
Abstract
A novel copolymer based on 6-cyanoindole (6CNIn) and 3,4-ethylenedioxythiophene (EDOT) was electrochemically synthesized in acetonitrile containing tetrabutylammonium tetrafluoroborate (TBATFB). The copolymer P(6CNIn-co-EDOT) was characterized by cyclic voltammetry, FT-IR, 1H NMR, SEM, and spectroelectrochemical analysis. According to the results of FT-IR and 1H NMR spectra, the electrochemical copolymerization located at C(2), C(3) position of 6CNIn. Spectroelectrochemical analysis indicates this copolymer film has distinct electrochromic properties, which can convert between brick-red in the reduced state and sky-blue in the oxidized state. Thus, an electrochromic device (ECD) based on P(6CNIn-co-EDOT) and poly(3,4-ethylenedioxythiophene) was also constructed, which had a color change from dark green in neutral state to dark blue in oxidized state. This ECD showed good optical contrast (30 % at 485 nm, 32 % at 610 nm), high coloration efficiency (216 cm2 C−1 at 485 nm, 223 cm2 C−1 at 610 nm), fast response time (0.7 s at 485 nm, 0.9 s at 610 nm), and long-term stability.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Electrochromism is a process in which the color of a material is changed through an electrochemical reaction controlled by an applied voltage [1, 2]. Substantially, electrochromic phenomenon is caused by electrochemically induced oxidation–reduction reaction, which generates changes of optical absorption bands at different regions of Vis–NIR region. Many kinds of materials, including inorganic (such as IrO2 and WO3) and organic (such as conducting polymers) materials, have been reported to show electrochromic properties, which can be further applied in electrochromic devices (ECDs) due to their specific qualities. Jensen et al. have reported an organic electrochromic material, which could be directly photopatterned and then utilized in ECDs [3]. Conducting polymers, which have higher coloration efficiency (CE) values, lower cost, and faster response time than the inorganic materials, have been widely studied as novel functional materials in applications including polymer-based light-emitting diodes (LEDs) [4, 5], field-effect transistors (FETs) [6, 7], photovoltaics [8, 9], and ECDs [10, 11]. Recently, the design, synthesis, and applications of novel electrochromic conducting polymers have been an area of intense interest since the properties of these materials can easily be tuned through rational design and modification of chemical structures [12].
Among conducting polymers, poly(3,4-ethylenedioxythiophene) (PEDOT), an important polythiophene derivative with two electron-donating oxygen atoms on 3, 4-positions of thiophene, have attracted considerable attention in recent years due to its interesting properties, such as low half-wave potential, small band gap, high conductivity [13], environmental stability upon cycling as well as air, good chemical and electrochemical properties [14, 15]. Furthermore, PEDOT, in particular, has stimulated much interest as a cathodic electrochromic material of ECDs due to fast switching between deep-blue and pale-blue hues, high transmissivity for visible radiation, and resistance to degradation in the doped form [16–18]. Now, PEDOT is becoming a representative electrochromic polymer [19, 20].
Polyindole is of much interest due to its fairly good thermal stability, high redox activity, and good solubility [21–24]. 6-Cyanoindole (6CNIn) is a significant indole derivative for the electron-withdrawing –C≡N group, which can decrease the band gap of corresponding conducting polymers by the decrease of LUMO energy level as an electron-withdrawing group substitution [25, 26]. In addition, they generally exhibit an electric field-induced switching between yellow and green colors, acquired in the dedoped and doped states, respectively, thus making them suitable as anodic colorants in electrochromic devices [27, 28]. The electrochemistry of poly(indole-3-carboxaldehyde) films doped by a conventional anion such as tetrafluoroborate and the electrorheological and electro-kinetic properties of chemically polymerized polyindole have been studied in the past [29]. In addition, poly(5-cyanoindole) were electrochemically synthesized and in situ Raman and spectroelectrochemical investigations were performed, which indicate that the films exhibit different absorption peaks at various applied potentials in UV–Vis and NIR regions [30]. However, there is no report of electrochromic properties of P6CNIn for some drawbacks have an effect on the applications of P6CNIn, such as the relatively low conductivity. As one of the important methods for overcoming the shortcomings of conducting polymers, the electrocopolymerization of different monomers can obtain corresponding copolymers with specific properties, thus producing diverse functional electrochromic materials [31–34]. In addition, electrocopolymerization for electrochromic materials is an effective method because it can be carried out at room temperature and homogeneous polymer films can be formed directly at the electrode surface, by which the film thickness is well controlled [35, 36]. At present, electrocopolymerizations of EDOT with a different monomer are a subject of intensive academic interest, such as EDOT-co-aniline [33, 37], EDOT-co-indole [38], EDOT-co-thiophene [39], and EDOT-co-pyrrole [40, 41]. The electrochemical properties of all these as-formed copolymers are significantly improved in comparison with the corresponding monomers due to the incorporation of EDOT units into the conjugated chain, which further proves electrocopolymerization is an excellent method.
In recent years, the need for cheap ECDs of larger areas of thin organic films has become an increasingly important goal within a number of research fields that have emerged from the idea that solution processing of organic electronics has the potential for just that [42]. In this context, roll-to-roll (R2R) vacuum-free processing on flexible substrates has always been the final goal [43]. Jensen et al. have reported a printable ECD that could be switched with low current density by roll coating methods and printable electrolytes [44]. ECDs can be constructed by several other methods. The bottom and up approach is a useful method [45], in which ITO-free substrate is used so that the ECDs can have fast switching time [46]. The lamination approach is another important way, in which the electrolyte system and the conductive substrate are crucial to the stability of the ECDs. Liquid electrolytes are difficult to process and the resulting ECDs need to be meticulously sealed to prevent leakage. However, using a solid electrolyte may compromise the switching time, which is dependent on the ionic mobility. To circumvent this, the electrolyte needs to be in the form of a gel. Jensen et al. have shown that the electrolyte layer can be efficiently deposited onto a polymeric layer by using an ionic liquid/PMMA-based polymeric gel [47]. Based on these discussion, different methods of constructing ECDs may have different impact on the performance of the device. The suitable method should be selected according to the practical needs of ECDs.
In this paper, a new copolymer P(6CNIn-co-EDOT) were obtained via electrocopolymerization of EDOT and 6CNIn monomers. The electrochemical properties, morphology, and electrochromic properties of as-prepared P(6CNIn-co-EDOT) films were studied. The structure and polymerization mechanism were elucidated by FT-IR and 1H NMR spectra. Additionally, its dual-type ECD was also constructed with PEDOT, which used the ITO electrodes separated by gel electrolyte. The specreoelectrochemistry, electrochromic switching, and stability of this device were also investigated.
Materials and methods
Materials
EDOT (Aldrich, 98 %) and 6CNIn (Acros Organics, 99 %) were used as received without further purification. Tetrabutylammonium tetrafluoroborate (TBATFB, Acros Organics, 95 %) was dried in vacuum at 80 °C for 24 h before use. Commercial HPLC grade acetonitrile (ACN, made by Shanghai Chemical Reagent Company) was dried and distilled before use. Propylene carbonate (PC) and poly(methyl metacrylate) (PMMA) were used as received.
Electrosynthesis of P(6CNIn-co-EDOT) films
Electrochemical syntheses and examinations were performed in a one-compartment cell with the use of a Model 263 potentiostat–galvanostat (EG and G Princeton Applied Research), which is under computer control strictly at room temperature. For electrochemical tests, the working and counter electrodes for cyclic voltammetric (CV) experiments were Pt wires and stainless-steel electrode each with a diameter of 0.5 mm placed 0.5 cm apart. Both were polished and cleaned by water and acetone successively before each examination. To obtain a sufficient amount of the polymer films for characterization, ITO-coated glasses and stainless-steel sheets with surface areas of 10 and 12 cm2 each were used as the working and counter electrodes, respectively. Stainless-steel sheets were carefully polished with abrasive paper (1500 mesh), subsequently cleaned successively with water and acetone, and finally dried in air before each experiment. An Ag/AgCl electrode directly immersed in the solution served as the reference electrode, which revealed sufficient stability during the experiments. All potentials mentioned in this work were referred to SCE. The polymers were deposited on an ITO-coated glass for UV–visible spectral measurements.
All the samples were grown in ACN solution containing 0.1 M TBATFB as a supporting electrolyte. All solutions were deaerated by a dry argon stream and maintained at a slight argon overpressure during experiments. The amount of polymer deposited on the electrode was controlled by the integrated charge passed through the cell. In order to remove the electrolyte and oligomer/monomer, the electropolymerized films were rinsed in mixtures of acetone and water containing increasing amounts of water and finally pure water and then dedoped with pure water for 3 days. Finally, the polymer was dried under vacuum at 60 °C for 2 days. The electrochemical polymerization of 6CNIn and EDOT monomer is illustrated in Scheme 1.

Electrochemical copolymerization route of 6CNIn and EDOT
Electrochromic device
To demonstrate the performance of P(6CNIn-co-EDOT) in a real ECD, a simple transmissive type of ECD was fabricated as shown in Scheme 2. PEDOT was coated on ITO electrodes (ITO, Delta Tech. 7–10 Ω, 0.7 × 6 cm) from ACN solution with 0.1 M TBATFB containing 0.05 M EDOT monomer. And then P(6CNIn-co-EDOT) was also obtained on ITO electrodes from ACN solution with 0.1 M TBATFB containing 0.02 M 6CNIn and 0.04 M EDOT. The device was constructed by using the electrochromic electrodes separated by gel electrolyte (TBATFB: ACN: PMMA: PC in the ratio of 3:70:7:20) [48].

Mechanism of the construction of the ECD and the color change
Characterizations
Infrared spectra were recorded using Nicolet 510P FT-IR spectrometer with KBr pellets. 1H NMR spectrum was recorded with a JEOL GAM-ECP600 NMR spectrometer in which CD3SOCD3 was used as the solvent. Scanning electron microscopy (SEM) measurements were taken by using a JEOL JSM-6700F scanning electron microscope. Spectroelectrochemical and kinetic studies were carried out on a Model 263 potentiostat–galvanostat (EG&G Princeton Applied Research) and a Cary 500 UV–Vis–NIR spectrophotometer under computer control.
The optical density (ΔOD) at a specific wavelength (λ max) was determined by using %T values of the electrochemically oxidized and reduced films, using the Eq. (1):
The CE is defined as the relation between the injected/ejected charge as a function of electrode area (Q d) and the change in optical density (ΔOD) at a specific dominant wavelength (λ max) as illustrated by the following Eq. (2) [49, 50]:
Results and discussion
Electrochemical copolymerization of EDOT and 6CNIn
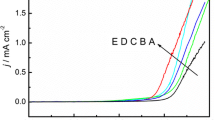
It is well known that successful electrochemical copolymerization of different monomers is due to the fact that the onset oxidation potential (E onset) of the monomers are close to each other [51, 52]. The closer the value of E onset of two monomers is (ΔE onset < 0.2 V), the more beneficial the copolymerization is. In order to investigate whether EDOT and 6CNIn can copolymerize successfully, electrochemical experiments were classified into different groups on the basis of feed ratios of two monomers. All the results of experiments showed that the E onset of EDOT was very close to that of 6CNIn, indicating that the electrochemical copolymerization can achieve. The typical anodic polarization curves taken in ACN containing 0.1 M TBATFB at a potential scanning rate of 25 mV s−1 are shown in Fig. 1. The working and counter electrodes were platinum wires and stainless-steel electrode, respectively. As shown in Fig. 1, the value of E onset of 6CNIn was initiated at 1.47 V (Fig. 1A) while that of EDOT began to oxidize at 1.50 V (Fig. 1C). The difference of the E onset between 6CNIn and EDOT is 0.03 V, which made it possible to realize the copolymerization. In the electrolytic solutions, containing 0.02 M 6CNIn and 0.04 M EDOT, the anodic curve B (Fig. 1B) was obtained, which was different from curves A and C. As can be seen from Fig. 1B, the E onset of this mixture was initiated at 1.48 V, which was intervenient between EDOT and 6CNIn. This may be partly due to some changes of the electrochemical environment caused by successive deposition of two monomers during the anodic polarization [53], or partly due to the influence of the low electrical conductivity accompanied by the incorporation of indole unit into the copolymer chain [54, 55]. In addition, the fact that the E onset of mixture was between the values of two monomers indicated that two monomers were oxidized alternately and the occurrence of copolymerization.

Anodic polarization curves of (A) 0.02 M 6CNIn, (B) 0.02 M 6CNIn + 0.04 M EDOT, and (C) 0.04 M EDOT in ACN/TBATFB solution. Scanning rates: 25 mV s−1
In order to ensure the suitable feed ratios of 6CNIn and EDOT, a series of experiments with different feed ratios were carried out, in which cyclic voltammetry (CV) method was used for electrochemical synthesis of copolymer [56]. The electrochemical copolymerization experiments of 0.02 M 6CNIn, 0.04 M EDOT, and the mixture with different feed ratios of two monomers were carried out in ACN containing 0.1 M TBATFB at a scan rate of 100 mV s−1 for 10 cycles. When the feed ratio is 0.04:0.02, the copolymer shows a couple of redox waves at 1.15 and 1.0 V, which is similar to 6CNIn. When the feed ratio becomes 0.02:0.06, the redox peaks locate at around 0.35 and 0.07 V, close to PEDOT. Compared with the other CVs of different feed ratios of two monomers, the CVs of 0.02 M 6CNIn and 0.04 M EDOT showed higher peak current density, which further proves that 0.02 M 6CNIn and 0.04 M EDOT is a suitable feed ratios of the copolymerization. As shown in Fig. 2, with the CV scan continuing, the peak current of the reversible redox couples increased, indicating that the electroactive polymer was formed on the surface of the working electrode. In addition, there was drastic change of current density between consecutive cycles. The polymerization of 6CNIn demonstrates the anodic peak is located at +1.0 V and a well-defined cathodic peak is at +1.2 V (Fig. 2a), while PEDOT can be reduced and oxidized between −0.3 and 0.5 V (Fig. 2c), which has broad redox waves, similar to the literature [57]. However, the oxidation and reduction waves of the mixture were different from both EDOT and 6CNIn, indicating the formation of a new copolymer P(6CNIn-co-EDOT) [58].

Cyclic voltammograms of (a) 0.02 M 6CNIn, (b) 0.02 M 6CNlIn + 0.04 M EDOT, and (c) 0.04 M EDOT in ACN/TBATFB, respectively. Scanning rates: 100 mV s−1
Electrochemistry of P(6CNIn-co-EDOT) films
P(6CNIn-co-EDOT) was washed with ACN to remove monomers from the film surface. Then CV curves of P(6CNIn-co-EDOT) film were investigated in monomer-free ACN containing 0.1 mol L−1 TBATFB solution between 0.03 and 1.63 V at different scan rates. As shown in Fig. 3, the reversible redox peaks can be observed and the film can be cycled repeatedly between the conducting (oxidized) and insulating (neutral) states without significant decomposition of materials, indicating high stability of the copolymer. Figure 3, inset illustrates the linear relation of the peak current densities as a function of the scan rates, which indicates that a well-adhered copolymer film is formed on the electrode surface and the redox process is non-diffusion controlled [59].

Cyclic voltammograms of P(6CNIn-co-EDOT) film at different potentials in monomer-free ACN/TBATFB solution. Scanning rates: (a) 250 mV s−1, (b) 200 mV s−1, (c) 150 mV s−1, (d) 100 mV s−1, (e) 50 mV s−1, and (f) 25 mV s−1
Structure characterizations
Vibrational spectra can provide structural information on neutral conducting polymers [60]. FT-IR spectra of 6CNIn, EDOT, and copolymer are shown in Fig. 4. According to the spectrum of pure EDOT (Fig. 4C), the bands at 1630, 1504, and 1350 cm−1 originated from the stretching modes of C=C and C–C in the thiophene ring [61]. In this region, the absorptions shifted in the spectra of copolymers due to the interaction of benzene ring and thiophene ring during the electrochemical copolymerization. The bands at 1135 and 1084 cm−1 (Fig. 4C) were assigned to the stretching of the C–O–C bond [62], which shifted downwards to 1125 and 1082 cm−1 in the spectra of the copolymer (Fig. 4B). This indicated that EDOT units were incorporated into the main chain of the copolymers. In the case of 6CNIn (Fig. 4A), the peaks at 1648, 1485, and 747 cm−1 are assigned to the stretching and bending modes of benzene ring, which can be found in the copolymers (Fig. 4B). This implies that the indole units are incorporated into the copolymer chain. The spectra of 6CNIn located at 620, 773, and 818 cm−1, assigned to the out-of-plane C–H vibrations of 1,2,4-trisubstituted benzene ring, were also present on the spectra of copolymer, indicating benzene ring incorporating into the copolymer. In addition, the narrow peak of N–H bond observed in the 6CNIn is broader and shifts to 3415 cm−1, which can also be seen in the spectra of copolymer (Fig. 4, inset). Based on all the features, it can be concluded that copolymerization happens at C(2), C(3) position of indole cycle (Scheme 1) [63].

FT-IR spectra of (A) 6CNIn, (C) EDOT, and (B) P(6CNIn-co-EDOT) film prepared at 1.5 V versus SCE from ACN/TBATFB solution containing 0.02 M 6CNIn and 0.04 M EDOT
To further investigate copolymer structure and polymerization mechanism of 6CNIn and EDOT, the 1H NMR spectra of 6CNIn, EDOT, and copolymer were also recorded. As shown in Fig. 5a, the spectrum of 6CNIn shows five groups of protons: δ 7.91 (H-7), δ 7.73 (H-4), δ 7.65 (H-5), δ 7.34 (H-2), δ 6.62 (H-3), and δ 11.71 (H-1), respectively [64, 65]. According to the spectrum of EDOT (Fig. 5c), there are two groups of protons: one is located at δ 4.21 (attributed to the aliphatic C–H), the other is at δ 6.50 (attributed to C–H of thiophene ring). The proton lines of copolymer (Fig. 5b) were much broader than the corresponding proton lines of 6CNIn (Fig. 5a) and EDOT monomer (Fig. 5c) due to the wide molar mass distribution of copolymer. As shown in Fig. 5b, some new peaks appeared after copolymerization and most of the peaks moved to the lower field, which was mainly due to the introduction of higher conjugation length in the copolymer main chain. It can be seen in Fig. 5b that there were four peaks of the copolymer spectra. The four peaks around δ 11.73 (H-1), δ 7.73 (H-4), δ 7.65 (H-5), and δ 7.91 (H-7) arose from the benzene ring of 6CNIn units. The peak around δ 4.3 was assigned to the aliphatic C–H of EDOT units. Furthermore, the two peaks around δ 7.34 (H-2), δ 6.62 (H-3) of 6CNIn cannot be seen in Fig. 5b, which can also reveal the occurence of copolymerization. Based on these considerations, the copolymerization mechanism of 6CNIn and EDOT was in accordance with the FT-IR results (Scheme 1).

1H NMR spectra of a 6CNIn, c EDOT, and b P(6CNIn-co-EDOT) prepared at 1.5 V versus SCE from ACN/TBATFB solution containing 0.02 M 6CNIn and 0.04 M EDOT. Solvent: CD3SOCD3
Morphology of P(6CNIn-co-EDOT) films
The film surface morphology of pure P6CNIn, pure PEDOT, and P(6CNIn-co-EDOT) were examined by scanning electron microscopy (SEM), which are illustrated in Fig. 6. All the films adhere tightly to the ITO substrate and washed by the monomer-free solution of ACN to remove monomers from the film surface. In this case, pure P6CNIn is compact (Fig. 6a). PEDOT is very regular with a growth of aggregates shaped as blossoms (Fig. 6c). The copolymer was obtained with a grain structure on the surface (Fig. 6b), which was different from two corresponding homopolymers, further confirmed that the electrocopolymerization of EDOT and 6CNIn units, which may be beneficial to extend the applications of P6CNIn.

SEM of a P6CNIn, c PEDOT, and b P(6CNIn-co-EDOT) deposited on the electrode surface prepared at 1.5 V versus SCE from ACN/TBATFB solution containing 0.02 M 6CNIn and 0.04 M EDOT
Spectroelectrochemical properties of P(6CNIn-co-EDOT) films
It is desired at this stage to gain a deeper insight into the relationship between polymer structure and electrochemical properties [66]. The spectroelectrochemistry is expected to give key properties of conducting polymers such as a band gap (E g) and the intergap states that happen upon doping. Therefore, the spectroelectrochemical measurements were performed in the UV–Vis and near-infrared (NIR) wavelength range using a quartz cell. The quartz cell includes three electrodes (working, counter, and reference) and the electrolytic solution (ACN/TBATFB solution). Before the measurement, P(6CNIn-co-EDOT) film, which was prepared potentiostatically at +1.5 V in ACN/TBATFB solution, had been deposited on the ITO-coated glass without the monomer/homopolymer under the same conditions. The absorbance of ITO glass is automatically subtracted from the spectroscopic data points by the potentiostat. During the spectroelectrochemical measurement, the working electrode potential (ITO) is swept from −0.5 to 1.1 V in steps of 0.1 V by the potentiostat.
Figure 7 shows the spectroelectrochemical analysis of P(6CNIn-co-EDOT) film at different voltages varying between −0.5 and 1.1 V. At an oxidation potential of 1.1 V, a broad absorption, corresponding to the bipolaron formation [67], is visible in the 700 nm, in which state it shows a sky-blue color (Fig. 7, inset). The intensity of this peak diminishes progressively as the oxidation potential is decreased from 1.1 to 0.2 V. At potentials lower than 0.2 V (−0.5 V < E (V) < 0.2 V), a distinct peak corresponding to π–π* transitions was observed at 475 nm, signaling the neutralization of bipolarons by electrons and formation of the reduced polymer. This peak gains intensity at the expense of the bipolaronic peak, as a function of increasing reduction potential. The absorption edge for the π–π* peak of the P(6CNIn-co-EDOT) film (E = −0.5 V) tends to show a brick-red shift (Fig. 7, inset). In order to investigate the photochemical stability of P(6CNIn-co-EDOT), P(6CNIn-co-EDOT) was irradiated by the high pressure mercury vapor lamp for 2 h. There is a slight decline in the absorbance of P(6CNIn-co-EDOT) after 2 h, attributed to the degradation of polymer chains, caused by the UV-irradiation [68]. This result indicated the good photochemical stability of P(6CNIn-co-EDOT).

Spectroelectrochemistry of P(6CNIn-co-EDOT) on ITO-coated glass in monomer-free ACN/TBATFB at applied potentials: (a) −0.5 V, (b) −0.4 V, (c) −0.3 V, (d) −0.2 V, (e) −0.1 V, (f) 0.0 V, (g) 0.1 V, (h) 0.2 V, (i) 0.3 V, (j) 0.4 V, (k) 0.5 V, (l) 0.6 V, (m) 0.7 V, (n) 0.8 V, (o) 0.9 V, (p) 1.0 V, (q) 1.1 V versus SCE. The inset is real photo of color change of copolymer films
Spectroelectrochemistry of ECD
It has been demonstrated that a dual-polymer ECD system using two complementary electrochromic electrodes can effectively improve electrochemical and electrochromic performances, such as optical contrast, response time, and optical memory [69]. A simple transmissive dual-type ECD consisting of PEDOT, as a complementary cathodic electrode, and P(6CNIn-co-EDOT) film was assembled as shown in Scheme 2, and its spectroelectrochemical behavior was also studied. Before composing the ECD, the anodically coloring polymer film P(6CNIn-co-EDOT) was fully reduced and the cathodically coloring polymer PEDOT was fully oxidized, which were both obtained in ACN containing 0.1 M TBATFB on ITO. This real ECD consisting of P(6CNIn-co-EDOT) and PEDOT deposited on transparent ITO, was placed in a position to face each other and a gel electrolyte was applied in between. The redox charges of these two complementary polymer films must be matched by chronocoulometry in order to get a balanced number of redox sites for switching. The spectroelectrochemical spectra of this ECD as a function of applied potential (between −1.8 and 1.0 V) are given in Fig. 8. The copolymer is in its neutral state at −1.8 V, where the absorption at 485 nm is due to π–π* transition of the copolymer. At that state, PEDOT does not reveal an obvious absorption at the UV–Vis region of the spectrum and device reveals dark green color (Scheme 2). As the applied potential increases, the copolymer layer starts to get oxidized and the intensity of the peak of π–π* transition decreased. Meanwhile, PEDOT layer is in its reduced state, which leads to a new absorption at 610 nm due to the reduction of PEDOT, and the dominated color of the device is dark blue (Scheme 2).

Spectroelectrochemistry of P(6CNIn-co-EDOT)/PEDOT ECD at applied potentials (V): (a) −1.8, (b) −1.6, (c) −1.4, (d) −1.2, (e) −1.0, (f) −0.8, (g) −0.6, (h) −0.4, (i) −0.2, (j) 0.0, (k) 0.2, (l) 0.4, (m) 0.6, (n) 0.8, (o) 1.0 versus SCE
For electrochromic applications, short switching time and high optical contrast (ΔT%) of the polymer between neutral and oxidized states are the reflections of its excellent electrochromic property. The color-bleach characteristics of the ECD recorded at 485 and 610 nm under a square wave potential input of −1.8 and 1.0 V at an optimized frequency of 0.1 Hz (equivalent to a step time of 5 s for each half cycle) are shown in Figs. 9 and 10, respectively. The switching kinetics was examined by evaluating the time taken for a 95 % transmission change corresponding to an absolute optical contrast for the films of same thickness and geometric area. At wavelengths of 485 nm, the optical contrast (ΔT%) is calculated to be 30 % (Fig. 9c), and the response time is found to be 0.7 s from the reduced to the oxidized state and 1.1 s from the oxidized to the reduced state. The transmission is found to be 60.8 % at 485 nm, as shown in Fig. 9c. It has to be noted that the switching stability of the ECD is satisfactory because the transmission was reduced by 0.7 % after ten cycles in the ECD switching experiment. While at wavelengths of 610 nm, the optical contrast (ΔT%) is calculated to be 32 % (Fig. 10c), the time required for a 95 % transmittance change (for %T to decrease from 95 to 0 %) is 1.2 s and the corresponding bleaching time (for %T raise from 0 to 95 %) is 0.9 s. As shown in Fig. 10c, the transmission is 41.7 % at 610 nm in original, which falls to 40.3 % when switched between −1.8 and 1.0 V after 10 times. The slight decline (1.4 %) indicates the good switching stability of ECD. CE is another key parameter for electrochromic materials as it describes the change in optical density at the wavelength of interest with per inserted charge [70]. For this device, the CE is calculated to be 216 cm2 C−1 (at 485 nm) and 223 cm2 C−1 (at 610 nm), respectively, which demonstrates that the device could be applied for large area devices.

a Potential–time, b current–time, and c transmittance–time profiles of P(6CNIn-co-EDOT)/PEDOT ECD at 485 nm under an applied square voltage signal between −1.8 V (the neutral state) and 1.0 V (the oxidized state)

a Potential–time, b current–time, and c transmittance–time profiles of P(6CNIn-co-EDOT)/PEDOT ECD at 610 nm under an applied square voltage signal between −1.8 V (the neutral state) and 1.0 V (the oxidized state)
The stability of this ECD toward redox switching usually limits the utility of electrochromic materials in ECD applications. Therefore, the stability of polymer is another crucially important feature in the practical use of the electrochromic material. For this reason, long-term stability of redox activity of this ECD was investigated to evaluate the stability of the devices. The potential was scanned between −1.1 and 2.3 V (Fig. 11). Form 1st to 10th, there was no obvious decrease of activity. After 1000 cycles, the CV curve of this ECD reveals the film retaining 95 % of its original electroactivity, and the changes in anodic (j pa) and cathodic peak current densities (j pc) are 8 and 6 %, respectively. When this ECD was scanned continuously up to 2000 cycles, there was no obvious lost of electroactivity. All these results imply that the ECD has a reasonable environmental and redox stability and could be as a promising candidate material for ECD.

Cyclic voltammograms of the device as a function of repeated with a scan rate of 100 mV s−1 between 1 and 2000 cycles
Conclusions
A novel electrochromic copolymer P(6CNIn-co-EDOT) based on 6CNIn and EDOT was successfully synthesized by electrocopolymerization of their monomers mixture. CV analyses confirmed that copolymer showed good redox activity and electrochemical properties. Both FT-IR and 1H NMR spectra revealed that electrochemical copolymerization located at C(2), C(3) position of 6CNIn. Furthermore, according to the spectroelectrochemical analyses, the copolymer film had good electrochromic property, which showed brick-red in its reduced state and sky-blue in its oxidized state. The dual-type ECD using P(6CNIn-co-EDOT) and PEDOT had good optical contrast (30 % at 485 nm, 32 % at 610 nm), high CE (216 cm2 C−1 at 485 nm, 223 cm2 C−1 at 610 nm), fast response time (0.7 s at 485 nm, 0.9 s at 610 nm), and long-term stability. In view of above results, the copolymer P(6CNIn-co-EDOT) can be useful as the active layers in electrochromic devices, which may extend the applications of both polyindole and PEDOT.
References
Ozyurt F, Gunbas EG, Durmus A et al (2008) Processable and multichromic polymer of bis-3-hexylthiophene substituted 4-tert-butylphenyl quinoxaline. Org Electron 9:296–302
Kaya İ, Yıldırım M, Aydın A (2011) A new approach to the Schiff base-substituted oligophenols: the electrochromic application of 2-[3-thienylmethylene] aminophenol based co-polythiophenes. Org Electron 12:210–218
Jensen J, Dyer AL, Shen DE et al (2013) Direct photopatterning of electrochromic polymers. Adv Funct Mater 23:3728–3737
Aurora R, Niclas S, Lars JL et al (2010) White light with phosphorescent protein fibrils in LEDs. Nano Lett 10:2225–2230
Lin Y, Chen Y, Chen Z et al (2010) Triphenylamine and quinoline-containing polyfluorene for blue light-emitting diodes. Eur Polym J 46:997–1003
Chua LL, Zaumseil J, Chang JF et al (2005) General observation of n-type field-effect behaviour in organic semiconductors. Nature 434:194–199
Zhen S, Xu J, Lu B et al (2014) Tuning the optoelectronic properties of polyfuran by design of furan-EDOT monomers and free-standing films with enhanced redox stability and electrochromic performances. Electrochim Acta 146:666–678
Krebs FC, Tromholt T, Jorgensen M (2010) Upscaling of polymer solar cell fabrication using full roll-to-roll processing. Nanoscale 2:873–886
Herrero-Carvajal D, de la Peña A, González Cano RC et al (2014) EDOT-based copolymers with pendant anthraquinone units: analysis of their optoelectronic properties within the “double-cable” context. J Phys Chem C 118:9899–9910
Sefer E, Koyuncu FB, Oguzhan E, Koyuncu S (2010) A new near-infrared switchable electrochromic polymer and its device application. J Polym Sci A 48:4419–4427
Nie G, Zhou L, Guo Q et al (2010) A new electrochromic material from an indole derivative and its application in high-quality electrochromic devices. Electrochem Commun 12:160–163
Qin L, Xu J, Lu B et al (2012) Synthesis and electrochromic properties of polyacrylate functionalized poly(3,4-ethylenedioxythiophene) network films. J Mater Chem 22:18345–18353
Hu D, Lu B, Duan X et al (2014) Synthesis of novel chiral l-leucine grafted PEDOT derivatives with excellent electrochromic performances. RSC Adv 4:35597–35608
O’Connell CD, Higgins MJ, Nakashima H et al (2012) Vapor phase polymerization of EDOT from submicrometer scale oxidant patterned by dip-pen nanolithography. Langmuir 28:9953–9960
Lu B, Zhang S, Qin L et al (2013) Electrosynthesis of poly(3,4-ethylenedithiathiophene) in an ionic liquid and its electrochemistry and electrochromic properties. Electrochim Acta 106:201–208
Yu W, Chen J, Fu Y et al (2013) Electrochromic property of a copolymer based on 5-cyanoindole and 3,4-ethylenedioxythiophene and its application in electrochromic devices. J Electroanal Chem 700:17–23
Elschner A, Kirchmeyer S, Lovenich W et al (2011) PEDOT: principles and applications of an intrinsically conductive polymer. Taylor & Francis Group, Boca Raton
Lu B, Zhen S, Zhang S et al (2014) Highly stable hybrid selenophene-3,4-ethylenedioxythiophene as electrically conducting and electrochromic polymers. Polym Chem 5:4896–4908
Deepa M, Awadhia A, Bhandari S (2009) Electrochemistry of poly(3,4-ethylenedioxythiophene)-polyaniline/Prussian blue electrochromic devices containing an ionic liquid based gel electrolyte film. Phys Chem Chem Phys 11:5674–5685
Jeong YS, Akagi K (2011) Control of chirality and electrochromism in copolymer-type chiral PEDOT derivatives by means of electrochemical oxidation and reduction. Macromolecules 44:2418–2426
Mackintosh JG, Redpath CR, Jones AC et al (1995) The electropolymerization and characterization of 5-cyanoindole. J Electroanal Chem 388:179–185
Talbi H, Humbert B, Billaud D (1998) FTIR and Raman spectroscopic investigations on the redox behaviour of poly(5-cyanoindole) in acidic aqueous solutions. Spectrochim Acta A 54:1879–1893
Talbi H, Billaud D (1998) Electrochemical properties of polyindole and poly(5-cyanoindole) in LiClO4–acetonitrile and in HCl and HClO4 solutions. Synth Met 93:105–110
Xu J, Hou J, Zhang S et al (2006) Electrosyntheses of high quality poly(5-methylindole) films in mixed electrolytes of boron trifluoride diethyl etherate and diethyl ether. Eur Polym J 42:1384–1395
Nie G, Cai T, Zhang S et al (2007) Electrodeposition of poly(indole-5-carboxylic acid) in boron trifluoride diethyl etherate containing additional diethyl ether. Electrochim Acta 52:7097–7106
Yuen OY, Choy PY, Chow WK et al (2013) Synthesis of 3-cyanoindole derivatives mediated by copper(I) iodide using benzyl cyanide. J Org Chem 78:3374–3378
Nie G, Zhou L, Yang H (2011) Electrosynthesis of a new polyindole derivative obtained from 5-formylindole and its electrochromic properties. J Mater Chem 21:13873–13880
Reddy BN, Deepa M (2013) Electrochromic switching and nanoscale electrical properties of a poly(5-cyano indole)-poly(3,4-ethylenedioxypyrrole) device with a free standing ionic liquid electrolyte. Polymer 54:5801–5811
Deletioğlu D, Hasdemir E, Solak AO et al (2010) Preparation and characterization of poly(indole-3-carboxaldehyde) film at the glassy carbon surface. Thin Solid Films 519:784–789
Talbi H, Billaud D, Louarn G et al (2000) UV-vis and Raman spectroelectrochemical investigation of the redox behavior of poly(5-cyanoindole) in acidic aqueous solutions. Spectrochim Acta A 56:717–728
Giovanella U, Botta C, Galeotti F et al (2013) Perfluorinated polymer with unexpectedly efficient deep blue electroluminescence for full-colour OLED displays and light therapy applications. J Mater Chem 1:5322–5329
Jeong YS, Akagi K (2011) Liquid crystalline PEDOT derivatives exhibiting reversible anisotropic electrochromism and linearly and circularly polarized dichroism. J Mater Chem C 21:10472–10481
Yang Z, Xue Z, Liao Y et al (2013) Hierarchical hybrids of carbon nanotubes in amphiphilic poly-(ethyleneoxide)-block-polyaniline through a facile method: from smooth to thorny. Langmuir 29:3757–3764
Bhattacharyya D, Gleason KK (2011) Single-step oxidative chemical vapor deposition of –COOH functional conducting copolymer and immobilization of biomolecule for sensor application. Chem Mater 23:2600–2605
Yen WC, Lee YH, Lin JF et al (2011) Effect of TiO2 nanoparticles on self-assembly behaviors and optical and photovoltaic properties of the P3HT-b-P2VP block copolymer. Langmuir 27:109–115
Bhatt MP, Sista P, Hao J et al (2012) Electronic properties-morphology correlation of a rod-rod semiconducting liquid crystalline block copolymer containing poly(3-hexylthiophene). Langmuir 28:12762–12770
Ouyang M, Fu Z, Lv X et al (2013) A multichromic copolymer based on 4-(9H-carbazol-9-yl)-N,N-diphenylaniline and 3,4-ethylenedioxythiophene prepared via electrocopolymerization. J Electrochem Soc 160:H787–H792
Yue R, Yao Z, Geng J et al (2013) Facile electrochemical synthesis of a conducting copolymer from 5-aminoindole and EDOT and its use as Pt catalyst support for formic acid electrooxidation. J Solid State Electron 17:751–760
Miozzo L, Battaglini N, Braga D et al (2012) Synthesis and characterization of all-conjugated copolymers of 3-hexyl-thiophene and EDOT by grignard metathesis polymerization. J Polym Sci A 50:534–541
Ma X, Ni X (2014) Copolymerization of EDOT with pyrrole on TiO2 semiconductor films by one-step reaction, structure-dependent electronic properties, and charge conduction models of the composite films. Langmuir 30:2241–2248
Algi MP, Öztaş Z, Tirkes S et al (2013) A new electrochromic copolymer based on dithienylpyrrole and EDOT. Org Electron 14:1094–1102
Søndergaard RR, Hösel M, Krebs FC (2013) Roll-to-roll fabrication of large area functional organic materials. J Polym Sci B 51:16–34
Søndergaard RR, Hösel M, Jørgensen M et al (2013) Fast printing of thin, large area, ITO free electrochromics on flexible barrier foil. J Polym Sci B 51:132–136
Jensen J, Dam HF, Reynolds JR et al (2012) Manufacture and demonstration of organic photovoltaic-powered electrochromic displays using roll coating methods and printable electrolytes. J Polym Sci B 50:536–545
Jensen J, Krebs FC (2014) From the bottom up–flexible solid state electrochromic devices. Adv Mater 26:7231–7234
Jensen J, Hösel M, Kim I et al (2014) Fast switching ITO free electrochromic devices. Adv Funct Mater 24:1228–1233
Jensen J, Madsen MV, Krebs FC (2013) Photochemical stability of electrochromic polymers and devices. J Mater Chem C 1:4826–4835
Ak M, Camurlu P, Yılmaz F et al (2006) Electrochromic properties and electrochromic device application of copolymer of N-(4-(3-thienyl methylene)-oxycarbonylphenyl) maleimide with thiophene. J Appl Polym Sci 102:4500–4505
Reeves BD, Grenier CRG, Argun AA et al (2004) Spray coatable electrochromic dioxythiophene polymers with high coloration efficiencies. Macromolecules 37:7559–7569
Beaujuge PM, Reynolds JR (2010) Color control in π-conjugated organic polymers for use in electrochromic devices. Chem Rev 110:268–320
Nie G, Qu L, Zhang Y et al (2008) Electrochemical copolymerization of 3,4-ethylenedioxythiophene and 5-methylindole and characterizations of the copolymers. J Appl Polym Sci 109:373
Nie G, Yang H, Wang S et al (2011) High-quality inherently organic conducting polymers electrosynthesized from fused-ring compounds in a new electrolytic system based on boron trifluoride diethyl etherate. Crit Rev Solid State Mater Sci 36:209–228
He Y, Guo WJ, Zhang GY et al (2011) Electrochemical polymerization of N-(9-fluorenylmethoxycarbony)-glycine and characterization of its polymers. Adv Mater 306:297–300
Camurlu P, Cirpan A, Toppare L (2004) Dual type complementary colored polymer electrochromic devices utilized by 3-ester substituted thiophenes. J Electroanal Chem 572:61–65
Zhang C, Xu Y, Wang NC et al (2009) Electrosyntheses and characterizations of novel electrochromic copolymers based on pyrene and 3,4-ethylenedioxythiophene. Electrochim Acta 55:13–18
Lu B, Wang J, Yue R et al (2012) Electrosynthesis and characterization of a polyfluorene derivative with green-light-emitting property. J Mater Sci 47:315–322
Turac E, Ak M, Sahmetlioglu E et al (2011) Synthesis and characterization of poly 2-[3-(1H-pyrrol-2-yl)phenyl]-1H-pyrrole and its copolymer with EDOT. Russ J Gen Chem 81:2510–2516
Zhi C, Cheng H (2011) Study on the electrochemical properties of zinc/polyindole secondary battery. J Power Sources 196:10731–10736
Berlin A, Zotti G, Zecchin S et al (2004) New low-gap polymers from 3,4-ethylenedioxythiophene-bis-substituted electron-poor thiophenes. The roles of thiophene, donor-acceptor alternation, and copolymerization in intrinsic conductivity. Chem Mater 16:3667–3676
Zhang S, Xu J, Lu B et al (2014) Electrochromic enhancement of poly(3,4-ethylenedioxythiophene) films functionalized with hydroxymethyl and ethylene oxide. J Polym Sci A 52:1989–1999
Seo KI, Chung IJ (2000) Reaction analysis of 3,4-ethylenedioxythiophene with potassium persulfate in aqueous solution by using a calorimeter. Polymer 41:4491–4499
Nie G, Qu L, Xu J et al (2008) Electrosyntheses and characterizations of a new soluble conducting copolymer of 5-cyanoindole and 3,4-ethylenedioxythiophene. Electrochim Acta 53:8351–8358
Nie G, Han X, Hou J et al (2007) Low-potential electrochemical polymerization of 5-fluoroindole and characterization of its polymers. J Electroanal Chem 604:125–126
Xu J, Hou J, Zhou W et al (2006) 1H NMR spectral studies on the polymerization mechanism of indole and its derivatives. Spectrochim Acta A 63:723–728
Chen Z, Wannere CS, Corminboeuf C et al (2005) Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem Rev 105:3842–3888
Unur E, Beaujuge PM, Ellinger S et al (2009) Black to transmissive switching in a pseudo three-electrode electrochromic device. Chem Mater 21:5145–5153
Celebi S, Balan A, Epik B et al (2009) Donor acceptor type neutral state green polymer bearing pyrrole as the donor unit. Org Electron 10:631–636
Kaczmarek H, Metzler M, Scigalski F (2014) Photochemical stability of poly(acrylic acid)/silver nanocomposite. Mater Lett 135:110–114
Kang JH, Oh YJ, Paek SM et al (2009) Electrochromic device of PEDOT-PANI hybrid system for fast response and high optical contrast. Sol Energy Mater Sol Cells 93:2040–2044
Özkut Mİ, Atak S, Önal AM et al (2011) A blue to highly transmissive soluble electrochromic polymer based on poly (3,4-propylenedioxyselenophene) with a high stability and coloration efficiency. J Mater Chem 21:5268–5272
Acknowledgements
This work was supported by National Natural Science Foundation of China (51373089), NSF of Shandong (ZR2011BM003), Specialized Research Fund for the Doctoral Program of Higher Education (20123719120006), Scientific and Technical Development Project of Qingdao (11-2-4-3-(10)-jch), National Training Programs of Innovation and Entrepreneurship for Undergraduates (201410426036).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, C., Liu, C., Shi, L. et al. Electrochemical copolymerization of 3,4-ethylenedioxythiophene and 6-cyanoindole and its electrochromic property. J Mater Sci 50, 1836–1847 (2015). https://doi.org/10.1007/s10853-014-8746-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8746-6