
Abstract
Low-cost and highly active water oxidation electrocatalysts are increasingly demanded to improve the efficiency of water splitting. Herein, for the first time, we developed a facile strategy to design a three-dimensional flower-like cobalt nickel phosphate as such an electrocatalyst. The cobalt nickel phosphate was directly supported on Ni foam ((Co0.5Ni0.5)3(PO4)2/Ni) via a simple two-step hydrothermal method. This method offers several advantages including low cost, facile synthesis and binder-free. Benefiting from its three-dimensional flower-like nanostructure with a big specific surface, high conductivity and the synergic effect of metal ions are the as-synthesized (Co0.5Ni0.5)3(PO4)2/Ni reveals excellent oxygen evolution reaction activity in alkaline electrolyte. It exhibits a low overpotential of 273 mV to reach a current density of 10 mA cm−2 and a Tafel slope of 59.3 mV dec−1 in 1.0 mol L−1 KOH. Notably, the (Co0.5Ni0.5)3(PO4)2/Ni also operates efficiently in 30 wt% KOH, capable of affording an anodic current of 10 mA cm−2 at a lower overpotential of 246 mV and retains its catalytic performance for at least 30 h.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Developing efficient electrocatalysts for oxygen evolution reaction (OER) is imperative for energy storage and conversion systems, including rechargeable metal–air batteries, reversible fuel cells and water splitting [1, 2]. Unfortunately, the OER is kinetically sluggish, needing large overpotential and high activation energy to form the O=O double bond [3,4,5]. Up to now, the noble metal oxides, such as RuO2 and IrO2, are still considered as the most active electrocatalysts to promote the OER, while the scarcity and prohibitive cost have heavily hindered their widespread commercialization [6,7,8]. Accordingly, many efforts are focused on exploring earth-abundant, cost-effective and highly efficient OER electrocatalysts.
Transition metal phosphates [9,10,11,12], phosphides [13,14,15], oxides [16,17,18], hydroxides [19,20,21] and sulfides [22,23,24] have exhibited a tremendous potential to replace those precious electrocatalysts for OER. For metal phosphates, they have PO4 3− or HPO4 2− to carry and accelerate the motion of protons (H+) and thereby increase the OER catalytic activity [4, 9]. In 2008, a Co–Pi reported by Kanan and Noreca [9], showing superior OER catalytic performance in neutral solution. After that, the earth-abundant and low-cost metal phosphates electrocatalysts have been extensively designed [25,26,27,28,29,30,31,32,33,34], such as NiCo2(PO4)2 [35], LiNi1−x Fe x PO4/mesoporous carbon (LiNi1−x Fe x PO4/C) [36], Co3(PO4)2/N-doped carbon layers (Co3(PO4)2@N–C) [37], Co- and Fe-doped nickel phosphate-Na-nanotubes (NiPNa(Co2Fe2)-NTs) [38], iron-doped nickel phosphate (Ni:Pi-Fe/NF) [39], nickel–iron phosphates (NiFe-P) [40], and so on. Nonetheless, the operations of these electrocatalysts usually employ the polymeric binders. These will reduce the conductivity, block the active sites and finally decrease the catalytic performance of electrocatalysts. In order to address this challenge, Sun et al. synthesized a Co–Pi nanoarray on Ti mesh (Co–Pi NA/Ti) by an electrodeposition method [41]. The Co–Pi NA/Ti showed excellent OER catalytic activity in neutral media. The authors concluded that the three-dimensional (3D) nanoarray configuration of Co–Pi NA/Ti not only exposed more active sites but also accelerated the diffusion of electrolytes and oxygen. Although the above excitingly advances have been reported, further studies of metal phosphates or the optimization of their OER catalytic performance are undoubtedly crucial. In this regard, an efficient method to prepare these materials is required. Recently, we have noticed that the hydrothermal anion exchange method is effective to synthesize materials [42]. Following this way, the products will retain the unique morphology of the corresponding precursors after an anions exchange process. However, it is still a challenge to utilize this method to prepare the metal phosphates.
In this work, we demonstrated a facile two-step hydrothermal method to synthesize 3D flower-like (Co0.5Ni0.5)3(PO4)2/Ni. This 3D flower-like (Co0.5Ni0.5)3(PO4)2/Ni combines the advantages of 3D porous architecture with a large surface area for fast mass transport, efficient electron transfer from conductive Ni foam to (Co0.5Ni0.5)3(PO4)2 and the synergistic effect between the optimized ratio of Co and Ni. It presents highly activity and stability for OER in alkaline medias, out-performing the majority reported non-noble metal materials and even comparable to the active RuO2 electrocatalyst.
Experimental sections
Materials
CoCl2·6H2O, NiCl2·6H2O, Na2HPO4·12H2O, urea and NH4F were purchased from Aladdin. Ni foam was purchased from Kunshan Electronic Limited Corporation. All the reagents were used without any further purification.
Synthesis of precursor Co0.5Ni0.5-LDH/Ni
The precursor Co0.5Ni0.5-LDH/Ni [XRD pattern is exhibited in Fig. S1, Supporting Information (SI)] was prepared by the following processes. Firstly, Ni foam (3 cm × 3 cm) was cleaned by sonication sequentially in 3 mol L−1 HCl solution and absolute ethanol for 15 min each. Then, 1.5 mmol CoCl2·6H2O, 1.5 mmol NiCl2·6H2O, 5.0 mmol urea and 0.5 mmol NH4F were fully dissolved in 50 mL distilled water to form a homogeneous solution under agitated stirring. The obtained solution was transferred into a Teflon-lined autoclave (80 mL), in which contained a cleaned Ni foam, sealed and kept at 120 °C for 6 h. After cooling down to room temperature, the Ni foam was taken out and washed with deionized water thoroughly before being dried at 60 °C.
Synthesis of (Co0.5Ni0.5)3(PO4)2/Ni
To obtain the (Co0.5Ni0.5)3(PO4)2/Ni, 5.0 mmol Na2HPO4·12H2O was mixed with 50 mL distilled water under stirring to form a homogeneous solution. Then, 3 mol L−1 HCl was dropped into the above solution to adjust the pH value to 8.5, which was tested by the pH meter. The obtained solution was placed into an autoclave, containing the precursor Co0.5Ni0.5-LDH/Ni. Moreover, the autoclave was heated at 180 °C for 12 h and cooled down to room temperature naturally. The product was severally washed by distilled water and ethanol for three times to remove the impurities on the surface and then dried at 60 °C overnight. The mass loading of (Co0.5Ni0.5)3(PO4)2/Ni is 4.4 mg cm−2. The sample synthesized without Ni foam was denoted as (Co0.5Ni0.5)3(PO4)2 nanoparticles.
Characterization of materials
Scanning electron microscopy (SEM) images were obtained by a Philips SEM-XL30S microscope operated at 15 kV. High-resolution transmission electron microscope (HRTEM, JEOL JEM-2100F) coupled with an energy-dispersive X-ray spectroscopy (EDS) analyzer was performed at an accelerating voltage of 200 kV. Nitrogen sorption isotherm was achieved by a Micromeritics TriStar 3000 analyzer at 77 K. The X-ray diffraction (XRD) pattern was conducted by a MSAL-XD2 X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). The inductively coupled plasma optical emission spectrometer (ICP-OES) was measured on Perkin-Elmer Optima 2000DV. The X-ray photoelectron spectroscopy (XPS) was tested by an ESCALab250.
Electrochemical measurements
Electrochemical measurements were conducted in a standard three-electrode system, in which the (Co0.5Ni0.5)3(PO4)2/Ni, Pt foil and Hg/HgO electrodes were severally used as the working, counter and reference electrodes. 1 mol L−1 KOH solution was employed as the electrolyte. All potentials were converted via the Nernst equation (E RHE = E Hg/HgO + (0.098 + 0.059pH) V). The polarization curves were obtained by linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1. The Tafel slope was obtained according to Tafel equation (ŋ = b log j + a, where a is a constant, j is the current density, b is the Tafel slope and ŋ is the overpotential). The overpotentials (ŋ) were calculated according to the equation (ŋ = E RHE − 1.23 V). Electrochemical impedance spectroscopy (EIS) was analyzed in the frequency range of 10 kHz to 10 mHz with an amplitude of 5 mV. Chronoamperometry was performed at certain potentials.
The roughness factor (R f) was determined from a ratio of the electrochemically active surface area (ECSA) to the electrode geometrical surface area (1.0 cm2) [43]. The ECSA was evaluated by the electrochemical double-layer capacitance (C dl) according to the equation (ECSA = C dl/C s, where C s is 40 uF cm−2, the specific electrochemical double-layer capacitance of an atomically smooth surface [43, 44]). The C dl was evaluated by cyclic voltammograms measured in a non-Faradaic region from 1.004 to 1.078 V versus RHE at scan rates ranging from 5 to 30 mV s−1. 2C dl is equal to the linear slope of capacitive currents versus scan rates [45].
Results and discussion
Structure and morphology of (Co0.5Ni0.5)3(PO4)2/Ni
The flower-like (Co0.5Ni0.5)3(PO4)2 grown on Ni foam is prepared through a simple two-step hydrothermal process, as shown in Scheme 1. In step I, the CO3 2− and OH− ions are released by the hydrolysis of urea and gradually co-precipitated with Co2+ and Ni2+ ions to form the Co0.5Ni0.5-LDH/Ni (yellow–green), which has a 3D flower-like structure. Furthermore, the (Co0.5Ni0.5)3(PO4)2/Ni (deep brown) is obtained through a facile in situ anions exchange process using the Co0.5Ni0.5-LDH/Ni as a sacrificial template (step II). In step II, the Na2HPO4 is employed as the source of PO4 3− and the related reactions may follow Eqs. (1)–(4):

Schematic diagrams of the synthesis of (Co0.5Ni0.5)3(PO4)2/Ni. The fuchsia, green, aquamarine blue, violet and yellow spheres severally represent the Co2+, Ni2+, urea, NH4F and Na2HPO4
From the SEM images revealed in Fig. 1a, b, the 3D flower-like structure has still retained after exchanging the Co0.5Ni0.5-LDH/Ni to (Co0.5Ni0.5)3(PO4)2/Ni. Figure 1c, d shows the “petals” of “flower” are composed of ultrathin nanosheets, while the thicker nanoplates grew on “petals” are also observed. Meanwhile, the structure of the whole “flower” is exhibited in Fig. S2, revealing the diameter of “flower” is about 3.6 um. Moreover, the N 2 adsorption–desorption isotherm (Fig. S3) analysis indicates the mesoporosity and a large specific area for (Co0.5Ni0.5)3(PO4)2/Ni of about 58 m2 g−1. The porous architecture of (Co0.5Ni0.5)3(PO4)2/Ni along with a large surface area will facilitate the mass diffusion/transport and expose a large number of catalytic active sites for OER [35, 36, 46, 47]. The HRTEM image in Fig. 1e clearly presents the lattice fringes of d = 0.344 nm, corresponding to the (111) crystal plane of (Co0.5Ni0.5)3(PO4)2/Ni. In Fig. 1f, the uniform distribution of elements of Co, Ni, P and O in (Co0.5Ni0.5)3(PO4)2/Ni is also observed.

a SEM images of Co0.5Ni0.5–LDH/Ni; b–e are severally the SEM, TEM and HRTEM images of (Co0.5Ni0.5)3(PO4)2/Ni; and f EDS element mappings of (Co0.5Ni0.5)3(PO4)2/Ni
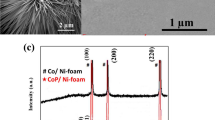
As the XRD pattern shown in Fig. 2a, the characteristic peaks of the as-synthesized sample are matched well with the standard diffraction patterns of monoclinic (Co0.5Ni0.5)3(PO4)2 (JCPDS No. 75-1328) and Ni foam (JCPDS No. 65-2865), suggesting the successful synthesis of (Co0.5Ni0.5)3(PO4)2/Ni after the phosphorylation. The EDS analysis of (Co0.5Ni0.5)3(PO4)2/Ni in Fig. 2b gives the mole ratio of Co2+ and Ni2+ is 48.6: 51.4 (~ 0.5:0.5), which can be further confirmed by the ICP results exhibited in Table S1, but the Cu element is coming from the copper mesh.

a XRD and b EDS patterns of (Co0.5Ni0.5)3(PO4)2/Ni
In addition, the XPS spectra in the Co 2p region (Fig. 3a) show two sharp peaks at 782.8 eV (2p 3/2) and 798.8 eV (2p 1/2) with two strong satellite peaks separately at 787.2 and 804.5 eV, indicating the presence of Co2+ [4, 37]. Simultaneously, Fig. 3b exhibits two strong peaks located at 858.1 and 875.7 eV in the Ni 2p region are severally corresponding to Ni 2p 3/2 and Ni 2p 1/2, attributed to Ni2+ [3]. Moreover, the peaks at 863.7 and 882.5 eV are the satellite peaks of Ni 2p 3/2 and Ni 2p 1/2. The oxidation states of Co and Ni presented in (Co0.5Ni0.5)3(PO4)2/Ni are benefited for OER. As shown in Fig. 3c, the peaks at 133.6 and 134.4 eV of P 2p are related to the pentavalent tetra-bonded phosphorous in (Co0.5Ni0.5)3(PO4)2/Ni [4, 37]. In Fig. 3d, the peaks at 531.6 and 532.9 eV are corresponding to the core levels of O 1s in phosphate species, adsorbed water and/or possibly adsorbed O2 [3, 37].

XPS spectra of (Co0.5Ni0.5)3(PO4)2/Ni
OER activity of (Co0.5Ni0.5)3(PO4)2/Ni
The above characterizations imply that the as-synthesized (Co0.5Ni0.5)3(PO4)2/Ni may be active for OER. Therefore, we further estimate its electrocatalytic properties for OER in 1 mol L−1 KOH solution by the LSV measurement at a scan rate of 1 mV s−1. For comparison, the (Co0.5Ni0.5)3(PO4)2 nanoparticles and commercial RuO2 loaded on Ni foam (all loading 4.4 mg cm−2, denoted as (Co0.5Ni0.5)3(PO4)2 Nps/Ni and RuO2/Ni [3]) and bare Ni foam were also tested. Figure 4a exhibits the polarization curves. The oxidation peaks at ~ 1.3 V versus reversible hydrogen electrode (RHE) are corresponding to the transition from M2+ (Co2+ and/or Ni2+) to M3+ (Fig. S4) [3, 6]. Owing to the large oxidation peaks, the onset potentials of (Co0.5Ni0.5)3(PO4)2/Ni, (Co0.5Ni0.5)3(PO4)2 Nps/Ni are inaccurate. Therefore, we report the overpotentials at 10 mA cm−2 here. As shown in Fig. 4a, the bare Ni foam has poor OER catalytic performance with an overpotential of 502 mV to achieve a current density of 10 mA cm−2 (ŋ 10), while (Co0.5Ni0.5)3(PO4)2 Nps/Ni is active for OER with a lower ŋ 10 of 305 mV. What should be noted is that the (Co0.5Ni0.5)3(PO4)2/Ni possesses significantly enhanced catalytic activity, because it just requires an overpotential of 273 mV to generate a current density of 10 mA cm−2, which even lower than 296 mV for RuO2/Ni. In addition, although the ŋ 10 of (Co0.5Ni0.5)3(PO4)2/Ni at 273 mV is larger than 220 mV for Ni:Pi-Fe/NF, it is superior to those of the Co3(PO4)2 (380 mV) [4], NiCo2(PO4)2 (347 mV) [35], LiNi1−x Fe x PO4/C (311 mV) [36], Co3(PO4)2/N–C (317 mV) [37], NiPNa(Co2Fe2)-NTs (300 mV) [38], Ni x Co2x (OH)6x @Ni (> 295 mV) [48], NiCo2O4/CNTs (> 320 mV) [49] and the other recently noble metal electrocatalysts in Table S2. Efficient performance of (Co0.5Ni0.5)3(PO4)2/Ni may be attributed to the uniquely 3D flower-like structure along with a large surface area. Interestingly, we found that the (Co0.5Ni0.5)3(PO4)2/Ni is not a simple mixture after synthesized, while the synergistic effect between the optimized ratio of Co and Ni can also improve the OER activity of this material, as shown in Fig. S5.

a Polarization curves (the inset exhibits the expanded region around the electrocatalytic onset of polarization curves); b Tafel plots; c EIS of electrocatalysts measured at a potential of 1.57 V; d chronoamperometric response of (Co0.5Ni0.5)3(PO4)2/Ni; and e, f SEM images after stability measurement in different magnifications
To further evaluate the OER kinetics of the as-prepared materials, the Tafel plots are modeled from the corresponding LSV curves and shown in Fig. 4b. It can be judged that the (Co0.5Ni0.5)3(PO4)2/Ni has more favorable OER catalytic kinetics and higher performance with a small Tafel slope of 59.3 mV dec−1 comparing to those of 119.0 mV dec−1 for (Co0.5Ni0.5)3(PO4)2 NPs/Ni and 87.0 mV dec−1 for RuO2/Ni. Moreover, the efficient OER activity of (Co0.5Ni0.5)3(PO4)2/Ni is supported by the electrochemical impedance spectroscopy (EIS) presented in Fig. 4c. The impedance responses of EIS are fitted by the equivalent circuit inset in Fig. 4c [50]. This equivalent circuit includes a solution resistance (R s) with two parallel comprised of constant phase elements (CPE1, CPE2) and resistors (R ct, R p). Especially, the R ct represents the charge transfer resistance, caused by redox reactions on the interfaces of electrocatalyst and electrolyte. It is observed that the (Co0.5Ni0.5)3(PO4)2/Ni possesses the lower R ct value (1.63 Ω) with respect to that of (Co0.5Ni0.5)3(PO4)2 Nps/Ni (3.76 Ω). This result implies the (Co0.5Ni0.5)3(PO4)2/Ni has better charge transfer ability in the electrochemical process of OER due to it directly grown on Ni foam with binder-free.
To further understand the superior performance of (Co0.5Ni0.5)3(PO4)2/Ni, the electrochemically active surface areas (ECSA) and roughness factors (R f) of the as-prepared samples were evaluated by the electrochemical double-layer capacitance (C dl, Fig. S6a–d). Figure S6e shows the ESCA and R f values of (Co0.5Ni0.5)3(PO4)2/Ni are severally 1385.0 cm2 and 1385.0, which are obviously larger than those of the (Co0.5Ni0.5)3(PO4)2 Nps/Ni (247.5 cm2 and 247.5), RuO2/Ni (550.0 cm2 and 550.0) and bare Ni foam (42.5 cm2 and 42.5). Therefore, it can be also concluded that the superior OER catalytic activity of (Co0.5Ni0.5)3(PO4)2/Ni is partially associated with its uniquely flower-like mesoporous architecture that possesses the large electrochemically active surface area to expose many active sites on the surface of the electrocatalyst.
The chronoamperometry measurement was carried out at the potential of 1.57 V versus RHE to probe the stability of (Co0.5Ni0.5)3(PO4)2/Ni in 1 mol L−1 KOH solution, as shown in Fig. 4d. One can see that the current density of (Co0.5Ni0.5)3(PO4)2/Ni just reveals a little decay after 30 h continuous measurements, demonstrating the superior stability of (Co0.5Ni0.5)3(PO4)2/Ni for OER. The great water oxidation stability of (Co0.5Ni0.5)3(PO4)2/Ni may benefit from its high structure durability, because the 3D flower-like morphology of (Co0.5Ni0.5)3(PO4)2/Ni reveals unobviously change even after long-term OER durability measurement, as SEM images shown in Fig. 4e, f.
Moreover, because the practical water electrolysis of electrocatalysts usually operates in 30 wt% KOH [3], the OER catalytic activity of (Co0.5Ni0.5)3(PO4)2/Ni was also evaluated under this circumstance. In Fig. 5a, b, the (Co0.5Ni0.5)3(PO4)2/Ni exhibits a larger OER current in 30 wt% KOH with respect to that of in 1 mol L−1 KOH. The ŋ 10 of (Co0.5Ni0.5)3(PO4)2/Ni in 30 wt% KOH is 246 mV, which is smaller than the 273 mV in 1 mol L−1 KOH. As shown in Fig. 5c, the observation of small OER activity decays for continuing OER electrolysis for 30 h at different operating potentials, suggesting the great durability of (Co0.5Ni0.5)3(PO4)2/Ni even under strongly alkaline solution. Simultaneously, Fig. 5d shows the volume–time curves reveal the Faradaic efficiency of (Co0.5Ni0.5)3(PO4)2/Ni is mostly 100% with the volume of O2 generating equal to the theoretical one. The high catalytic activity and remarkable stability imply the considerable potential of (Co0.5Ni0.5)3(PO4)2/Ni for practical application.

a Polarization curves; b the expanded region around the electrocatalytic onset of the polarization curves; c chronoamperometric response of (Co0.5Ni0.5)3(PO4)2/Ni tested at potentials of 1.48 and 1.54 V in 30 wt% KOH solutions; and d Faradic efficiency of oxygen production of (Co0.5Ni0.5)3(PO4)2/Ni
In general, the excellent OER activity and stability of (Co0.5Ni0.5)3(PO4)2/Ni in alkaline electrolytes could be involved the following factors: (1) the obtained (Co0.5Ni0.5)3(PO4)2/Ni has a unique 3D flower-like mesoporous architectures and big specific surface area, which not only allow the random motion of the electrons and electrolytes, but also accelerate the oxygen molecules evolution/transportation [35, 51]; (2) the (Co0.5Ni0.5)3(PO4)2/Ni was directly grown on the high conductivity Ni foam substrate along with an open 3D network, which affords a stable structure to promote the durability of materials and results in low charge transfer resistance in OER electrolysis process; and (3) the synergistic effect between the optimized ratio of Co and Ni is believed to be an another factor for the efficient OER catalytic performance of (Co0.5Ni0.5)3(PO4)2/Ni.
Conclusions
In summary, we have demonstrated a facile hydrothermal method to synthesize a (Co0.5Ni0.5)3(PO4)2/Ni. This method is low cost and effective. The (Co0.5Ni0.5)3(PO4)2/Ni displays highly catalytic activity toward OER in a basic electrolyte with a low overpotential of 273 mV to drive an anodic current of 10 mA cm−2 and a Tafel slope of 59.3 mV dec−1 in 1.0 mol L−1 KOH. More importantly, this novel electrocatalyst even reveals higher catalytic activity in 30 wt% KOH, demonstrating considerable potential for the commercial application. It is believed that this work not only offers us a cost-effectively, active and stable OER electrocatalyst, but also opens up a new avenue to prepare metal phosphates and the other electrocatalysts, which have excellent activity toward water electrolysis and/or electrochemical devices.
References
Walter MG, Warren EL, McKone JR, Boettcher SW, Mi Q, Santori EA, Lewis NS (2010) Solar water splitting cells. Chem Rev 110:6446–6473
Sun H, Chen G, Zhu Y, Liu B, Zhou W, Shao Z (2017) B-site cation ordered double perovskites as efficient and stable electrocatalysts for oxygen evolution reaction. Chem Eur J 23:5722–5728
Cheng N, Liu Q, Asiri AM, Xing W, Sun X (2015) A Fe-doped Ni3S2 particle film as a high-efficiency robust oxygen evolution electrode with very high current density. J Mater Chem A 3:23207–23212
Pramanik M, Li C, Imura M, Malgras V, Kang YM, Yamauchi Y (2016) Ordered mesoporous cobalt phosphate with crystallized walls toward highly active water oxidation electrocatalysts. Small 12:1709–1715
Zhao Q, Zhong D, Liu L, Li D, Hao G, Li J (2017) Facile fabrication of robust 3D Fe-NiSe nanowires supported on nickel foam as a highly efficient, durable oxygen evolution catalyst. J Mater Chem A 5:14639–14645
Masa J, Weide P, Peeters D et al (2016) Amorphous cobalt boride (Co2B) as a highly efficient nonprecious catalyst for electrochemical water Splitting: oxygen and hydrogen evolution. Adv Energy Mater 6:1502313
Fang W, Liu D, Lu Q, Sun X, Asiri AM (2016) Nickel promoted cobalt disulfide nanowire array supported on carbon cloth: an efficient and stable bifunctional electrocatalyst for full water splitting. Electrochem Commun 63:60–64
Liu W, Du K, Liu L et al (2017) One-step electroreductively deposited iron-cobalt composite films as efficient bifunctional electrocatalysts for overall water splitting. Nano Energy 38:576–584
Kanan MW, Nocera DG (2008) In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 321:1072–1075
Liu Y, Wang H, Lin D et al (2015) Electrochemical tuning of olivine-type lithium transition-metal phosphates as efficient water oxidation catalysts. Energy Environ Sci 8:1719–1724
Zhan Y, Lu M, Yang S, Liu Z, Lee JY (2016) The origin of catalytic activity of nickel phosphate for oxygen evolution in alkaline solution and its further enhancement by iron substitution. ChemElectroChem 3:615–621
Cobo S, Heidkamp J, Jacques PA et al (2012) A janus cobalt-based catalytic material for electro-splitting of water. Nat Mater 11:802–807
Wang M, Lin M, Li J et al (2017) Metal-organic framework derived carbon-confined Ni2P nanocrystals supported on graphene for an efficient oxygen evolution reaction. Chem Commun 53:8372–8375
Li D, Baydoun H, Kulikowski B, Brock SL (2017) Boosting the catalytic performance of iron phosphide nanorods for the oxygen evolution reaction by incorporation of manganese. Chem Mater 29:3048–3054
Das D, Das A, Reghunath M, Nanda KK (2017) Phosphine-free avenue to Co2P nanoparticle encapsulated N, P co-doped CNTs: a novel non-enzymatic glucose sensor and an efficient electrocatalyst for oxygen evolution reaction. Green Chem 19:1327–1335
Antoni H, Xia W, Masa J, Schuhmann W, Muhler M (2017) Tuning the oxidation state of manganese oxide nanoparticles on oxygen- and nitrogen-functionalized carbon nanotubes for the electrocatalytic oxygen evolution reaction. Phys Chem Chem Phys 19:18434–18442
Favaro M, Yang J, Nappini S et al (2017) Understanding the oxygen evolution reaction mechanism on CoO x using operando ambient-pressure X-ray photoelectron spectroscopy. J Am Chem Soc 139:8960–8970
Deng X, Ozturk S, Weidenthaler C, Tuysuz H (2017) Iron-induced activation of ordered mesoporous nickel cobalt oxide electrocatalyst for the oxygen evolution reaction. ACS Appl Mater Interfaces 9:21225–21233
Tkalych AJ, Zhuang HL, Carter EA (2017) A density functional + U assessment of oxygen evolution reaction mechanisms on β-NiOOH. ACS Catal 7:5329–5339
Zaffran J, Stevens MB, Trang CDM et al (2017) Influence of electrolyte cations on Ni(Fe)OOH catalyzed oxygen evolution reaction. Chem Mater 29:4761–4767
Liu W, Bao J, Guan M et al (2017) Nickel-cobalt-layered double hydroxide nanosheet arrays on Ni foam as a bifunctional electrocatalyst for overall water splitting. Dalton Trans 46:8372–8376
Yang D, Gao L, Yang J-H (2017) Facile synthesis of ultrathin Ni(OH)2–Cu2S hexagonal nanosheets hybrid for oxygen evolution reaction. J Power Sources 359:52–56
Jiang J, Yan C, Zhao X, Luo H, Xue Z, Mu T (2017) A PEGylated deep eutectic solvent for controllable solvothermal synthesis of porous NiCo2S4 for efficient oxygen evolution reaction. Green Chem 19:3023–3031
Zhang J, Hu Y, Liu D, Yu Y, Zhang B (2017) Enhancing oxygen evolution reaction at high current densities on amorphous-like Ni–Fe–S ultrathin nanosheets via oxygen incorporation and electrochemical tuning. Adv Sci 4:1600343
Kanan MW, Surendranath Y, Nocera DG (2009) Cobalt–phosphate oxygen-evolving compound. Chem Soc Rev 38:109–114
Kanan WM, Yano J, Surendranath Y, Dinca˘ M, Yachandra VK, Nocera DG (2010) Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy. J Am Chem Soc 132:13692–13701
Wang Y, Wang Y, Jiang R, Xu R (2012) Cobalt phosphate-ZnO composite photocatalysts for oxygen evolution from photocatalytic water oxidation. Ind Eng Chem Res 51:9945–9951
Zhong DK, Cornuz M, Sivula K, Grätzel M, Gamelin DR (2011) Photo-assisted electrodeposition of cobalt–phosphate (Co-Pi) catalyst on hematite photoanodes for solar water oxidation. Energy Environ Sci 4:1759–1764
Samal A, Swain S, Satpati B, Das DP, Mishra BK (2016) 3D Co3(PO4)2-reduced graphene oxide flowers for photocatalytic water splitting: a type II staggered heterojunction system. Chemsuschem 9:3150–3160
Irshad A, Munichandraiah N (2017) Ir-phosphate cocatalyst for photoelectrochemical water oxidation using α-Fe2O3. RSC Adv 7:21430–21438
Cui L, Liu D, Hao B et al (2017) In situ electrochemical surface derivation of cobalt phosphate from a Co(CO3)0.5(OH)·0.11H2O nanoarray for efficient water oxidation in neutral aqueous solution. Nanoscale 9:3752–3756
Costentin C, Porter TR, Saveant JM (2016) Conduction and reactivity in heterogeneous-molecular catalysis: new insights in water oxidation catalysis by phosphate cobalt oxide films. J Am Chem Soc 138:5615–5622
Yang X, Tang H, Xu J, Antonietti M, Shalom M (2015) Silver phosphate/graphitic carbon nitride as an efficient photocatalytic tandem system for oxygen evolution. Chemsuschem 8:1350–1358
Gonzalez-Flores D, Sanchez I, Zaharieva I et al (2015) Heterogeneous water oxidation: surface activity versus amorphization activation in cobalt phosphate catalysts. Angew Chem 54:2502–2506
Zhang J, Yang Y, Zhang Z, Xu X, Wang X (2014) Rapid synthesis of mesoporous Ni x Co3−x (PO4)2 hollow shells showing enhanced electrocatalytic and supercapacitor performance. J Mater Chem A 2:20182–20188
Ma S, Zhu Q, Zheng Z, Wang W, Chen D (2015) Nanosized LiNi1−x Fe x PO4 embedded in a mesoporous carbon matrix for high-performance electrochemical water splitting. Chem Commun 51:15815–15818
Yuan C-Z, Jiang Y-F, Wang Z et al (2016) Cobalt phosphate nanoparticles decorated with nitrogen-doped carbon layers as highly active and stable electrocatalysts for the oxygen evolution reaction. J Mater Chem A 4:8155–8160
Liu H, Li H, Wang X (2016) Electrostatic interaction-directed growth of nickel phosphate single-walled nanotubes for high performance oxygen evolution reaction catalysts. Small 12:2969–2974
Li Y, Zhao C (2016) Iron-doped nickel phosphate as synergistic electrocatalyst for water oxidation. Chem Mater 28:5659–5666
Xing J, Li H, Ming-Cheng Cheng M, Geyer SM, Ng KYS (2016) Electro-synthesis of 3D porous hierarchical Ni–Fe phosphate film/Ni foam as a high-efficiency bifunctional electrocatalyst for overall water splitting. J Mater Chem A 4:13866–13873
Xie L, Zhang R, Cui L et al (2017) High-performance electrolytic oxygen evolution in neutral media catalyzed by a cobalt phosphate nanoarray. Angew Chem 129:1084–1088
Ma L, Hu Y, Chen R et al (2016) Self-assembled ultrathin NiCo2S4 nanoflakes grown on Ni foam as high-performance flexible electrodes for hydrogen evolution reaction in alkaline solution. Nano Energy 24:139–147
Fang M, Gao W, Dong G et al (2016) Hierarchical NiMo-based 3D electrocatalysts for highly-efficient hydrogen evolution in alkaline conditions. Nano Energy 27:247–254
Zhang Y, Zhou X, Zhang F, Tian T, Ding Y, Gao H (2017) Design and synthesis of Cu modified cobalt oxides with hollow polyhedral nanocages as efficient electrocatalytic and photocatalytic water oxidation catalysts. J Catal 352:246
Shen JQ, Liao PQ, Zhou DD et al (2017) Modular and stepwise synthesis of a hybrid metal-organic framework for efficient electrocatalytic oxygen evolution. J Am Chem Soc 139:1778–1781
Li S, Cheng P, Luo J et al (2017) High-performance flexible asymmetric supercapacitor based on CoAl-LDH and rGO electrodes. Nano Micro Lett 9:31
Li R, Zhou D, Luo J et al (2017) The urchin-like sphere arrays Co3O4 as a bifunctional catalyst for hydrogen evolution reaction and oxygen evolution reaction. J Power Sources 341:250–256
Liu Z-Q, Chen G-F, Zhou P-L, Li N, Su Y-Z (2016) Building layered Ni x Co2x (OH)6x nanosheets decorated three-dimensional Ni frameworks for electrochemical applications. J Power Sources 317:1–9
Cheng H, Su Y-Z, Kuang P-Y, Chen G-F, Liu Z-Q (2015) Hierarchical NiCo2O4 nanosheet-decorated carbon nanotubes towards highly efficient electrocatalyst for water oxidation. J Mater Chem A 3:19314–19321
Yue X, Jin Y, Shen PK (2017) Highly stable and efficient non-precious metal electrocatalysts of tantalum dioxyfluoride used for the oxygen evolution reaction. J Mater Chem A 5:8287–8291
Li J, Xu W, Li R et al (2016) A tremella-like Ni76Co24 layered double hydroxides nanosheets as an efficient catalyst for oxygen evolution reaction. J Mater Sci 51:9287–9295
Acknowledgements
The authors wish to acknowledge financial support from the National Natural Science Foundation of China (21376105, 21576113).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, J., Xu, W., Zhou, D. et al. Synthesis of 3D flower-like cobalt nickel phosphate grown on Ni foam as an excellent electrocatalyst for the oxygen evolution reaction. J Mater Sci 53, 2077–2086 (2018). https://doi.org/10.1007/s10853-017-1631-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1631-3