
Abstract
This study investigated inclusion formation and the physicochemical properties of naringin/cyclodextrin through a combined computational and experimental approach. Molecular dynamics simulations were applied to investigate the thermodynamics and geometry of naringin/cyclodextrin cavity docking. The complexes were investigated by UV, FT-IR, DSC, XRD, SEM, 2D-NOSEY and 1H-NMR analyses. Clearly visible protons belonging to naringin and chemical shift displacements of the H3 and H5 protons in cyclodextrin were anticipated in the formation of an inclusion complex. Naringin solubility increased linearly with increasing cyclodextrin concentration (displaying an AL profile). The simulations indicated that the phenyl group of naringin was located deep within the cyclodextrin cavity, while the glycoside group of naringin was on the plane of the wider rim of cyclodextrin. The simulation and molecular modeling results indicate that (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD) provided the more stable inclusion complex. This result was also in good concordance with the stability constants that had been determined by the phase solubility method. The consistency of the computational and experimental results indicates their reliability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1. Introduction
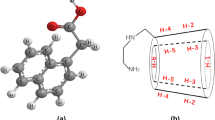
Naringin is an interesting class of flavonoid glycoside that is an active constituent of Rhizoma Drynariae (Traditional Chinese Medicine; Fig. 1) [1]. Naringin has rich bioactivity as an antioxidant and reactive oxygen species (ROS) scavenger, and possesses neuro-protective, anti-inflammatory [2], antiapoptotic [3], antiatherogenic and antihyperlipidemic agent [4]; it has also been shown to have effects on fat metabolism [5]. Naringin has been found to be effective for the treatment of osteoporosis. Naringin has shown promise as a preventive care product and to possess a wide variety of health benefits. However, to the best of our knowledge, a common constraint in the use of flavonoids in many applications is their poor water solubility and stability [6]. The poor solubility (S0 = 0.6502 mg/ml) [7], oral bioavailability and undesirable bitter taste of naringin require the development of a suitable delivery system.

Chemical structure of naringin (a) and cyclodextrins (b)
The Biopharmaceutics Classification System (BCS) predicts that oral drug absorption is based on the active drug ingredient’s water solubility and intrinsic permeability in the gastrointestinal mucosa [8]. The formation of a naringin/cyclodextrin inclusion complex is one way to enhance its solubility [9]. Native cyclodextrins (CD) are a group of cyclic oligosaccharides formed by six (α-CD), seven (β-CD) or eight (γ-CD) α-1,4-linked glucose units that are produced from starch by cyclodextrin glycosyl transferase catalysis [10]. CD structures show a truncated conical shape with a relatively hydrophilic outer surface and hydrophobic internal cavity due to the steric arrangement of glucose units [11]. The depth of the CD cavity is consistent (7.8 Å), but the diameters of α-, β-, and γ-CD cavities are different at 5.7, 7.8 and 9.5 Å [12], respectively. The bucket-like structure of CD allows for the formation of non-covalent inclusion complexes, where lipophilic compounds are non-covalently bound within the molecule. In recent years, numerous researches have been reported to improve physical–chemical properties of bioactive compounds [13–16]. Recently, our group investigated the inclusion complexation of Epothilone A [17] and satraplatin [18] with various CDs to improve drug solubility. Therefore, the aqueous solubility of hydrophobic molecules can be improved by inclusion complex formation between a guest molecule and a suitable CD.
In addition to experimental investigations, molecular dynamics (MD) simulations are important tools to predict the preferable binding mode of host-guest-type compounds at the molecular level. As MD simulations can effectively rationalize experimental phenomena and make predictions concerning the results of future experiments, this method is commonly used by researchers as a valuable addition to existing studies [19, 20]. Presently, MD simulations have been used as a tool for making a thorough inquiry into CD inclusion complexes in aqueous solution. In this contribution, this study investigated the intermolecular interactions and performed modeling calculations on naringin/cyclodextrin inclusion complexes by the MD approach.
In this work, inclusion complexes of naringin have been developed for the first time in combination with β-CD and HP-β-CD to increase the aqueous solubility of naringin. To enhance the practicability, a simple freeze-drying method has been employed. UV–Vis spectroscopy was measured to evaluate the naringin/CDs complexes in aqueous solutions. In order to verify the formation of the inclusion complexes, drug/CDs interactions in the solution were investigated using molecular modeling, thermodynamic parameter and molecular dynamics simulations analyses, and interactions in the solid state were characterized using many other technical analyses (FT-IR, DSC, XRD and SEM analyses). The goals of the study were to evaluate, characterize, and research possible molecular mechanisms that might explain the observed differences of two naringin-CD inclusion complexes. Both laboratory experiments and computational modeling were surrealistically employed in this study.
2. General materials and methods
Materials
Naringin (FW = 580.53 g/mol, purity >99%), β-CD (FW = 1135.00 g/mol, purity >99%) and HP-β-CD (FW = 1541.54 g/mol, purity >99%) were obtained from Adamas Reagent Co. Ltd.
The naringin/CD inclusion complexes were prepared by the freeze-drying method. Naringin (0.0725 g; 0.125 mmol) and β-CD (0.1420 g; 0.125 mmol) were added to a 50 ml round bottom flask, and completely dissolved in solution (30 ml; 1:4 C2H5OH:H2O) in 1:1 molar ratios with the assistance of ultra-sonication. The suspension was stirred for 48 h at room temperature under an N2 atmosphere and away from light. Insoluble substances were removed by filtration through a 0.45-μm microporous membrane to obtain a yellow solution. Finally, the solution was lyophilized in a freeze-dryer for 24 h to obtain solid complexes. Physical mixtures were prepared by mixing the powders at a 1:1 molar ratio.
Methods
1H NMR spectra and 2D ROESY spectra
1H NMR spectra and 2D ROESY spectra were acquired on a Bruker Avance 500 spectrometer (Germany) at 25.0 (±0.1) °C with a mixing time of 300 ms.
SEM, XRD, FTIR, DSC
All samples were analyzed with a JSM 500 LV scanning electron microscope, using secondary electron emission [21]. The methods used for solid state characterization, including X-ray powder diffractometry, FTIR spectroscopy, thermogravimetry and differential scanning calorimetric analyses are provided in the supplementary information.
UV–Vis analysis
The solutions were evaluated at 282 nm using a UV spectrophotometer (UV-2401, Shimadzu UV-2401, Japan). The phase-solubility experiment was performed according to the Higuchi-Connors method [22]. Detailed experimental operations were carried out according to previous work in our group. The phase-solubility diagram was used for the calculation of apparent stability constants (K C) [22, 23] using the Higuchi-Connors Eq. (1):
Molecular modeling and thermodynamic parameter
Gaussian 03 W [24] was usually used to perform the theoretical calculations [25]. The initial geometry of naringin and CDs was constructed with Chem3D Ultra 10.0 software (Cambridge soft) and then completely optimized by the DFT method [26] without any symmetry limit. The optimized structure of the naringin molecule docked in the CD cavity, and demonstrated computational efficiency in calculating CD systems.
Molecular dynamics (MD) simulations
The initial molecular structure of β-CD and HP-β-CD were retrieved from the Cambridge crystallographic databases. Given the absence of a naringin structure in this database, the initial geometry of naringin was constructed with Chem 3D Ultra software [27]. The initial orientations were prepared using Accelrys DS Visualizer 2.0. Partial atomic charges were generated with the AM1-BCC method and GAFF parameters as implemented in the program Antechamber. All MD simulations were solvated with TIP3P for all water models using the AMBER 14.0 simulation package. Langevin dynamics were used for the NVT and NPT stages of equilibration. A Berendsen barostat with the pressure set to 1 bar and the relaxation time set to 2 ps was used during the NPT stage of equilibration. Subsequently, it was operated under conditions of an MD simulation of 100 ps at a constant volume and temperature, and considering periodic boundary conditions [28].
3 Results and discussion
NMR analysis
The complexes were firstly investigated by FT-IR, DSC, TG and XRD analyses; the relevant results were showed in supplementary materials. NMR was used to assess the possible inclusion modes of naringin. It has been reported that 1H NMR can provide evidence for the inclusion complexation between aromatic substances and CD. The hydrogens on the inner surface (H3 and H5) rather than the hydrogens on the outer surface (H1, H2, H4) change in response to naringin-CD inclusion complexation. Meanwhile, the corresponding hydrogens on each glucose unit should obtain identical shielding contributions from the included guest molecule [29].
The 1H NMR spectra of pure naringin, as well as β-CD, HP-β-CD and the inclusion complexes are illustrated in Fig. 2. The chemical shifts of naringin were as follows: δ 7.092 (d, H1 of naringin), δ 6.742 (d, H2 of naringin), δ 5.969 (d, H3a of naringin), δ 5.877 (s, H3b of naringin), δ 4.000 (s, H6 of naringin), δ 3.900 (s, H4 of naringin) and δ 1.140 (s, H5 of naringin). The observed trends in Fig. 2 can be rationalized if it is assumed that the naringin moiety was oriented in the CD cavity. The H3 and H5 protons of CD shifted upfield, which confirmed the presence of naringin in the cavity of CD. It was expected that H3 and H5 located above the plane of the aromatic group were shielding by naringin. Thus, the upfield shifts shown for H3 and H5 when the inclusion complex was formed in aqueous solution was reasonable. All chemical shift changes may suggest an inclusion complexation of naringin and CD.

1H NMR spectra of naringin, β-CD,HP-β-CD and inclusion complex in D2O at 25 °C
The Δδ in the 1H chemical shift for naringin and CDs resulting from the complexation, were calculated by applying Eq. (2) as follows:
As showed in Table 1, the chemical shifts of H-1, H-2, H-4 and H-6 in the CD 1H NMR spectra showed only comparatively weak variation between the uncomplexed and complexed forms. In contrast, after the formation of the inclusion complex, the H3 and H5 protons of CD shifted upfield, which may have been caused by the H-3 and H-5 of β-CD lying in a region where a shielding by a reflection of close-contact interactions of naringin, while protons H-1, H-2, and H-4 were outside of the cavity. Here, the H-3 and H-5 experience large changes in shielding which they may be affected by the strong ring-current effects when the benzene ring of naringin is deeply embedded inside the cyclodextrin cavity. All of the chemical shift changes upon naringin complexation suggested a partial inclusion of naringin into the CD cavity.
A two-dimensional spectrum was used to obtain further conformational information. The intermolecular dipolar cross correlation (Fig. 3) provided evidence for host–guest inclusion complexation. The 2D ROESY pattern for the naringin/β-CD inclusion complex showed a strong cross-peak (green arrow) formed between the H3 and H5 protons of β-CD and the aromatic ring group of protons (H1, H2 and H3) of naringin (Fig. 3). An appreciable correlation of the H1, H2, H3, H4, H5 and H6 protons of naringin with the H3 and H5 protons of β-CD and HP-β-CD were demonstrated by the ROESY spectra. This result is in excellent agreement with the results obtained by 1H-NMR spectroscopy. Based on these observations, a favorable naringin/HP-β-CD inclusion mode was proposed (Fig. 3b).

ROESY spectrum of inclusion complexes in D2O at 25 °C, a naringin/β-CD complex, b naringin/HP-β-CD complex
SEM analysis
Naringin crystals showed a rod-like microcrystalline structure in the SEM images (Fig. 4). The surface morphology of β-CD presented a loose, irregular and uneven surface, whereas pure HP-β-CD particles showed a microsphere having a hollow structure. In contrast, the inclusion complexes appeared as irregular particles with variable sizes. The comparison of these images can be taken as a confirmation of the formation of inclusion complexes after molecular encapsulation.

SEM: A (a) naringin, (b) β-CD, (c) naringin/β-CD inclusion complex, (d) naringin/β-CD physical mixture; B (a) naringin, (b) HP-β-CD, (c) naringin/HP-β-CD inclusion complex, (d) naringin/HP-β-CD physical mixture
Phase solubility studies
The stoichiometry was studied by the phase solubility method. The obtained linear relationship of the naringin-CD complexes were classified as a typical AL-type. The regression equations were as follows (Fig. 5a, b):
where Y is the concentration (mM) of naringin and X is the concentration (mM) of CD. These results can be considered as evidence of guest–host inclusion formation at a 1:1 ratio. The thermodynamic parameters were calculated from the Gibbs Eq. (3) [23]:

Phase solubility diagrams of the naringin/β-CD complex and naringin/HP-β-CD complex
The formation of a guest–host system depends on molecular suitability and other factors, such as van der Waals interactions, hydrogen bonding, electrostatic interactions and hydrophobic interactions [30]. The calculated K c values of the naringin/β-CD and naringin/HP-β-CD complexes were 194.12 and 653.05 M−1, respectively. The higher K c value suggests that HP-β-CD could form more stable inclusion complexes with naringin in aqueous solutions. Furthermore, the Gibbs free energy (∆G) upon the formation of the naringin-CD system was calculated by Eq. (3); the values were −13,100 and −16,160 J/mol, respectively. The negative ∆G value suggests an energetically favorable complexation process.
Molecular dynamics (MD) simulations
Four different orientations (Fig. 6) significantly contributing to the molecular complexation of CD with naringin are discussed in this paper. For orientation a, the glycoside group of naringin was included in gravity from the wider rim of CD. For orientation b, the phenyl group of naringin was included in gravity from the wider rim of CD. In contrast, the location of the glycoside group was close to the wider rim of CD in orientation c. The location of the phenyl group was close to the wider rim of CD in orientation d.

The orientations of naringin-CD inclusion complexes a naringin/β-CD complex. b Naringin/HP-β-CD complex
Figure 7 shows the distances between naringin and an individual CD molecule in the four orientations (a, b, c and d) shown in Fig. 6, which indicates the motion of the host–guest molecules during 55 ns of simulation time. The distances were measured from the center of mass of naringin to the center of mass of CD. The center of mass of β-CD was at approximately 3 Å since the height of the optimized skeletal structure of β-CD was 6 Å. In this study, distances of <3 Å were seen as an inclusion formation in which the phenyl group of naringin remained inside the CD cavity. However, inclusion complexes could also be constituted at a distance >3 Å and <6 Å [27].

Distances between center of mass of naringin and CD molecules in four orientations a naringin/β-CD complex. b Naringin/HP-β-CD complex. (\(\overline{\text{x}}\) is the average distance and σ is the corresponding standard deviation)
Figure 7A shows the distances between naringin and β-CD. The plot shows that the distances between the host and guest molecules in orientation a, b, c and d were 5.03 Å, 5.12 Å, 6.78 Å and 5.39 Å, respectively. In contrast, the distances between naringin and HP-β-CD in Fig. 7b were closer, from 1.94 to 5.38 Å. These results show that orientation b, in which the phenyl group of naringin remains inside the CD cavity was most favorable. An inclusion complex was successfully formed with orientation b, where the phenyl group remained inside the HP-β-CD cavity every time, with a distance between the molecules of approximately 2 Å.
The computational results provided evidence that CDs and naringin could constitute stable host–guest inclusion complexes in aqueous solution. Figure 7 shows that naringin can form inclusion complexes with β-CD and HP-β-CD molecules, where orientation b is the most favorable orientation. The simulation indicated that the phenyl group of naringin was inserted in the cavity of the wider rim of HP-β-CD. The lower Kc value of naringin/β-CD agreed well with the simulation. Moreover, the experimental data obtained from the NMR, DSC, UV and FTIR analyses also indicated a similar geometric structure of the naringin/CDs inclusion complexes.
Hydrogen bonding has been extensively studied to investigate the structure of biomolecules at the atomic level. MD simulations have played an important role in delineating the hydrogen bonding pattern of a host–guest molecular system in aqueous solution [31]. The number of hydrogen bonds (H-bonds) in the naringin/CD complex were calculated by the common geometric restrictions with the definition of the distance between the donor and the acceptor (rHB) <0.35 nm and a deviation from linearity of <30° [32, 33]. Thus, the number of hydrogen bonds was calculated with the conditions of 3.0 Å distance and an angle of 20°. Figure 8 shows the number of hydrogen bonds in the last 55 ns and 1 μs for the naringin/β-CD and naringin/HP-β-CD systems. The results show that complexation increased the number of hydrogen bonds from 1 to 3, and suggest that the naringin/HP-β-CD complex is much more stable because of the greater the number of hydrogen bonds (from 1 to 3) caused by the persistent hydrogen bonds during complexation between CD and naringin in orientation b.

The numbers of hydrogen bonds of the naringin-CD complexes over the simulation time a naringin/β-CD complex. b Naringin/HP-β-CD complex
Figure 9 shows the populations of 10 clusters which were evaluated by RMSD in cluster analysis. Figure 10 illustrates the lowest energy structures of the complexes extracted from the highest clustering analyses in orientations a, b, c and d. The structures were selected in accordance with the highest population obtained from the MDS trajectories. In orientations a, c, and d, naringin was not well-defined inside the CD cavity, while for both inclusion complex structures in orientation b, the naringin molecules were located in the CD cavity. The illustration also agrees with the phase-solubility results (Fig. 5) in that orientation b had the best possible configuration. Thus, only orientation b was considered further in this study.

Populations of each cluster from the clustering analysis using root mean square deviation (RMSD) of naringin-CD complexes a naringin/β-CD complex. b Naringin/HP-β-CD complex

The highest populated clusters of four orientations of naringin-CD complexes a naringin/β-CD complex. b Naringin/HP-β-CD complex
The inclusion complexation mechanisms of naringin with β-CD and HP-β-CD were investigated using MS simulations. The distances and number of H-bonds indicate that the naringin/HP-β-CD complex has excellent stability and that orientation b is the best guest/host binding mode. All MD simulation results indicated that naringin/β-CD and naringin/HP-β-CD existed in a 1:1 stoichiometry in aqueous solution. Additionally, it also suggested that the naringin-HP-β-CD complex is more stable because of the effect of the hydroxypropyl group at the C6 position on the glucopyranose of HP-β-CD in orientation b.
Molecular modeling and thermodynamic parameter studies
As described above, different analytical methods were employed to investigate the interactions between naringin and CD. Quantum mechanical (density functional theory; DFT) calculations were executed to assess the binding affinities of the naringin/CD system. The optimized HOMO and LUMO structures of the naringin molecule and the stable inclusion complexes are shown in Figs. S3 and 4. The structures obtained from molecular docking (Fig. S3) are consistent with the results of NMR (Fig. 3).
The energy gap (E HOMO − E LUMO) reveals the activity and stability of a molecular fragment. A negative energy gap (−4.53 eV) revealed that the naringin/HP-β-CD complex was stable. This conclusion was consistent with the phase-stability results. The HOMO and LUMO energy orbital diagrams of the naringin molecule are shown in Fig. S4.
The ΔE, ΔH, ΔG and ΔS values of the inclusion complexes extracted from the optimization process were calculated using Eqs. 4, 5, 6 and 7.
Using the DTF method, ΔE, ΔH, ΔG and ΔS for the most stable complexes (naringin/β-CD, and naringin/HP-β-CD) are shown in Table 2. The negative value for the binding energy indicates a thermodynamically favorable complexation process. The difference in binding energies (ΔΕ) for the 1:1 inclusion complexes was 18.62 kcal/ mol, indicated that the naringin/HP-β-CD complex was more stable than the naringin/β-CD complex. The negative values of ΔH and ΔS indicate that inclusion formation was an exothermic reaction and an enthalpy-driven process.
The ∆G values obtained from UV–Vis analysis were both negative, while positive value of ΔG was obtained for naringin/β-CD system in molecular modeling. The ∆G values obtained from UV–Vis analysis were both negative, the more negative ∆G value suggested an energetically favorable complexation process, and indicated that HP-β-CD could form more stable inclusion complexes with naringin in aqueous solutions. The ∆G values obtained from molecular modeling, the more negative ∆G value for naringin/HP-β-CD system also suggested that the naringin/HP-β-CD complex was more stable than the naringin/β-CD complex. To a certain extent, the DFT calculation results were consistent with the UV–Vis experimental results. The negative ∆G value suggests an energetically favorable complexation process. However, since the exact experimental conditions (for instance, pH, temperature, measured techniques etc.) are difficult to be completely simulated in computational studies, it is reasonable that the molecular modeling result has the quantitative difference with the experimental result. The positive value of ΔG may indicate that naringin/β-CD formation was a non-spontaneous process.
Solubilization
Additionally, UV–Vis analysis were carried out to investigate the solubilization effect of cyclodextrin [34]. An excess amount of naringin/CD inclusion complex was dissolved in distilled water at room temperature until equilibrium was reached. After removal of the solid particles by filtration and dilution of the filtrate, the amount of naringin was determined by UV spectrophotometry at 282 nm. The apparent solubility of naringin/CD inclusion complex was calculated by the standard curve of naringin (A = 18.86C + 0.04). The apparent solubility of naringin (S0 = 0.6502 mg/ml) was remarkably increased to 4.03 mg/ml (naringin/β-CD), 6.43 mg/ml (naringin/HP-β-CD) respectively. This result indicated that the water-soluble CD improved the aqueous solubility of naringin effectively.
4. Conclusion
In conclusion, experimental and computational studies were conducted to characterize naringin/cyclodextrin inclusion complexation behavior. The experimental results indicate that the phenyl group of naringin is entrapped in the CD cavity with 1:1 stoichiometry. In comparison to native β-CD, HP-β-CD was found to increase the solubility of naringin effectively in aqueous solution. The obtained orientations of naringin inside CD cavities were compared with available experimental data to verify the simulation models. In addition, it was found that the insertion of a phenyl ring inside the cavity of CD was more favorable, although there were various possible inclusion geometries of the naringin/cyclodextrin complexes. The MD simulations and the HOMO and LUMO orbital investigations confirmed the orientation and stability of the inclusion complexes. The combined experimental and computational methods represent a prospective approach for further studies.
References
Ji, Y., Wang, L., Watts, D.C., Qiu, H., You, T., Deng, F., Wu, X.: Controlled-release naringin nanoscaffold for osteoporotic bone healing. Dent. Mater. 30, 1263 (2014)
Leem, E., Nam, J.H., Jeon, M., Shin, W., Won, S., Park, S., Choi, M., Jin, B.K., Jung, U.J., Kim, S.R.: Naringin protects the nigrostriatal dopaminergic projection through induction of GDNF in a neurotoxin model of Parkinson’s disease. J. Nutr. Biochem. 25, 801 (2014)
Özyürek, M., Akpınar, D., Bener, M., Türkkan, B., Güçlü, K., Apak, R.: Novel oxime based flavanone, naringin-oxime: synthesis, characterization and screening for antioxidant activity. Chem-Biol. Interact. 212, 40 (2014)
Cao, X., Lin, W., Liang, C., Zhang, D., Yang, F., Zhang, Y., Zhang, X., Feng, J., Chen, C.: Naringin rescued the TNF-α-induced inhibition of osteogenesis of bone marrow-derived mesenchymal stem cells by depressing the activation of NF-кB signaling pathway. Immunol. Res. 62, 357 (2015)
Shpigelman, A., Shoham, Y., Israeli-Lev, G., Livney, Y.D.: β-Lactoglobulin–naringenin complexes: nano-vehicles for the delivery of a hydrophobic nutraceutical. Food Hydrocolloid 40, 214 (2014)
Sangpheak, W., Kicuntod, J., Schuster, R., Rungrotmongkol, T., Wolschann, P., Kungwan, N., Viernstein, H., Mueller, M., Pongsawasdi, P.: Physical properties and biological activities of hesperetin and naringenin in complex with methylated beta-cyclodextrin. Beilstein. J. Org. Chem. 11, 2763 (2015)
Lee, S.J., Kim, J., Kim, M.J., Kitaoka, M., Park, C.S., Lee, S.Y., Ra, M., Moon, T.W., Robyt, J.F., Park, K.H.: Transglycosylation of naringin by bacillus stearothermophilus maltogenic amylase to give glycosylated naringin. J. Agric. Food. Chem. 47, 3669 (1999)
Felton, L.A., Popescu, C., Wiley, C., Esposito, E.X., Lefevre, P., Hopfinger, A.J.: Experimental and computational studies of physicochemical properties influence NSAID-cyclodextrin complexation. AAPS PharmSciTech 15, 872 (2014)
Fernandes, A., Ivanova, G., Bras, N.F., Mateus, N., Ramos, M.J., Rangel, M., de Freitas, V.: Structural characterization of inclusion complexes between cyanidin-3-O-glucoside and beta-cyclodextrin. Carbohydr. Polym. 102, 269 (2014)
Kicuntod, J., Khuntawee, W., Wolschann, P., Pongsawasdi, P., Chavasiri, W., Kungwan, N., Rungrotmongkol, T.: Inclusion complexation of pinostrobin with various cyclodextrin derivatives. J. Mol. Graph. Model. 63, 91 (2016)
Sangpheak, W., Khuntawee, W., Wolschann, P., Pongsawasdi, P., Rungrotmongkol, T.: Enhanced stability of a naringenin/2,6-dimethyl beta-cyclodextrin inclusion complex: molecular dynamics and free energy calculations based on MM- and QM-PBSA/GBSA. J. Mol. Graph. Model. 50, 10 (2014)
Whang, H.S., Vendeix, F.A., Gracz, H.S., Gadsby, J., Tonelli, A.: NMR studies of the inclusion complex of cloprostenol sodium salt with beta-cyclodextrin in aqueous solution. Pharm. Res. 25, 1142 (2008)
Savic, I.M., Savic-Gajic, I.M., Nikolic, V.D., Nikolic, L.B., Radovanovic, B.C., Milenkovic-Andjelkovic, A.: Enhencemnet of solubility and photostability of rutin by complexation with β-cyclodextrin and (2-hydroxypropyl)-β-cyclodextrin. J. Incl. Phenom. Macro. 86, 33 (2016)
Savic-Gajic, I., Savic, I.M., Nikolic, V.D., Nikolic, L.B., Popsavin, M.M., Kapor, A.J.: Study of the solubility, photostability and structure of inclusion complexes of carvedilol with β-cyclodextrin and (2-hydroxypropyl)-β-cyclodextrin. J. Incl. Phenom. Macro. 86, 7 (2016)
Savic, I.M., Nikolic, V.D., Savic-Gajic, I., Nikolic, L.B., Radovanovic, B.C., Mladenovic, J.D.: Investigation of properties and structural characterization of the quercetin inclusion complex with (2-hydroxypropyl)-β-cyclodextrin. J. Incl. Phenom. Macro. 82, 383 (2015)
Tačić, A., Savić, I., Nikolić, V., Savić, I., Ilić-Stojanović, S., Ilić, D., Petrović, S., Popsavin, M., Kapor, A.: Inclusion complexes of sulfanilamide with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin. J. Incl. Phenom. Macro. 80, 113 (2014)
Xiao, C., Li, K., Huang, R., He, G., Zhang, J., Zhu, L., Yang, Q., Jiang, K., Jin, Y., Lin, J.: Investigation of inclusion complex of epothilone A with cyclodextrins. Carbohydr. Polym. 102, 297 (2014)
Zhang, J., Li, K., Jiang, K., Cong, Y., Pu, S., Xie, X., Jin, Y., Lin, J.: Development of an oral satraplatin pharmaceutical formulation by encapsulation with cyclodextrin. RSC Adv. 6, 17074 (2016)
Jiao, A., Zhou, X., Xu, X., Jin, Z.: Molecular dynamics simulations of cyclodextrin–cumene hydroperoxide complexes in water. Comput. Theor. Chem. 1013, 1 (2013)
Boonyarattanakalin, K., Wolschann, P., Toochinda, P., Lawtrakul, L.: Molecular dynamics simulations of UC781-cyclodextrins inclusion complexes in aqueous solution. Eur. J. Pharm. Sci. 47, 752 (2012)
Dandawate, P., Vemuri, K., Khan, E.M., Sritharan, M., Padhye, S.: Synthesis, characterization and anti-tubercular activity of ferrocenyl hydrazones and their beta-cyclodextrin conjugates. Carbohydr. Polym. 108, 135 (2014)
Higuchi, T., Connor, K.A. (1965) Phase-solubility techniques. In: Reilly, C.N. (ed.) Advances in Analytical Chemistry and Instrumentation. Wiley, New York
Madan, J., Baruah, B., Nagaraju, M., Abdalla, M.O., Yates, C., Turner, T., Rangari, V., Hamelberg, D., Aneja, R.: Molecular cycloencapsulation augments solubility and improves therapeutic index of brominated noscapine in prostate cancer cells. Mol. Pharm. 9, 1470 (2012)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.A., Vreven, J.T., Kudin, K.N., Burant, J.C. (eds.): Gaussian 03, Revision C.02. Gaussian, Inc, Wallingford (2004)
Rajendiran, N., Siva, S.: Inclusion complex of sulfadimethoxine with cyclodextrins: preparation and characterization. Carbohydr. Polym. 101, 828 (2014)
Siva, S., Kothai Nayaki, S., Rajendiran, N.: Fabrication of cyclodextrins-procainamide supramolecular self-assembly: shape-shifting of nanosheet into microtubular structure. Carbohydr. Polym. 122, 123 (2015)
Boonyarattanakalin, K., Viernstein, H., Wolschann, P., Lawtrakul, L.: Influence of ethanol as a co-solvent in cyclodextrin inclusion complexation: a molecular dynamics study. Sci. Pharm. 83, 387 (2015)
Chiang, P.C., Shi, Y., Cui, Y.: Temperature dependence of the complexation mechanism of celecoxib and hydroxyl-beta-cyclodextrin in aqueous solution. Pharmaceutics. 6, 467 (2014)
Wood, D.J., Hruska, F.E., Saenger, W.: 1 H NMR study of the inclusion of aromatic molecules in α-cyclodextrin. J. Am. Chem. Soc. 99 (1977)
Liu, M., Chen, A., Wang, Y., Wang, C., Wang, B., Sun, D.: Improved solubility and stability of 7-hydroxy-4-methylcoumarin at different temperatures and pH values through complexation with sulfobutyl ether-beta-cyclodextrin. Food Chem. 168, 270 (2015)
Khuntawee, W., Wolschann, P., Rungrotmongkol, T., Wong-ekkabut, J., Hannongbua, S.: Molecular dynamics simulations of the interaction of beta cyclodextrin with a lipid bilayer. J. Chem. Inf. Model. 55, 1894 (2015)
Koehler, J.E., Saenger, W., van Gunsteren, W.F.: On the occurrence of three-center hydrogen bonds in cyclodextrins in crystalline form and in aqueous solution: comparison of neutron diffraction and molecular dynamics results. J. Biomol. Struct. Dyn. 6, 181 (1988)
Tekpinar, M., Yildirim, A., Wassenaar, T.A.: Molecular dynamics study of the effect of active site protonation on Helicobacter pylori 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Eur. Biophys. J. 44, 685 (2015)
Lee, S.J., Kim, J.C., Kim, M.J., Kitaoka, M., Park, C.S., Lee, S.Y., Ra, M.J., Moon, T.W., Robyt, J.F., Park, K.H.: Transglycosylation of naringin by Bacillus stearothermophilus maltogenic amylase to give glycosylated naringin. J. Agric. Food. Chem. 47, 3669 (1999)
Acknowledgements
This work was supported by the Program for Chang jiang Scholars and Innovative Research Team in University (No. IRT13095), the National Natural Science Foundation of China (Nos. 21442006, 21262043 and 20902079). The authors thank the High Performance Computing Center at Yunnan University for use of the high performance computing platform.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Hui-Huan Yan, Jian-Qiang Zhang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yan, HH., Zhang, JQ., Ren, SH. et al. Experimental and computational studies of naringin/cyclodextrin inclusion complexation. J Incl Phenom Macrocycl Chem 88, 15–26 (2017). https://doi.org/10.1007/s10847-017-0704-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-017-0704-x