mater Res 50:8–15
Goel A, Tulyaganov Du, Pascual MJ et al (2010) Development and performance of diopside based glass-ceramic sealants for solid oxide fuel cells. J Non Cryst Solids 356:1070–1080
Reddy AA, Tulyaganov DU, Mather GC et al (2014) Effect of strontium-to-calcium ratio on the structure, crystallization behavior and functional properties of diopside-based glasses. Int J Hydrog Energy 39:3552–3563
Reddy AA, Tulyaganov DU, Kharton VV, Ferreira JMF (2015) Development of bilayer glass-ceramic SOFC sealants via optimizing the chemical composition of glasses—a review. J Solid State Electrochem 19:2899–2916
Chou CC, Feng KC, Raevski IP et al (2017) Part Ι: effects of two-stage heat treatment on densification, microstructural features and dielectric properties of CaO–MgO–SiO2 glass-ceramics with ZrO2 nucleating agents. Mater Res Bull 96:66–70
Feng KC, Chou CC, Chu LW, Chen H (2012) Zirconia nucleating agent on microstructural and electrical properties of a CaMgSi2O6 diopside glass-ceramic for microwave dielectrics. Mater Res Bull 47:2851–2855
Zanotto ED (2011) A bright future for glass ceramics. Am Ceram Soc Bull 89:609–612
Davis MJ, Zanotto ED (2017) Glass-ceramics and realization of the unobtainable: property combinations that push the envelope. MRS Bull 42:195–199
Deubener J, Allix M, Davis MJ et al (2018) Updated the definition of glass-ceramics. J Non Cryst Solids 501:3–10
Colombrini R, Zanotto ED, Craievich AF (1981) Vitrocerâmicos a partir de materias primas naturais. Ceramica 27:213–218
Zhang S, Zhang Y, Wu T (2018) Effect of Cr2O3 on the crystallization behavior of synthetic diopside and characterization of Cr-doped diopside glass ceramics. Ceram Int 44:10119–10129
Ray CS, Zhang T, Reis ST, Brow RK (2007) Determining kinetic parameters for isothermal crystallization of glasses. J Am Ceram Soc 90:769–773
Zhang T, Brow RK, Reis ST, Ray CS (2008) Isothermal crystallization of a solid oxide fuel cell sealing glass by differential thermal analysis. J Am Ceram Soc 91:3235–3239
Marotta A, Buri A, Branda F (1981) Nucleation in glass and differential thermal analysis. J Mater Sci 16:341–344
Donald IW (1995) The crystallization kinetics of a glass based on the cordierite composition studied by DTA and DSC. J Mater Sci 30:904–915
Fokin VM, Zanotto ED (1999) Surface and volume nucleation and growth in TiO2-cordierite glasses. J Non Cryst Solids 246:115–127
Fokin VM, Zanotto ED, Yuritsyn NS, Schmelzer JWP (2006) Homogeneous crystal nucleation in silicate glasses: a 40 years perspective. J Non Cryst Solids 352:2681–2714
Prasad NS, Varma KBR (2005) Crystallization kinetics of the LiBO2–Nb2O5 glass using differential thermal analysis. J Am Ceram Soc 88:357–361
Choi HW, Kim YH, Rim YH, Yang YS (2013) Crystallization kinetics of lithium niobate glass: determination of the Johnson-Mehl-Avrami-Kolmogorov parameters. Phys Chem Chem Phys 15:9940–9946
Christian JW (1965) The theory of transformations in metals and alloys: an advanced textbook in physical metallurgy, vol 1, 1st edn. Pergamon Press, Oxford
Bansal NP, Doremus RH, Bruce AJ, Moynihan CT (1983) Kinetics of crystallization of ZrF4-Ba2-LaF3 glass by differential scanning calorimetry. J Am Ceram Soc 66:233–238
Pietrzak TK, Wasiucionek M, Nowiński JL, Garbarczyk JE (2013) Isothermal nanocrystallization of vanadate-phosphate glasses. Solid State Ionics 251:78–82
Acknowledgements
We are thankful to the São Paulo Research Foundation (FAPESP) for funding this research, under the grant number 2013/07793-6 (CEPID). P.S. Bayer would also like to thank the Federal Institute of Science, Technology and Education of Santa Catarina for granting him a leave of absence to complete his doctoral studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mastelaro, V.R., Bayer, P.S. & Zanotto, E.D. Crystallization mechanism and kinetics of a Fe-diopside (25CaO·25MgO·50SiO2) glass–ceramic. J Mater Sci 54, 9313–9320 (2019). https://doi.org/10.1007/s10853-019-03572-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03572-y