
Abstract
A range of nanocomposites based on a polyethylene polymer and hexagonal boron nitride (hBN) filler have been explored in this study. The dielectric properties of the nanocomposites, which consisted of 2, 5, 10, 20 and 30 wt% of hBN, have been compared to the dielectric properties of the unfilled polyethylene blend. Scanning electron microscopy revealed that the hBN was uniformly distributed in the polyethylene matrix, although large amounts of agglomerates were present in the nanocomposites containing more than 10 wt% of hBN. The incorporation of hBN into polyethylene resulted in a highly disordered morphology in comparison with the unfilled polyethylene, in which this effect was more pronounced with increasing hBN content. This is consistent with the increasing crystallisation temperature as the hBN content increases, as shown by differential scanning calorimetry, where the hBN acted as a highly effective nucleating due to the strong interactions between the polyethylene and the hBN. This strong interaction is again reflected in the thermal decomposition temperature which similarly increases with increasing hBN content. The study demonstrates the remarkable electrical properties of the prepared nanocomposites, where the breakdown strength monotonically increased as a function of hBN content, even with a very high 30 wt% of hBN. The improvement in electrical properties, even at high hBN concentrations, is contradictory to the reported results in the literature and is mainly attributed to the hydrophobic surface of the hBN particles.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The availability of reliable and affordable supplies of energy that do not lead to significant climate change is one of the most pressing challenges of present times and is one that affects both developed and developing economies [1]. This is forcing a paradigm shift in the way we generate electricity, from large centralised power stations, which frequently rely upon fossil fuels, to a much more diversified—both in terms of their nature and location—sources. For example, the adoption of renewable generation involving offshore wind necessarily requires the transmission of large amounts of energy from the point of production to centres of demand. As transmission distances increase, a change from conventional alternating current (AC) to direct current (DC) becomes increasingly attractive [2]. Indeed, this approach also enables the interconnection of non-synchronous, national power systems, thereby enabling the more efficient use of resources.
All electrical systems rely upon insulation and, as the demands placed on power systems increase, for example, through a desire to reduce insulation systems in order to aid heat dissipation or to exploit high voltage DC transmission, so do the demands placed on the insulation. The critical enabling role played by dielectric materials in facilitating the evolution of next-generation and transmission infrastructures has led to significant-related research efforts, and a topic that has received particular attention is that of nanocomposite dielectrics—nanodielectrics [3]. The potential of nanodielectrics was first highlighted by Lewis in 1994 [4] albeit that such systems had received much attention for many years in connection with other applications. Lewis proposed that the addition of nanometric sized particles in a polymer would form interfaces that highly influence the dielectric properties of the resulting nanodielectrics material.
The potential of nanodielectrics as high-performance insulation systems has been demonstrated in many studies. For example, the addition of only 1 wt% of magnesium oxide nanoparticles in polyethylene has been shown to decrease the electrical conductivity of polyethylene by 30 times [5] which is a very useful property for high voltage cable applications. However, such improvements are not always reported as the effects of incorporating nanoparticles in polymers can have huge drawbacks. Wang et al. [6] reported an increase in the breakdown strength in LDPE/alumina nanocomposites from 317 to 344 and 361 kV/mm as 0.1 and 0.5 wt% of alumina is added, respectively; however, further addition of alumina including 1–10 wt% reduced the breakdown strength. However, Hillborg et al. [7] reported no change in the breakdown strength in polyethylene/alumina nanocomposites with alumina content up to 3 wt% although the breakdown strength behaviour was shown to gradually decrease with increasing alumina content from 3 to 15 wt%. In polyamide-imide/alumina nanocomposites, the breakdown strength increased with increasing alumina content up to 5 wt%, followed by a decrease of breakdown strength with increasing alumina content up to 10 wt% [8]. Similarly, Rytoluoto et al. [9] reported a similar pattern in polypropylene/silica nanocomposites with a decrease in breakdown strength as the amount of silica is increased. Tian et al. [10] reported an increase in breakdown strength in LDPE/ZnO nanocomposites with 0.1 wt% of ZnO followed by a decrease in breakdown strength with increasing ZnO content up to 7 wt%. The breakdown strength in epoxy/organoclay nanocomposites gradually increased up to 5 wt% of clay followed by a sudden decrease in breakdown strength with 10 wt% of clay content [11].
When effects of percolation are considered, the work of Grabowski et al. [12] has shown that adding up to 45 vol% of silica in polymethyl methacrylate (PMMA) significantly reduced the breakdown strength from ~ 800 to ~ 340 kV/mm. An increase in breakdown strength was reported by Siddabattuni et al. [13] in 5 vol% epoxy/TiO2 and epoxy/BaTiO3 nanocomposites, containing surface modified titania and barium titanate nanoparticles followed by a drastic reduction in breakdown strength at percolation when 15 and 30 vol% of particles were added. The breakdown strength of other BaTiO3-based nanocomposites has been shown to be drastically reduced at the percolation threshold [14]; the addition of 20 vol% BaTiO3 in poly(vinylidene fluoride-co-hexafluoropropylene) reduced the breakdown strength from ~ 380 to ~ 230 kV/mm. The decrease in breakdown strength reported by many researchers was almost always attributed to the presence of large agglomerates at high filler loading levels that form conductive pathways for the charge carriers, although no clear correlation between the two properties has yet been established. On another hand, the presence of agglomerates forming a continuous percolating network could highly improve the thermal conductivity of the system: a topic that is also of great technological importance and, consequently, the potential of composite systems as a route to improved material characteristics has been studied by many workers [15, 16]. However, increasing the thermal conductivity in composite system requires the formation of a percolating network of particles with high thermal conductivity, which as seen from the discussion above, comes at the expense of a reduced dielectric breakdown strength.
This work set to consider the electrical behaviour of systems based upon hexagonal boron nitride (hBN) in polyethylene. This choice of system was based upon two factors: first, polyethylene is of massive technological importance in power cable application; second, hexagonal boron nitride is characterised by high thermal conductivity and, in a number of reports, has been shown to facilitate composites with desirable electrical characteristics most likely due to its hydrophobic nature. The rationale behind this study is that the electrical behaviour is not dominated by the aggregation state of the nanoparticles—a state which normally enhances charge transport. The specific objectives of this study were to: examine the effect of hBN on the morphology of polyethylene by scanning electron microscopy (SEM), study the melting and crystallisation behaviour of the resulting nanocomposites by differential scanning calorimetry (DSC), confirm the hBN content and investigate the thermal decomposition behaviour by thermogravimetric analysis (TGA), and understand the effect of hBN on the electrical breakdown strength.
Experimental
Materials
The hexagonal boron nitride filler (NX-1 from Momentive: mean lateral particle size, 900 nm; specific surface area, 20 m2/g) was used in this study. Low-density polyethylene (LDPE LD100BW from ExxonMobil: density, 0.923 g/cm3; melt index at 190 °C and 2.16 kg, 2 g/10 min) and high density polyethylene (HD Rigidex HD5813EA from BP Chemicals: density 0.958 g/cm3; melt index at 190 °C and 2.16 kg 13 g/10 min) were used to prepare the host polymer. The polymer matrix was prepared from a combination of 80% LDPE and 20% HDPE blend. This choice of matrix polymer was based upon a desire to eliminate potential effects related to the nucleating effect of the hBN. It has long been appreciated that inter-spherulitic boundaries constitute sites of electrical weakness in semicrystalline polymers [17] and, therefore, the inclusion of additives that affect nucleation density can indirectly influence breakdown performance, through changing the morphology of the host matrix such that the influence of inter-spherulitic boundaries is mitigated. Blend systems, such as the one used here, undergo a two-stage isothermal crystallisation process [18,19,20] that involves the initial formation of a skeleton of lamellae composed, primarily, of HDPE, followed by the subsequent crystallisation of the remaining LDPE-rich fraction of the system within the interstitial regions between the pre-existing HDPE-rich lamellar framework. This both prevents concentration of impurities at inter-spherulitic boundary (they remain solvated within the molten LDPE-rich phase prior to quenching) and uniformly distributes throughout the system the mechanical stresses which are associated with the reduction in specific volume that occurs on crystallisation. In this way, the breakdown strength of such blends systems has been shown to be independent of nucleation-related effects [21], such that the true, direct influence of any additives (hBN here) is apparent.
Material preparation
The desired amount of LDPE, HDPE and hexagonal boron nitride (hBN) was weighed out to produce six different materials containing different hBN concentrations. In this study, a solvent blending procedure was used to prepare all the materials. The LDPE and HDPE, which were supplied in the form of pellets, were first added to a round-bottomed flask containing 200 mL of xylene; a small amount of xylene solvent was added to the hBN powder in a small container to create a suspension, which was then added directly to the polymer/xylene mixture. The polymer/hBN/xylene mixture was then heated to the boiling point of xylene (~ 140 °C) and stirred simultaneously using a magnetic stirrer bar. Once the xylene started to boil, the heat was lowered to allow the mixture to boil gently. After all the polymer had dissolved in the xylene, the hot polymer/hBN/xylene mixture was quickly poured into a beaker containing 300 mL of the cold non-solvent, methanol, with vigorous stirring, resulting in the precipitation of the polymer/hBN system as a white gel. The resultant material was then filtered, left to dry in a fume cupboard for 5 d and, finally, dried in a vacuum oven at 60 °C for 3 days to remove the residual xylene solvent. The resulting dry polyethylene/hBN product was then melt pressed with a 5-ton load at 180 °C to make the material into a more compact form, which eased the subsequent production of specimens for testing.
The samples of the appropriate geometry for the required tests, as described in the sections below, were prepared by melt pressing with a 5-ton load at 180 °C. For isothermal crystallisation, the samples were then directly placed from the melt in an oil bath maintained at 115 °C for 1 h, which were then directly quenched into water; this thermal treatment was chosen to facilitate the process of morphological evolution described above. Samples were stored under vacuum conditions prior to any testing to prevent any moisture absorption from the environment.
Sample formulations
The materials listed in Table 1 are denoted by “PEBN/hBN Content/Crystallisation method” where the “PEBN” refers to a polyethylene boron nitride nanocomposite, the “hBN Content” is the weight per cent of the used hBN and the “crystallisation method” is 115 due to the isothermal crystallisation at that temperature.
Scanning electron microscopy
The structure and morphology of all the materials were examined using scanning electron microscopy (SEM), following permanganic etching. Potassium permanganate was gradually added to an acidic mixture composed of five parts sulphuric acid, two parts phosphoric acid and one part water, which was then stirred for 15 min without heat to give a 1% solution. Samples, 200 µm in thickness, were transferred to empty vials where ~ 10 mL of the etchant mixture was added and the vials were shaken for 4 h. After etching was complete, a solution of 25% hydrogen peroxide and 75% dilute sulphuric acid (two parts sulphuric acid and seven parts water) was added, before the samples were rinsed in distilled water, then acetone and then left to dry. Prior to examination in the SEM, the etched surfaces were gold coated using an Emitech K550X sputter coater. A JEOL Model JSM-6500F scanning electron microscope was used to examine the specimens at an applied voltage of 15 kV.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was conducted using a Perkin Elmer DSC-7 to measure the effect of the filler on the melting and crystallisation behaviour of the different nanocomposites. High purity indium was routinely used to calibrate the equipment before use. Samples weighing ~ 5 mg were placed in an aluminium pan with the heat flow measured relative to an empty aluminium reference pan. The samples were heated in dry nitrogen from 40 to 160 °C at a rate of 10 °C/min to analyse the melting behaviour then the samples were cooled from 160 to 40 °C at the same rate to analyse the crystallisation behaviour (Table 2).
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was conducted in air using a Perkin Elmer 1 TGA, which determined both the true loading level of hBN in each system and revealed the effect of the filler on the decomposition behaviour of the system. Samples weighing ~ 5 mg were placed in an aluminium pan and heated, in air, from 40 to 600 °C at a rate of 10 °C/min. The hBN concentration was determined as the mass of the residue at the final temperature of 600 °C.
Dielectric breakdown strength
The AC dielectric breakdown strength of all the materials was measured, to examine the effect of the filler content on this parameter. Thin film samples, ~ 70 µm in thickness, were prepared as described above. The samples were placed between two opposing 6.3 mm steel ball bearing electrodes immersed in Dow Corning 200/20cs silicone fluid to prevent surface flashover. A 50 Hz AC voltage, increasing at a ramp rate of 50 V/s, was applied until the sample failed. A total of 20 breakdown measurements were taken for each formulation, and the resulting data were analysed, assuming a two-parameter Weibull distribution.
Results and discussion
Scanning electron microscopy
Figure 1 shows low and high magnification micrographs of selected materials. The SEM micrographs of the unfilled polyethylene, the 5 wt% and the 30 wt% nanocomposites are shown to compare the structure and morphology of the nanocomposites with low and high content of hBN. Low magnifications micrographs of the unfilled polyethylene show space-filling banded spherulites, developed by the isothermally crystallised HDPE phase, separated by featureless regions of the rapidly quenched LDPE phase. These morphological features are typical of quenched and isothermally crystallised, at 115 °C, polyethylene systems which have been reported elsewhere in the literature [22, 23].

Low magnification micrographs of a 0 wt%, b 5 wt%, c 30 wt% and high magnification micrographs of d 0 wt%, e 5 wt%, and f 30 wt%
Figure 1e reveals a significant degree of agglomeration of the hBN, where the agglomerates are up to 10 µm in size. The structure of nanocomposites with low and high filler content appears fundamentally different. In the low filler content nanocomposites, there are discrete hBN inclusions in a continuous matrix, whereas continuous percolating hBN networks are evident at high filler contents. These micrographs clearly show that the hBN exists in a range of several different sizes. As the hBN content increases, the variation in the particles sizes becomes larger. The hBN particles are generally not well dispersed in the matrix in all nanocomposites and at very high loading levels, e.g. 20 and 30 wt%, and hBN aggregates of sizes near 10 µm are present due to particle agglomeration. High magnification SEM micrographs of the nanocomposites reveal that the aggregates are made up of smaller particles, or hBN platelets, that are agglomerated at different orientations. Despite the agglomeration, the hBN in all of the nanocomposites, which consists of some aggregated instead of exfoliated sheets, is uniformly distributed in all space in the matrix.
Low magnification SEM micrographs of the isothermally crystallised nanocomposites show that the banded spherulites, that were previously seen in the unfilled polyethylene, are no longer visible even after the addition of very small amounts (2 wt%) of hBN. The whole texture of the polymer matrix is different to the unfilled polyethylene blend, where there is no slight evidence of banded spherulites. High magnification SEM micrographs show that there is a much more disordered polymer morphology, relative to the unfilled polyethylene, in the isothermally crystallised nanocomposites. In the low magnification micrographs of the nanocomposites, there is little evidence of the banded spherulitic morphology that characterises the unfilled system; the higher magnification images reveal a much more disordered morphology. This is consistent with the work of Green et al. [24], who reported that dispersing MMT platelets into polyethylene resulted in system characterised by enhanced nucleation and a disordered lamellar texture.
It is speculated that the change in the morphology upon the addition of hBN particles is due to a very strong nucleating effect brought about by the addition of hBN particles. The incorporation of hBN particles, which act as nucleating sites and nucleating agents, increases the rate at which spherulites develop where they seem to impinge upon each other, thus limiting further growth. This results in an undeveloped banded spherulitic growth in the morphology, where the spherulites are not circular like the banded spherulites in the pure polyethylene matrix; this effect becomes more pronounced with increasing hBN loading levels. This could be attributed to the stronger interaction between the boron nitride and the polymer matrix as the hBN content increases, due to the increasing nucleating effect. Green et al. [25] found similar observations where they found that the addition of MMT particles into polyethylene resulted in a system with a highly disordered morphology due to the inhibition of crystal growth, similar to the system studied here. Similar results have been reported by Chan et al. [26] in polypropylene/calcium carbonate showing SEM images of polypropylene with spherulites 40 µm in size whereas the nanocomposites containing 9.2 vol% of filler showed no spherulites and a completely different morphology. As the extent of morphological disorder in the polyethylene matrix increases with increasing hBN content, the polymer becomes almost featureless at high hBN content and is unrecognisable from the isothermally crystallised unfilled polyethylene, as seen from the high magnification SEM micrographs. This highly disordered morphology is a result of the ability of hBN to promote nucleation which in turn limits the ordered growth of banded spherulites where they are not clearly present.
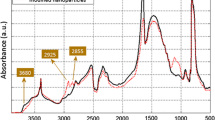
The sheets of hexagonal boron nitride are composed of hexagonal ring structures made up of boron and nitrogen atoms which, both being trivalent, would be fully satisfied in such a structure. Consequently, while idealised basal surfaces should contain hexagons composed of boron and nitrogen alone, it is likely that the edges of hBN sheets will contain species such as hydroxyl groups. Indeed, examination of the as-supplied hBN power used here by Fourier-transform infrared spectroscopy (FTIR) revealed the presence of weak absorption bands centred at 3131 and 3420 cm−1, which can be associated the stretching of O–H and N–H bonds, as reported elsewhere [27,28,29,30,31]. However, if the true structure were to differ markedly from the ideal, then the presence of many polar surface groups would be expected to lead to hydrogen bonds with any labile polar species present in the system, most notably, water. Many studies of water uptake and accumulation at nanoparticle/polyethylene interfaces have been undertaken [32,33,34], which have demonstrated that the presence of the resulting bound water is readily seen in the dielectric spectrum and that the presence of this water has a marked detrimental influence on electrical performance. In contrast, the systems considered here interact extremely weakly with water, which implies the presence of few accessible hydroxyl groups at hBN/polyethylene interfaces [35]; this is discussed fully elsewhere [36]. Another consequence of this lies in the critical role that hydroxyl moieties play in commonly used surface functionalisation strategies [37]. While surface treatment could facilitate the separation of the hBN layers, the introduction of additional hydroxyl groups would be detrimental to the electrical properties of the nanocomposites and would counteract one of the principal motives of using such a highly hydrophobic particle in this study—minimising interactions with environmental water. In contrast, a layered particle such as montmorillonite clay is highly polar as it contains metal cations in between the clay layers, which makes it hard to interact with non-polar polymers. Therefore, it is necessary to replace these cations with organic groups such that the polymer becomes intercalated in between the clay layers [38]. Similarly, the surfaces of graphene oxide have an abundance of functional groups, which easily interact with the solvent and aid in the separation of the layers [39]. Furthermore, the polar B-N bonds in hBN result in stronger interlayer forces than in graphite despite both particles having the same interlayer distance. This property makes the hBN layers more difficult to separate in comparison with graphite, especially when a high filler loading is used, as reported elsewhere [40, 41].
Differential scanning calorimetry
Figure 2 contains DSC crystallisation and melting data obtained, as explained above, by a three-step process: isothermal crystallisation at 115 °C, followed by quenching; melting by heating at 10 °C/min, with subsequent cooling at 10 °C/min. Despite the actual experiment time sequence used, consider first the crystallisation data shown in Fig. 2a and then the melting data shown in Fig. 2b.

a Non-isothermal (cooling at 10 °C/min) DSC crystallisation traces and b DSC melting traces for the polyethylene/hBN nanocomposites
The effect on hBN loading level of the non-isothermal crystallisation behaviour of the polyethylene blend used here is shown in Fig. 2a. In the absence of hBN, two exotherms are present, which correspond, essentially, to the sequential crystallisation of the HDPE (at about 116 °C) and the LDPE (around 95 °C). On including hBN, two features become evident. First, the higher temperature exotherm is displaced, progressively, to higher temperatures as the hBN loading level increases. This phenomenon is commonly observed in systems where enhanced nucleation occurs. For example, the use of both nucleating agents and nanoparticles has been studied by Zaman et al. [42], who investigated the crystallisation behaviour of polyethylene blends composed of 50 wt% LDPE and 50 wt% HDPE containing small amounts of dialkyl peroxide (DAP) plus up to 15 wt% of nanocalcium carbonate. The authors found that the crystallisation temperature of the polyethylene blend was 114 °C, which increased to 117 and 121 °C, for the nanocomposite containing 10 wt% of filler with and without DAP, respectively. The nucleating effect was attributed to the presence of the nanoparticles. Similarly, Cui et al. [43] characterised polyethylene nanocomposites based on different compositions of MMT. They reported that at MMT loading levels of up to 7.7 wt% the nanocomposites exhibited higher crystallisation temperatures than the pure polyethylene. Kuila et al. [44] studied the physical properties of linear low-density polyethylene (LLDPE)/graphene nanocomposites and found that the crystallisation temperature of the nanocomposites again increased with increasing graphene content. Jiang et al. [45] found that the crystallisation temperatures of HDPE/graphene nanocomposites significantly increased at low graphene loading levels and, thereafter, increased progressively with increasing graphene content. As in the above studies, we therefore associate the elevation in the crystallisation temperature of all the systems containing hBN compared with the unfilled blend as stemming from the nucleating ability of the hBN. Indeed, this assertion is consistent with the work of Puente et al. [46], who reported that the addition of only 0.2 wt% hBN to poly (3-hydroxybutyrate) increased the crystallisation temperature from 91 to 121 °C. As in Fig. 2a, the observed crystallisation temperature was found to increase progressively with increasing hBN content. Boron nitride particles have also been reported to behave in the same way in poly(3-hydroxybutyrate-co-3-hydroxyvalerate) [47] and in poly(3-hydroxybutyrate-co-4-hydroxybutyrate) [48], where they similarly act as highly effective nucleating agents.
The second notable feature of the data shown in Fig. 2a is the evidence for crystallisation occurring at temperatures in excess of 128 °C. This manifests itself most obviously as a crystallisation exotherm close to 128 °C, which increases in enthalpy, but not temperature, as the hBN content of the system increases. In addition, close examination of the crystallisation trace of the system indicated PEBN/30/115 reveals the presence of an extremely small crystallisation peak at about 133 °C. Such features are highly unusual and, we suggest, are associated with the way in which the hBN and the linear polyethylene sequences present in the polymer interact. Clearly, the temperature at which a polymer is seen to crystallise under non-isothermal conditions is determined by the time necessary to form a stable crystal nucleus at the rate at which this can grow to form sufficient material to be detected. Since the rate of crystal growth of polyethylene at temperatures in excess of 128 °C is extremely slow [36] and, in this case, the time available is less than 100 s (DSC cooling at 10 °C/min and the thermodynamic equilibrium melting temperature of polyethylene is close to 142 °C [49]), the most credible interpretation of the origin of these features relates to the thermodynamic characteristics of the polyethylene/hBN surface interactions. Muchova and Lednicky [50] proposed that secondary nucleation, as occurs here, is determined by interfacial interactions. This analysis assumes that the size of the critical nucleus is determined thermodynamically (including any specific surface energy terms) and that, since the associated energy barrier can only be overcome through random thermal fluctuations in the melt, this will determine the time necessary for a critical nucleus to form. During this dynamic process, molecular segments attach and detach from the surface of the nucleus until a critical dimension is exceeded. This defines the induction time which, consequently, may contain both the time necessary to lay down the initial molecular layer on any foreign surface, plus the time for this to grow to stability. Based upon this analysis, which is entirely general, it would seem that hBN surfaces favour rapid formation of stable crystalline structures. They manifest themselves as high-temperature crystalline exotherms in the DSC cooling traces as shown in Fig. 2a. Indeed, comparable features have been reported in the work of Zhang et al. [51], who used a scanning probe thermal analysis to determine the melting behaviour of the polyethylene immediately adjacent to their hBN. Elevated melting temperatures were seen, indicating that, despite being unexpected, this effect is reproducible and an intrinsic feature of polyethylene/hBN interactions.
The DSC melting behaviour of all the materials is shown in Fig. 2b. In general, data from the unfilled system take the expected form and reflect the fact, that two components are present in the blend, and that some degree of co-crystallisation occurs between the HDPE with the more linear molecular sequences in the LDPE. Since this has been fully discussed elsewhere [22, 23], the basic form of the melting traces will not be considered further here. However, as discussed above, the addition of hBN leads to the formation of a minor lamellar population that melts at an unexpectedly high temperature, around 135 °C. On kinetic grounds, this value is reasonable, given that the melting traces were obtained from samples that were crystallised to completion [22, 23] at 115 °C over a prolonged period. Whereas the data shown in Fig. 2a resulted from a crystallisation regime, where relatively little time was available for crystal formation from the melt.
Finally, some comment is worthwhile concerning the transition enthalpies values obtained from the data presented in Fig. 2. In this, all the data were normalised with respect to the total sample mass, which includes both polymeric and inorganic components. As such, as the percentage of hBN increases, the residual fraction of the material that crystallises/melts is reduced. This explains the variation in the apparent sizes of the various features shown; peak integration and renormalisation with respect to just the mass of the polymer reveal no systematic variations in the transition enthalpies.
Thermogravimetric analysis
Figure 3a shows the TGA curves for all the samples. The unfilled polyethylene sample retained 100% of its initial mass until the temperature reached around 383 °C, where the sample started to degrade, at which then the sample lost 50% of its initial mass at 467 °C and lost almost all of its mass at 600 °C with a residue of 0.25% of its initial mass. Polyethylene undergoes a thermal decomposition process dominated by random chain scission of the polymer chain which is usually initiated at weak link sites in the polyethylene backbone structure such as peroxides, carbonyl groups and chain branches [52]. The chain scission of the polyethylene backbone results in the formation of free radicals which eventually decompose into volatile products consisting of polyethylene fragments with lower molecular weight. The degradation products consist of a large number of hydrocarbons with varying chain lengths which contain anywhere between 1 and 70 carbon atoms, of which propene and 1-hexene are the most abundant products [53].

TGA measurements showing a mass change with temperature and b degradation temperatures at different filler content
For comparative purposes, taking into account the temperature at which 10% (T10) and 50% (T50) of the material’s initial mass have degraded is a good indication of the material’s thermal stability. The TGA results that are shown in Fig. 3 indicate that the unfilled sample, PEBN/0/115, starts to degrade at an earlier temperature than all the nanocomposites; the higher the hBN content, the higher the onset of degradation in both T10 and T50. Figure 3b shows that the degradation temperature at which 10% of the mass is lost is greatly affected in low filler content where it stops to be dependent on the filler content when percolation is reached. However, the degradation temperature at which 50% of the mass is lost appears to increase constantly with increasing filler content even when the percolation limit is reached. Li et al. [54] reported the increase in the onset degradation temperature with increasing hBN content in polyimide/hBN nanocomposites where the degradation temperature increased from 460 °C in the unfilled polyimide to 461, 466 and 474 °C in the 10, 20 and 30 wt% nanocomposites, respectively. In layered nanocomposites containing graphene, Fim et al. [55] reported an increase in the maximum degradation temperature from 480 °C in the unfilled polyethylene to 510 °C in the 15.3 wt% nanocomposite whereas the onset degradation temperature increased from 442 °C in the unfilled polyethylene to 472 °C in the 6.6 wt% nanocomposite. However, the onset degradation temperature decreased to 463 °C in the 15.3 wt% nanocomposite. They suggest that graphene is more stable and stiffer than polyethylene which causes the polyethylene to become rigid thus decreasing the chain mobility which hinders the degradation process. Kuila et al. [44] found that the onset temperature in LLDPE/graphene nanocomposites decreased from 317 °C in the unfilled LLDPE to 302 °C in the 8 wt% nanocomposite, due to the degradation of the dodecyl amine groups of the functionalised graphene, while the T50 lost increases with increasing graphene content from 443 to 464 °C. In layered nanocomposites containing clay, Panupakorn et al. [56] reported an increase maximum degradation temperature polyethylene/clay nanocomposite from 463 °C in the unfilled polyethylene to 481 °C in the 5 wt% nanocomposite. However, the degradation temperature decreased to 452 °C and 449 °C in the 10 and 20 wt% nanocomposites, respectively. Similar trends in the thermal decomposition behaviour of clay nanocomposites have been reported elsewhere [56], which show an increase in thermal degradation temperature at low loading levels followed by a decrease in thermal degradation temperature at higher loading levels of clay. For low clay content, the nanocomposites degraded slower than the pure polyethylene due to the barrier properties caused by the nanoclays, which prevented heat transfer into the bulk. At higher clay content, the nanocomposite degrades at lower temperatures than the pure polyethylene due to the acidic sites from degradation of the organic surface modifiers, which acts as catalysts for the polymer chain degradation. Therefore, clay layers act as barriers which enhance thermal stability. But as the clay content increases, it acts as a catalyst which reduces the thermal stability.
In this study, as the temperature at which the samples lose 10 and 50% of their initial mass both increases with increasing filler content, this suggests that the addition of more boron nitride particles increases the thermal stability of the system as the material degrades at higher temperatures. There is no presence of functional groups or surface modifiers on the surface of hBN as found in graphene or clay that would promote the thermal degradation, and therefore the presence of hBN, even at very high loading levels, serves only to enhance the thermal stability of the system. During the thermal degradation of the nanocomposites, the layers of the hBN act as barriers which limit the diffusion of gases into and out of the polymer nanocomposite. This barrier which is formed by the hBN layers on the surface limits the transport of oxygen and heat which delays the thermal degradation of the nanocomposite. Dash et al. [57] experimentally found that there is a significant reduction in oxygen permeability in soy/BN nanocomposites with increasing BN content which confirms the oxygen barrier properties of the hBN layers. The hBN layers on the surface limit oxygen and heat flow, to assist in the formation of char. As a result, the layers consisting of boron nitride, which are impermeable to gases, and a thermally stable char are formed on the surface. Their combined effect, which prominently increases with increasing hBN content, further restricts both the diffusion of the volatile products evolved during the degradation of the polymer into the air and the oxygen to the nanocomposites, which effectively reduces the rate of the thermal degradation [58].
Dielectric breakdown strength
Figure 4 shows the breakdown strength of all the materials, which reveals a monotonic increase in breakdown strength with increasing filler content up to 30 wt%. Scale and shape parameters of all samples can be found in Table 3. Although we are not aware of any comparable studies of the breakdown behaviour of polyethylene/hBN systems, examination of the reported behaviour of other nanofilled polymers demonstrates that this is entirely contrary to expectations. For example, Tsekmes et al. [59, 60] found that the AC and DC breakdown behaviour in epoxy/hBN nanocomposites are similar, where the breakdown strength increases in the nanocomposites containing 1 vol% of hBN and starts to decrease at higher filler contents. They attribute the decrease in breakdown strength at mainly to the structural imperfections created by nanocomposites at higher filler loading, which introduce voids due to the increase in interfacial area and mass density. Gao et al. [61] tested the dielectric breakdown strength of epoxy/silica nanocomposites, where they observed an increase in breakdown strength with increasing silica content up to 5 wt%. This was followed by a significant decrease with further addition of silica, attributed to percolation effects. They suggested that the overlapping of the double layer associated to the interfaces at higher silica content allows charge carriers to move easily through the system, thereby reducing the breakdown strength. Singha et al. [62] found that adding 0.1 wt% of titania and alumina fillers in epoxy significantly reduces the breakdown strength, compared to the pure epoxy. An interesting observation is that the breakdown strength in the titania nanocomposites decreases with titania content up to 0.5 wt%, with subsequent increase with increasing filler content up to 10 wt%, although it remains lower than the breakdown strength of the pure epoxy. Grabowski et al. [12] investigated the dielectric strength of silica nanocomposites, based on four different amorphous polymers with up to 45 vol% of silica. Where the breakdown strength increased initially in some of the nanocomposites, the breakdown strength of all nanocomposites decreased after the addition of 15 vol% and higher, again due to reaching the percolation threshold. Due to the high aspect ratio of hBN, it can reach percolation threshold at lower volume fractions compared to spherical particles with low aspect ratio.

a Weibull plots of the polyethylene/hBN nanocomposites and b Weibull scale parameter as a function of hBN content
The SEM images clearly show that there is a percolating network at high hBN content and almost all studies in the literature have reported that percolating systems always result in the deterioration in electrical properties, as charge transport easily occurs through the connected interfacial layers. This is peculiar, since the results clearly show that the percolation of hBN does not have a negative effect on the breakdown strength, where the charge transport is expected to occur in a system with continuous interfaces touching each other. The breakdown strength improves, although the system is agglomerated and percolating pathways exist. Hence, the breakdown strength and charge transport have less to do with the distribution of the nanofiller, and much more with how the nanofiller interacts with the charge carriers.
It has also been suggested that the change in morphology of the polymer upon the addition of the nanoparticles highly affects the interfacial regions in the material, which can increase the density of charge traps and effectively change the breakdown behaviour [63]. However, the work of Vaughan et al. [64, 65] showed that the addition of 10 wt% of MMT in polyethylene increased the breakdown strength by 20% relative to the unfilled polymer, where the presence MMT had no effect on the morphology of the PE. However, in another system containing a different grade of MMT, no change of the breakdown strength of the nanocomposite was observed, but the MMT was found to have a strong nucleating effect on the polyethylene, which inhibited crystal growth and disrupted the polymer’s morphology. Conversely, the work of Lau et al. [66] showed that while the presence of silica in polyethylene enhanced nucleation, it did not affect crystal growth and morphology. No significant change in breakdown strength was found in these composites.
While the breakdown strength in this study may have been affected by the changes in morphology, there does not appear to be a precise relationship between the two properties. Improved electrical properties in nanocomposites which exhibit large volume resistivity, larger breakdown strength and reduced charge mobility relative to the unfilled polymer, have largely been attributed to the charge trapping capabilities of the nanofiller [67]. When the material is placed between the two metal electrodes, the charge carriers injected from the electrodes are captured by the traps present at the electrode-dielectric interface, which subsequently suppresses any further charge injection from the electrodes to limit the charge carrier movement in the polymer nanocomposite [68]. Therefore, the presence of the hBN platelets could suppress the electrode charge injection to trap the charges near the electrode. This is thought to increase the energy required for charge injection, which effectively increases the electric field required for the sample to reach breakdown. However, the origin of these traps is usually associated with the chemical interaction between the surface functional groups of the nanoparticles, and the polymer at the interface due to surface functionalisation of the nanoparticle [69]. While the hBN particles could effectively trap the charge carriers, the limited number of surface functional groups on hBN would therefore suggest that the increase in breakdown strength may not be solely due to the presence of charge traps.
While nanoparticle can effectively trap charge carriers, nanoparticles can also act as scattering centres whereby they effectively reduce the mobility of the charge carriers which results in a large breakdown strength [70]. The platelet structure of hBN, as opposed to the spherical structure of many ceramic nanoparticles, could therefore act as effective scattering sites for charge carriers, causing a reduction in the conduction current which, in turn, increases the energy required for breakdown. The lack of functional groups on the surface of hBN and its platelet structure suggests that a charge scattering effect is more dominant than a charge trapping effect, which is what ultimately results in increased breakdown values. The lack of surface functional groups also prevents the formation of an electrically conductive pathway during percolation in the nanocomposites containing a high hBN content. Thus, a decrease in breakdown strength is not observed in this study, as opposed to the many studies discussed earlier, which were incorporating fillers at a high loading level (> 10 wt%), which always reported a decrease in breakdown strength, especially when the percolation threshold is reached.
Conclusions
A range of polyethylene hBN nanocomposites have been prepared with dielectric properties suitable for high voltage applications. The hBN particles strongly interact with the polyethylene matrix to produce a highly disordered morphology. This is consistent with the boron nitride acting as an effective nucleating agent, as evidenced from the DSC results. Although the hBN in the nanocomposites is uniformly distributed in the polyethylene matrix, there is still a reasonable degree of agglomeration, without any evidence of intercalation or exfoliation. The TGA results revealed that the hBN particles enhance the thermal stability of the system, in which the thermal degradation temperature is proportional to the hBN content. Similarly, there is a monotonic increase in breakdown strength with increasing hBN content. This trend persists even at 30 wt% of hBN in the polymer matrix, despite the existence of a percolating hBN network. This suggests that the charge transport in this system is limited even at the percolation limit. Therefore, electrical properties, such as the charge transport and breakdown mechanisms, are primarily dominated by how the nanofiller interacts with the charge carriers, rather than how the nanofillers are distributed in the polymer. The breakdown data are unexpected, since the opposite effect is mostly reported in the published literature. The classical analysis assumes that once nanoparticles are added into a polymer, they tend to agglomerate. Once they agglomerate, it is further assumed that the electrical properties of the nanocomposite deteriorate, which is why particles have to be as small as possible and at low filler concentrations. However, the work presented here has clearly shown that this is not the case of hBN. Main reason is the surface chemistry of hBN, which is characterised by a hydrophobic surface with lack of available hydroxyl groups, which dominates the charge transport dynamics of the system. The results presented in this work clearly demonstrate that the dispersion of nanoparticles is not the main factor to consider when trying to improve the electrical properties in nanocomposite. It is the nature of the surfaces that need to carefully studied, and the interactions at the polymer/particle interface, which would enable the use of the right nanoparticle for the appropriate application. It has been speculated that the presence of water shells around nanoparticles is the mechanism by which electrical properties are limited, and the nature of the systems in this work suggests that investigating the water absorption capabilities in hBN nanocomposites would be helpful.
References
Gielen D, Boshell F, Saygin D (2016) Climate and energy challenges for materials science. Nat Mater 15:117–120. https://doi.org/10.1038/nmat4545
Lakshmanan P, Liang J, Jenkins N (2015) Assessment of collection systems for HVDC connected offshore wind farms. Electr Power Syst Res 129:75–82. https://doi.org/10.1016/j.epsr.2015.07.015
Nelson JK (2007) Overview of nanodielectrics: insulating materials of the future. In: Electrical insulation conference and electrical manufacturing expo, Nashville, TN, pp 229–235
Lewis TJ (1994) Nanometric dielectrics. IEEE Trans Dielectr Electr Insul 1:812–825
Pourrahimi AM, Pallon LKH, Liu D et al (2016) Polyethylene nanocomposites for the next generation of ultralow-transmission-loss HVDC cables: insulation containing moisture-resistant MgO nanoparticles. ACS Appl Mater Interfaces 8:14824–14835. https://doi.org/10.1021/acsami.6b04188
Wang W, Li S (2014) Correlation between trap parameters and breakdown strength of polyethylene/alumina nanocomposites. In: International symposium on electrical insulating materials, pp 73–76
Li W, Hillborg H, Gedde UW (2015) Influence of process conditions and particle dispersion on the ac breakdown strength of polyethylene–aluminium oxide nanocomposites. IEEE Trans Dielectr Electr Insul 22:3536–3542. https://doi.org/10.1109/TDEI.2015.005366
Calebrese C, Hui L, Schadler LS, Nelson JK (2011) A review on the importance of nanocomposite processing to enhance electrical insulation. IEEE Trans Dielectr Electr Insul 18:938–945. https://doi.org/10.1109/TDEI.2011.5976079
Rytoluoto I, Lahti K, Karttunen M et al (2015) Large-area dielectric breakdown performance of polymer films—part II: interdependence of filler content, processing and breakdown performance in polypropylene-silica nanocomposites. IEEE Trans Dielectr Electr Insul 22:2196–2206. https://doi.org/10.1109/TDEI.2015.004764
Tian F, Lei Q, Wang X, Wang Y (2012) Investigation of electrical properties of LDPE/ZnO nanocomposite dielectrics. IEEE Trans Dielectr Electr Insul 19:763–769
Therlakkadan AS, Coletti G, Guastavino F, Fina A (2012) Thermomechanical and electrical characterization of epoxy-organoclay nanocomposites. Polym Eng Sci 52:1037–1046. https://doi.org/10.1002/pen
Grabowski CA, Fillery SP, Westing NM et al (2013) Dielectric breakdown in silica-amorphous polymer nanocomposite films: the role of the polymer matrix. ACS Appl Mater Interfaces 5:5486–5492. https://doi.org/10.1021/am4005623
Siddabattuni S, Schuman TP, Dogan F (2013) Dielectric properties of polymer–particle nanocomposites influenced by electronic nature of filler surfaces. ACS Appl Mater Interfaces 5:1917–1927
Kim P, Doss NM, Tillotson JP et al (2009) High energy density nanocomposites based on surface-modified BaTiO3 and a ferroelectric polymer. ACS Nano 3:2581–2592
Tsekmes IA, Kochetov R, Morshuis PHF, Smit JJ (2013) Thermal conductivity of polymeric composites: a review. IEEE Int Conf Solid Dielectr 2013:678–681. https://doi.org/10.1109/ICSD.2013.6619698
Zhi C, Bando Y, Terao T et al (2009) Towards thermoconductive, electrically insulating polymeric composites with boron nitride nanotubes as fillers. Adv Funct Mater 19:1857–1862. https://doi.org/10.1002/adfm.200801435
Kolesov SN (1980) The influence of morphology on the electric strength of polymer insulation”. IEEE Trans Electr Insul 15(5):382–388
Vaughan AS (1992) On morphology and polymer blends—polystyrene and polyethylene. Polymer 33(12):2513–2521. https://doi.org/10.1016/0032-3861(92)91132-L
Brana MTC, Sainz JII, Terselius B, Gedde UW (1989) Morphology of binary blends of linear and branched polyethylene—transmission electron-microscopy. Polymer 30(3):410–415
Norton DR, Keller A (1984) On the morphology of blends of linear and branched polyethylene. J Mater Sci 19(2):447–456. https://doi.org/10.1007/BF00553568
Hosier IL, Vaughan AS, Swingler SG (2002) The effects of measuring technique and sample preparation on the breakdown strength of polyethylene. IEEE Trans Dielectr Electr Insul. 9(3):353–361. https://doi.org/10.1109/TDEI.2002.1007697
Hosier IL, Vaughan AS, Swingler SG (1997) Structure–property relationships in polyethylene blends: the effect of morphology on electrical breakdown strength. J Mater Sci 2:4523–4531. https://doi.org/10.1023/A:1018617200285
Hosier IL, Vaughan AS, Swingler SG (2000) On the effects of morphology and molecular momposition on the electrical strength of polyethylene blends. J Polym Sci Part B Polym Phys 38:2309–2322
Green CD, Vaughan AS (2007) Morphology and crystallisation kinetics of polyethylene/montmorillonite nanocomposites. In: IEEE international conference solid dielectrics. IEEE, pp 368–371
Green CD, Vaughan AS (2007) Morphology and crystallisation kinetics of polyethylene/montmorillonite nanocomposites. In: Annual report—conference on electrical insulation and dielectric phenomena, CEIDP, pp 635–638. https://doi.org/10.1109/CEIDP.2007.4451507
Chan C-M, Wu J, Li J-X, Cheung Y-K (2002) Polypropylene/calcium carbonate nanocomposites. Polymer (Guildf) 43:2981–2992. https://doi.org/10.1016/S0032-3861(02)00120-9
Yu B, Xing W, Guo W, Qiu S, Xin W, Lo S, Hu Y (2016) Thermal exfoliation of hexagonal boron nitride for effective enhancements on thermal stability, flame retardancy and smoke suppression of epoxy resin nanocomposites via sol–gel process. J Mater Chem A 4:7330–7340
Hod O (2012) Graphite and hexagonal boron-nitride have the same interlayer distance. why? J Chem Theory Comput 8(4):1360–1369. https://doi.org/10.1021/ct200880m
Huang MT, Ishida H (2005) Surface study of hexagonal boron nitride powder by diffuse reflectance Fourier transform infrared spectroscopy. Surf Interfaces Anal 37(7):621–627
Li J, Xiao X, Xu X, Lin J, Huang Y, Xue Y, Jin P, Zou J, Tang C (2013) Activated boron nitride as an effective adsorbent for metal ions and organic pollutants. Sci Rep 3:3208. https://doi.org/10.1038/srep03208
Gautam C, Tiwary CS, Jose S, Brunetto G, Ozden S, Vinod S, Raghavan P, Biradar S, Galvao DS, Ajayan PM (2015) Synthesis of low-density, carbon-doped, porous hexagonal boron nitride solids. ACS Nano 9(12):12088–12095
Zou C, Fothergill JC, Rowe SW (2008) The effect of water absorption on the dielectric properties of epoxy nanocomposites. IEEE Trans Dielectr Electr Insul. 15(1):106–117. https://doi.org/10.1109/T-DEI.2008.4446741
Hui L, Schadler LS, Nelson JK (2013) The influence of moisture on the electrical properties of crosslinked polyethylene/silica nanocomposites. IEEE Trans Dielectr Electr Insul. 20(2):641–653. https://doi.org/10.1109/TDEI.2013.6508768
Hosier IL, Praeger M, Vaughan AS, Swingler SG (2017) The effects of water on the dielectric properties of silicon-based nanocomposites. IEEE Trans Nano 16(2):169–179. https://doi.org/10.1109/TNANO.2016.2642819
Ayoob R, Andritsch T, Vaughan AS (2016) Water absorption behaviour in polyethylene boron nitride nanocomposites. IEEE Int Conf Dielectr 1–2:784–787
Bassett DC, Hodge AM (1981) On the morphology of melt-crystallized polyethylene 1 Lamellar profiles. Proc R Soc Lond A 377(1768):25–37. https://doi.org/10.1098/rspa.1981.0113
Weng Q, Wang X, Wang X et al (2016) Functionalized hexagonal boron nitride nanomaterials: emerging properties and applications. Chem Soc Rev 45:3989–4012. https://doi.org/10.1039/C5CS00869G
Malucelli G, Ronchetti S, Lak N et al (2007) Intercalation effects in LDPE/o-montmorillonites nanocomposites. Eur Polym J 43:328–335
Nicolosi V, Chhowalla M, Kanatzidis MG et al (2013) Liquid exfoliation of layered materials. Science 340:1226419. https://doi.org/10.1126/science.1226419
Lin Z, Mcnamara A, Liu Y et al (2014) Exfoliated hexagonal boron nitride-based polymer nanocomposite with enhanced thermal conductivity for electronic encapsulation. Compos Sci Technol 90:123–128. https://doi.org/10.1016/j.compscitech.2013.10.018
Vaughan AS, Hosier IL (2008) The effect of dibenzylidene sorbitol on the crystallization behaviour of polyethylene. J Mater Sci 43:2922–2928. https://doi.org/10.1007/s10853-007-1801-9
Zaman HU, Khan MA, Khan RA, Beg MDH (2014) Effect of nano-CaCO3 on the mechanical and crystallization behavior of HDPE/LDPE/nano-CaCO3 ternary blend. J Thermoplast Compos Mater 27:1701–1710. https://doi.org/10.1177/0892705712475010
Cui L, Cho HY, Shin JW et al (2007) Polyethylene-montmorillonite nanocomposites: preparation, characterization and properties. Macromol Symp 260:49–57. https://doi.org/10.1002/masy.200751408
Kuila T, Bose S, Mishra AK et al (2012) Effect of functionalized graphene on the physical properties of linear low density polyethylene nanocomposites. Polym Test 31:31–38. https://doi.org/10.1016/j.polymertesting.2011.09.007
Jiang X, Drzal LT (2012) Multifunctional high-density polyethylene nanocomposites produced by incorporation of exfoliated graphene nanoplatelets 2: crystallization, thermal and electrical properties. Polym Compos 33:636–642. https://doi.org/10.1002/pc.22187
Puente JAS, Esposito A, Chivrac F, Dargent E (2013) Effect of boron nitride as a nucleating agent on the crystallization of bacterial poly(3-hydroxybutyrate). J Appl Polym Sci 128:2586–2594. https://doi.org/10.1002/app.38182
Liu WJ, Yang HL, Wang Z et al (2002) Effect of nucleating agents on the crystallization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate). J Appl Polym Sci 86:2145–2152. https://doi.org/10.1016/j.polymer.2015.03.061
Wang L, Wang X, Zhu W et al (2010) Effect of nucleation agents on the crystallization of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) (P3/4HB). J Appl Polym Sci 116:1116–1123. https://doi.org/10.1002/app
Hoffman JD (1985) Growth-rate of extended-chain crystals—the lateral surface free-energy of pure N-C94H190 and a fraction C207H416. Macromolecules 18(4):772–786. https://doi.org/10.1021/ma00146a032
Muchova M, Lednicky F (1996) Investigation of heterogeneous nucleation using the induction time of crystallization: 1. Theory of induction time. Polymer 37(14):3031–3036. https://doi.org/10.1016/0032-3861(96)89401-8
Zhang X, Wu H, Guo S, Wang Y-Z (2015) Understanding in crystallization of polyethylene: the role of boron nitride (BN) particles. RSC Adv 5:99812–99819. https://doi.org/10.1039/C5RA19982D
Corrales T, Catalina F, Peinado C et al (2002) Photooxidative and thermal degradation of polyethylenes: interrelationship by chemiluminescence, thermal gravimetric analysis and FTIR data. J Photochem Photobiol A Chem 147:213–224. https://doi.org/10.1016/S1010-6030(01)00629-3
Peterson JD, Vyazovkin S, Wight CA (2001) Kinetics of the thermal and thermo-oxidative degradation of polystyrene, polyethylene and poly(propylene). Macromol Chem Phys 202:775–784. https://doi.org/10.1002/1521-3935(20010301)202:6<775:AID-MACP775>3.0.CO;2-G
Li T-L, Hsu SL-C (2011) Preparation and properties of thermally conductive photosensitive polyimide/boron nitride nanocomposites. J Appl Polym Sci 121:916–922. https://doi.org/10.1002/app
Fim FC, Basso NRS, Graebin AP, Azambuja DS (2013) Thermal, electrical, and mechanical properties of polyethylene–graphene nanocomposites obtained by in situ polymerization. J Appl Polym Sci 128:2630–2637
Panupakorn P, Chaichana E, Praserthdam P, Jongsomjit B (2013) Polyethylene/clay nanocomposites produced by in situ polymerization with zirconocene/MAO catalyst. J Nanomater 2013:1–9
Dash S, Swain SK (2013) Effect of nanoboron nitride on the physical and chemical properties of soy protein. Compos Sci Technol 84:39–43. https://doi.org/10.1016/j.compscitech.2013.05.004
Leszczyńska A, Njuguna J, Pielichowski K, Banerjee JR (2007) Polymer/montmorillonite nanocomposites with improved thermal properties. Part II. Thermal stability of montmorillonite nanocomposites based on different polymeric matrixes. Thermochim Acta 454:1–22. https://doi.org/10.1016/j.tca.2006.11.003
Tsekmes IA, Kochetov R, Morshuis PHF, Smit JJ (2015) AC breakdown strength of epoxy-boron nitride nanocomposites: trend and reproducibility. In: Electrical insulation conference, pp 446–449
Tsekmes IA, Kochetov R, Morshuis PHF, Smit JJ (2015) DC breakdown strength of epoxy-boron nitride nanocomposites: trend and reproducibility. In: Conference electrical insulation dielectric phenomena, pp 479–482
Gao M, Zhang P, Wang F (2013) Effect of percolation and interfacial characteristics on breakdown behavior of nano-silica/epoxy composites. In: 8th international forum strategic technology, vol 1, pp 120–123. https://doi.org/10.1109/IFOST.2013.6616957
Singha S, Thomas M (2008) Dielectric properties of epoxy nanocomposites. IEEE Trans Dielectr Electr Insul 15:12–23. https://doi.org/10.1109/T-DEI.2008.4446732
Smith RC, Liang C, Landry M et al (2008) The mechanisms leading to the useful electrical properties of polymer nanodielectrics. IEEE Trans Dielectr Electr Insul 15:187–196. https://doi.org/10.1109/T-DEI.2008.4446750
Green CD, Vaughan AS, Mitchell GR, Liu T (2008) Structure property relationships in polyethylene/montmorillonite nanodielectrics. IEEE Trans Dielectr Electr Insul 15:134–143. https://doi.org/10.1109/T-DEI.2008.4446744
Vaughan AS, Swingler SG, Zhang Y (2006) Polyethylene nanodielectrics: the influence of nanoclays on structure formation and dielectric breakdown. IEEJ Trans Fundam Mater 126:1057–1063
Lau KY (2013) Structure and electrical properties of silica-based polyethylene nanocomposites. PhD Thesis, University of Southampton
Li S, Yin G, Chen G et al (2010) Short-term breakdown and long-term failure in nanodielectrics: a review. IEEE Trans Dielectr Electr Insul 17:1523–1535. https://doi.org/10.1109/TDEI.2010.5595554
Li S, Min D, Wang W, Chen G (2016) Linking traps to dielectric breakdown through charge dynamics for polymer nanocomposites. IEEE Trans Dielectr Electr Insul 23:2777–2785. https://doi.org/10.1109/TDEI.2016.7736837
Wang W, Min D, Li S (2016) Understanding the conduction and breakdown properties of polyethylene nanodielectrics: effect of deep traps. IEEE Trans Dielectr Electr Insul 23:564–572. https://doi.org/10.1109/TDEI.2015.004823
Roy M, Nelson JK, MacCrone RK, Schadler LS (2007) Candidate mechanisms controlling the electrical characteristics of silica/XLPE nanodielectrics. J Mater Sci 42:3789–3799. https://doi.org/10.1007/s10853-006-0413-0
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ayoob, R., Alhabill, F., Andritsch, T. et al. Enhanced dielectric properties of polyethylene/hexagonal boron nitride nanocomposites. J Mater Sci 53, 3427–3442 (2018). https://doi.org/10.1007/s10853-017-1786-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1786-y