
Abstract
Background
While the triggers for ventricular fibrillation (VF) are well-known, the substrate required for its maintenance remains elusive. We have previously demonstrated dynamic spatiotemporal changes across VF from electrical induction of VF to asystole. Those data suggested that VF drivers seemed to reside in the distal RV and LV. However, signals from these areas were not recorded continuously. The aim of this study was to map these regions of significance with stationary basket electrodes from induction to asystole to provide further insights into the critical substrate for VF rhythm sustenance in canines.
Methods
In six healthy canines, three multipolar basket catheters were positioned in the distal right ventricle (RV), RV outflow tract, and distal left ventricle (LV), and remained in place throughout the study. VF was induced via direct current application from an electrophysiologic catheter. Surface and intracardiac electrograms were recorded simultaneously and continuously from baseline, throughout VF, and until asystole, in order to get a complete electrophysiologic analysis of VF. Focused data analysis was also performed via two defined stages of VF: early VF (immediately after induction of VF to 10 min) and late VF (after 10 min up to VF termination and asystole).
Results
VF was continuously mapped for a mean duration of 54 ± 9 min (range 42–70 min). Immediately after initiation of VF in the early phase, the distal LV region appeared to drive the maintenance of VF. Towards the terminal stage of VF, the distal RV region appeared to be responsible for VF persistence. In all canines, we noted local termination of VF in the LV, while VF on surface ECG continued; conversely, subsequent spontaneous termination of VF in the RV was associated with termination of VF on surface ECG into a ventricular escape rhythm. Continuous mapping of VF showed trends towards an increase in peak-to-peak ventricular electrogram cycle length (p = 0.06) and a decrease in the ventricular electrogram amplitude (p = 0.06) after 40 min. Once we could no longer discern surface QRS activity, we demonstrated local ventricular myocardial capture in both the RV and LV but could not reinitiate sustained VF despite aggressive ventricular burst pacing.
Conclusions
This study describes the evolution of VF from electrical initiation to spontaneous VF termination without hemodynamic support in healthy canines. These data are hypothesis-generating and suggest that critical substrate for VF maintenance may reside in both the distal RV and LV depending on stage of VF. Further studies are needed to replicate these findings with hemodynamic support and to translate such findings into clinical practice.
Graphical abstract
Ventricular fibrillation maintenance may be dependent on critical structures in the distal RV. ECG: electrocardiogram; LV: left ventricle; RV: right ventricle; RVOT: right ventricular outflow tract; VF: ventricular fibrillation

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Ventricular fibrillation (VF) remains a common cause of sudden cardiac death (SCD) [1]. Of the approximately 300,000–450,000 annual cases of SCD in the USA, up to one-third are attributed to VF [2,3,4]. Because the critical substrate required for VF maintenance remains elusive, current strategies for ablating VF are primarily focused on foci that serve as triggers for VF [5, 6]. Unfortunately, prior research on VF mapping and ablation done either in animals or humans has not been translatable into the clinical realm. Thus, a better understanding of VF in an intact large-scale animal model is needed. We previously described a novel means of VF mapping from induction to asystole in a healthy canine model. These data support a regional and temporal organization throughout, with the endocardial distal right ventricular (RV) and left ventricular (LV) regions harboring potential drivers for VF. The dominant regions of electrical activity were noted to change over the course of VF, based on sequential endocardial and epicardial mapping at different time points [7]. One of the shortcomings of the prior study was the lack of consistent and continuous mapping of VF activity in these critical regions (endocardial aspect of the distal RV and LV).
The lack of stationary mapping in these regions has left our knowledge of these critical regions incomplete. Thus, the aim of this study is to provide further insights into the critical substrate for VF rhythm sustenance.
2 Methods
2.1 Canine model for studying VF
The study conforms to the Guide for the Care and Use of Laboratory Animals and was approved by the Mayo Clinic Institutional Animal Care and Use Committee. A total of six healthy mongrel dogs (weight 25–40 kg) underwent VF mapping with the use of general anesthesia for deep sedation. One additional dog died due to a tear in the aortic arch with the placement of an 8.5-Fr Agilis NxT (Abbott, St Paul, MN) steerable sheath. Intravenous ketamine (10 mg/kg) and diazepam (0.5 mg/kg) were used for induction of anesthesia, isoflurane (1–3%) was used for maintenance of anesthesia with volume-cycled ventilation, and intramuscular buprenorphine (0.03 mg/kg) was used for analgesia. After obtaining vascular access, the canines were anticoagulated using intravenous heparin targeting an activated clotting time goal of 400–450 s (a higher goal was targeted due to extensive mapping and lack of circulation during VF). VF was induced using a standard EPT Blazer catheter (Boston Scientific, Marlborough, MA) connected to a 9-V battery. No attempt was made to resuscitate or provide hemodynamic support after induction of VF. This was done purposefully in order to provide novel insights into “real-life” in vivo VF which leads to hemodynamic instability and death. A surface electrocardiogram (ECG) was used to monitor global cardiac electrical activity and was continuously recorded during the experiments.
2.2 Mapping of VF
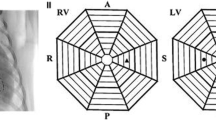
Multielectrode basket catheters were positioned endocardially in both ventricles for mapping and remained in the same position throughout the entire duration of all experiments (Fig. 1 A). These catheters were positioned as deep into the ventricles to permit contact along the complex intracavitary structures and its closely associated and overlapping His-Purkinje tissue. These regions that we refer to as the distal RV and LV are regions that we define as including the complex compartment of these ventricular regions including the RV moderator band, right- and left-sided papillary muscles, false tendons, and the distal Purkinje tissue. The locations of the mapping catheters were confirmed by fluoroscopy: a 38 mm Constellation catheter (Boston Scientific, Marlborough MA—64 electrodes on eight splines) was placed in the distal RV, a 60 mm Constellation catheter (Boston Scientific, Marlborough MA—64 electrodes on eight splines) was positioned in the distal LV, and an Orion catheter (Boston Scientific, Marlborough MA—64 electrodes on eight splines) was positioned in the RVOT (Fig. 1 C). A retrograde aortic approach was used to access the LV in all canines.

A and B Gross anatomy specimen of the canine heart showing placement of the multielectrode basket mapping catheters in the distal right ventricle (RV), the right ventricular outflow tract (RVOT), and the distal left ventricle (LV). Note the complex anatomy of these distal regions including the moderator band, false tendons, papillary muscle, and Purkinje tissue (labeled in the figure). C Fluoroscopic image (LAO view) illustrating the placement of the multielectrode basket mapping catheters. D Position of the multielectrode basket mapping catheters as displayed on the electroanatomical (EAM) map created using the Rhythmia EAM system
An electroanatomic mapping system (EAM) (Rhythmia, Boston Scientific, Natick, MA; Fig. 1 D) and an electrophysiology recording system (Prucka Cardiolab recording system, GE Healthcare, Wauwatosa, WI) were used to record the distribution of electrical signals. Bipolar electrograms (EGMs) were obtained using a high-pass filter of 0.05 Hz and a low-pass filter of 1000 Hz, and notch filtering was turned off, all in order to optimize the recording of low amplitude ventricular EGMs and high-frequency His-Purkinje signals.
In all canines, a comprehensive baseline voltage map in sinus rhythm was performed to demonstrate normal voltage in both ventricles prior to the VF study. Catheters were then placed and confirmed using fluoroscopy, echocardiography, and an EAM system to be in the distal RV, distal LV, and RVOT. Once catheters were in position, they were not moved throughout the duration of the study. VF was induced using a separate EPT Blazer catheter (Boston Scientific, Natick, MA) positioned in the RV and connected to a 9-V battery. The basket catheters remained in position in the RVOT, and distal RV and LV in order to permit simultaneous and continuous recording of VF in all studies. Intracardiac EGMs were recorded continuously from baseline, throughout VF, and until asystole occurred.
2.3 Definition of early and late stage of VF
Focused data analysis was also performed via two defined stages of VF: early VF (immediately after induction of VF to 10 min) and late VF (after 10 min up to VF termination and asystole). These cutoffs were implemented in order to evaluate and compare both early and later stages of VF. In addition, a comparison of the first 10 min to the terminal portion of VF was performed to appreciate rhythm dynamics at an early stage, where ischemia and metabolic derangements were expected to be less pronounced (early VF), while still capturing the full evolution of VF throughout a “real-life” course of complete mapping of this rhythm.
2.4 Data measurements and signal analysis
Full Rhythmia and Prucka data were available in five animals. In one animal, the data was not completely stored due to a technical problem and was not included in the signal analysis. Signal measurements were performed on the Prucka software by three authors (FME, ZJ, and JT). Both ventricular and His-Purkinje system (HPS) signals were noted throughout VF. For HPS signals analysis, the electrodes with the clearest Purkinje signals in each ventricle were used. Purkinje EGMs were easily discerned in all animals immediately after VF induction only. It was difficult to discern each HPS signal to obtain an accurate cycle length of these signals unless clearly dissociated from the ventricular EGMs. Thus, given the inability to discern accurately the HPS signals from the ventricular signals consistently, we used the ventricular EGMs for cycle length, amplitude, and regularity index measurements. The ventricular cycle length (CL) was measured from at least one set of 10 consecutive ventricular EGMs obtained from 4 regions in the LV (distal LV, LV septum, LV anterior wall, and LV inferior wall), and 4 regions in the RV (distal RV, RV septum, RV free wall, and RVOT).
The CLs were measured as the interval between two consecutive positive or negative deflections that departed from the baseline. Ventricular amplitude was measured from the peak or trough of the ventricular signal to baseline; whichever was higher. A regularity index (RI) was calculated and used to assess the regularity of the CL. The RI was defined as the percentage of ventricular signals of which the CL was within one standard deviation (SD) of the means of the mean CL for each specific region [8].
2.5 Statistical analysis
Continuous variables were expressed as mean ± SD. The ventricular CLs and voltage amplitudes were compared throughout VF to the baseline CLs and voltage amplitudes using the Wilcoxon signed-rank test. In 5 to 10-min intervals, the ventricular CLs and voltage amplitudes in the RVOT and distal RV and LV were compared using the Wilcoxon sum-rank test. A p-value less than 0.05 was considered statistically significant. Statistical analysis was done using BlueSky Statistics – version 7.4 (BlueSky Statistics LLC, Chicago, IL, USA).
3 Results
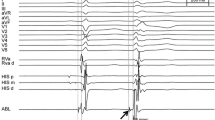
In six healthy canines, VF mapping was performed continuously using multielectrode mapping basket catheters placed in the RVOT and distal RV and LV. The mean duration from induction of VF to asystole was 54 ± 9 min (range 42–70 min). In all six canines, VF terminated in the LV prior to the RV. Despite local termination of VF with the LV being in a ventricular escape rhythm, VF persisted in the RV and on the surface ECG (Fig. 2, middle panel). The mean duration of VF activity in the LV was 49 ± 6 min, whereas the mean duration of VF activity in the RV was 54 ± 9 min (Table 1). With spontaneous termination of VF activity in the RV to a ventricular escape rhythm, there was a cessation of VF with an escape rhythm noted on the surface ECG (Fig. 2, bottom panel). Post-VF termination, ventricular capture with pacing in both the RV and LV was demonstrated. However, despite aggressive ventricular burst pacing at cycle lengths from 400 ms down to 200 ms in both the RV and LV, we were unable to reinduce sustained VF (Fig. 3).

Electrograms from a representative canine study of simultaneous endocardial biventricular mapping during ventricular fibrillation (VF). Top panel: electrograms at the start of VF. Middle panel: VF termination in the left ventricle (LV). Bottom panel: VF termination in the right ventricle (RV)

After termination of ventricular fibrillation (VF) – right ventricular (RV) (top panel) and left ventricular (LV) (middle panel) high-output pacing with local myocardial capture. No reinduction of VF with aggressive burst pacing from the RV (bottom panel)
3.1 Ventricular CL variability
3.1.1 Early VF
The regional differences of ventricular CL during VF are summarized in Table 2 and Fig. 4 A. Immediately after induction of VF, the shortest ventricular CL occurred in the distal LV (92 ± 6 ms). The ventricular CLs were not significantly different in the RVOT, nor the other RV and LV regions, when compared to the distal LV (Table 2). After 5 min of VF, the shortest ventricular CL occurred in the RV free wall (79 ± 7 ms), and the CL of VF was significantly slower in the distal LV (91 ± 5 ms, p = 0.016), the LV septum (90 ± 6 ms, p = 0.016), the LV inferior wall (110 ± 24 ms, p = 0.016), and the RVOT (101 ± 14 ms, p = 0.012), comparatively. After 10 min of VF, the shortest ventricular CL occurred in the RV free wall (72 ± 6 ms), and the CL of VF was significantly slower in the distal LV (83 ± 5 ms, p = 0.016), the LV septum (87 ± 7 ms, p = 0.008), the LV anterior wall (84 ± 3 ms, p = 0.012), and the RVOT (110 ± 25 ms, p = 0.008).

Variation of the average ventricular cycle length (ms) (A) and average ventricular voltage (mV) (B) with time (minutes)
3.1.2 Late VF
Across the rest of VF, the shortest ventricular CL was in the RV septum (82 ± 15 ms) after 20 min, the distal RV (99 ± 22 ms) after 30 min, the RV free wall (130 ± 21 ms) after 40 min, and the distal RV (111 ± 2 ms) after 50 min of VF. There was no significant difference in the ventricular CLs in the RVOT and the different LV and RV regions as compared to the regions with the shortest CLs at each time interval during this period (20 to 50 min of VF).
There was a trend towards an increase in the ventricular CLs over time (Table 2 and Fig. 4 A) from an average of 95 ± 8 ms in all 5 canines immediately after VF induction to an average of 154 ± 33 ms after 40 min of VF (p = 0.063). Mapping data for individual animal experiments are available in Supplemental Tables.
3.2 Purkinje CL variability
Purkinje CL variability after VF induction in the right and left ventricles is summarized in Table 3. Immediately after VF induction, Purkinje fibers had a faster firing rate in the LV as compared to the RV in all animals. The mean Purkinje CL in the LV was 87.3 ± 7.9 ms, whereas the mean Purkinje CL in the RV was 97.5 ± 12.1 ms (Table 3). Data about Purkinje CL variability throughout the rest of VF is summarized in Supplemental Table VII.
3.3 Regionality of RI
The regional differences of RI during VF are summarized in Table 4. The ventricular signals were most regular in early VF in the LV inferior wall and RV septum immediately after induction of VF (RI = 74%), in the RVOT after 5 min of VF (RI = 78%) and 10 min of VF (RI = 80%). In late VF, the LV anterior wall after 20 min of VF (RI = 74%), in the LV inferior wall after 30 min of VF (RI = 78%), and in the distal LV after 40 min of VF (RI = 76%).
3.4 Ventricular amplitude
Prior to the induction of VF, comprehensive mapping in sinus rhythm demonstrated normal biventricular voltage in all canines. In early VF, the largest voltage amplitude was in the LV anterior wall immediately after VF induction (10.60 ± 6.2 mV), in the distal LV after 5 min of VF (4.75 ± 2.5 mV), and in the LV septum after 10 min of VF (3.45 ± 2.5 mV). In late VF, the largest voltage amplitude was in the RV free wall after 20 min of VF (1.96 ± 1.7 mV), in the distal LV after 30 min of VF (1.44 ± 0.7 mV), and in the RV free wall after 40 and 50 min of VF (1.37 ± 1.6 mV and 1.88 ± 1.8 mV, respectively).
The ventricular voltage amplitudes were not significantly different in the RVOT, nor the other RV and LV regions, when compared to the region with the largest voltage amplitude at every time interval except after 30 min of VF, when the RVOT signals were significantly smaller in amplitude as compared the distal LV signals (0.35 ± 0.5 versus 1.44 ± 0.7 mV, p = 0.032). There was a trend towards a decrease in the amplitude of ventricular signals over time (Table 5 and Fig. 4 B) from an average of 7.3 ± 1.59 mV in all 5 canines immediately after VF induction to an average of 1.09 ± 0.3 mV after 40 min of VF (p = 0.063).
4 Discussion
We report simultaneous and continuous comprehensive endocardial mapping of the RVOT, and distal RV and LV from induction of VF to asystole without hemodynamic support or attempt to resuscitate. This study provides novel insights into the evolution of VF in order to understand its dynamics as it leads to death. These data suggest that in healthy canines, the regions that are responsible for the maintenance of VF may be in the distal LV in early VF and in the distal RV in late VF.
The novel findings from our study are summarized as follows:
-
1.
Immediately after VF induction, the distal LV appeared to be driving the maintenance of VF. Whereas, the distal RV was responsible for the maintenance of the later stage of VF to termination.
-
2.
In all canines, local termination of VF occurred in the LV prior to the distal RV. Despite the termination of VF to a ventricular escape rhythm in the LV, VF continued based on the surface ECG and locally in the distal RV. Only when VF terminated locally in the distal RV, did VF termination on surface ECG occur.
-
3.
Despite a long period of continuous VF, after the rhythm terminated to a ventricular escape rhythm, we were still able to demonstrate the capture of ventricular myocardium in both the distal RV and LV, suggestive of at least some excitable ventricular myocardium. Despite aggressive burst pacing, we could no longer induce sustained VF.
These data may suggest that the substrate necessary for VF maintenance in healthy canines may include the distal LV in the initial stages of VF as well as the distal RV in the late stages of VF. We postulate that a critical portion of the HPS and/or ventricular myocardium are necessary for the sustenance of VF, and it is possible that hypoperfusion and ensuing ischemia eliminated these critical regions and resulted in spontaneous termination of VF. Furthermore, after elimination of the critical regions, VF could no longer be induced despite aggressive ventricular burst pacing from either ventricle, supporting this potential association.
4.1 Dynamic spatiotemporal changes during VF in canines without hemodynamic support or attempts at resuscitation
In our previous VF mapping studies, we used a process of sequential rather than simultaneous mapping, in order to collect data from both ventricles, in both endocardial and epicardial fashion. We noted that the regions with the shortest ventricular CLs were in the endocardial aspect of the RV during the early stages of VF (0–30 min) followed by a transition to the endocardial aspect of the LV during the later stages of VF (30–60 min) (7). Thus, focusing our attention on the endocardium rather than the epicardium, we performed biventricular endocardial mapping in this study and found that the shortest cycle lengths similarly were located in the RV during the early stages of VF (0–30 min); however, this also persisted into the later stages of VF (30–50 min) in contrast to our prior study (Table 2). These findings are to be interpreted in the context of different mapping methods, with the previous study utilizing sequential mapping with continued movement, and the current study utilizing continuous and simultaneous mapping with catheters remaining in place throughout the study.
4.2 Potential role of the right ventricle and His-Purkinje system in VF maintenance
Historically, the RV has received less attention with the LV being the focus during VF mapping studies. Krummen et al. [9] have suggested that VF is sustained by LV rotors with RV rotors being rare and unstable. However, most dogs in that study underwent LV mapping only. Similar to our study, mapping was done in vivo using multielectrode basket catheters in the LV and RV. However, in the prior study mapping was done for the first 10 s of VF or until VF terminated, whichever came first. This contrasts with our study in which VF was mapped for much longer periods (almost an hour) and until asystole.
Several explanations support the role of the RV in the genesis and maintenance of VF. Embryologically, the right bundle branches and right Purkinje fibers are derived from the second heart field similar to the sinoatrial node and the RVOT [10], and may be one reason for their increase in arrhythmogenicity as compared to the left bundle branches and left Purkinje fibers which are derived from the first heart field.
From a gross anatomic perspective, the increased trabeculation and the presence of the moderator band as a bridge between the RV free wall and the septum allowing for reentry can also support the role of the RV in maintaining VF. Furthermore, the anterior papillary muscle in the RV can facilitate the initiation of reentry and anchoring of the reentrant wavefront due to source-sink mismatch. Kim et al. [11] showed that tissue mass reduction and procainamide converted VF to VT in isolated swine RVs by preventing spontaneous wave breaks and allowing the anchoring of a single reentrant wavefront to the papillary muscle. Trimming of the RV papillary muscle to the level of the endocardium resulted in the inability to induce VT [11].
The HPS of the mammalian heart consists of specialized Purkinje cells with properties that are distinct from ventricular myocytes and allow for rapid transmission of action potentials [12]. Previous studies have implicated the Purkinje system as a potentially crucial component in the initiation and propagation of ventricular arrhythmias [5, 8, 13,14,15]. Supportive data has shown that the destruction of parts of the HPS impacted the maintenance of VF [16,17,18,19]. The HPS has also been shown to be relatively refractory (or resistant) to ischemia, maintaining conduction during longer periods of hypoxia compared to other cardiac tissue [20] and exhibiting persistent action potentials after infarction of surrounding ventricular tissue [21].
Premature ventricular contractions (PVCs) have been implicated in the initiation of VF both in normal hearts and ischemic or infarcted hearts [6, 22]. Malignant PVCs have been mapped to originate in the papillary muscles of both ventricles and the RV moderator band [13, 23,24,25], and when mapped, often exhibit early Purkinje signals. Moreover, limited studies in patients with idiopathic VF have shown local abnormal electrograms as VF drivers most commonly in the RV [26]. Our current study is consistent with HPS electrical activity as potentially being a driver or at least necessary for VF maintenance and alludes to the notion that the distal RV may contain the critical substrate for the late stages of VF.
4.3 Clinical implications and translation into the clinical arena
These data provide novel insights into VF dynamics which we attempted to study without hemodynamic support in a more “real-life” setting as it occurs in nature and leads to death. These data are significant as they inform us about the evolution of VF compared to our prior work using simultaneous rather than sequential mapping of both ventricles. Such insights may be exploited with the use of hemodynamic support so that VF can be mapped and intervened in clinical practice. Such studies are currently being performed in our lab in order to make such an approach more clinically feasible.
5 Limitations
Our study is best to be interpreted in the context of several limitations. Due to the animal committee restrictions associated with hypothesis-generating experiments, a limited number of canine experiments were able to be performed. From an experimental standpoint, we could not define with certainty which cardiac walls/regions were in contact with the catheters due to the small chamber size of the ventricles in canines. This will require larger animal hearts and improved mapping resolution techniques. The spatial resolution of the 64-electrode basket catheters may miss activation patterns around or within complex anatomical geometry such as the papillary muscles and may not be positioned appropriately to record Purkinje potentials [27].
To avoid catheter movement as well as difficulty in catheter manipulation without blood flow, reinduction of VF at the end of the experiments was done via aggressive burst pacing using one of the mapping catheters whereas VF induction at the beginning of the experiments was done by using an EPT Blazer catheter positioned in the RV and connected to a 9-V battery. Using different methods for VF induction at the beginning and end of the experiments may also be a limitation. No attempt was made to resuscitate or perfuse the animals; hence, the VF patterns were certainly affected by ischemia. This was purposefully done to give novel insights into VF dynamics in the natural setting, albeit in a canine model under deep sedation. Ischemia and significant cell damage may have also limited reinduction of VF at the end of the experiments. However, this experimental setup does give us the rare ability to study VF as would occur in a patient that suffered SCD where VF leads to ischemia and continues until asystole. A cardiac circulatory bypass model or a left ventricular assist device would be necessary to mitigate this effect.
General anesthesia may have also affected the pattern of VF; however, it is required per humane euthanasia guidelines. The choice of the animal model may have also impacted the VF pattern particularly due to the different cardiac anatomy in canines. Therefore, validation in other animal species would be important. Lastly, even though the use of in vivo animal models for VF mapping facilitates the translation of the research findings into clinical practice, the results cannot be extrapolated to humans because spontaneous VF may have a different pattern compared to that of induced VF [28]. Thus, whether these findings are consistent with human VF remains undetermined. However, given the lethality of VF and the extreme difficulty of attempting to perform full mapping studies, these data are essential to advance our knowledge of VF as we work towards understanding VF in humans. Despite these limitations, we believe these novel studies will provide a significant addition to the field and literature to continue our quest to better understand and potentially treat VF.
6 Conclusions
This study describes the evolution of VF from electrical initiation to spontaneous VF termination without hemodynamic support in healthy canines. These data are hypothesis-generating and suggest that critical substrate for VF maintenance may reside in both the distal RV and LV depending on the stage of VF. Further studies are needed to replicate these findings with hemodynamic support and to translate such findings into clinical practice.
Data availability
The data that support the findings of this paper are available from the corresponding author, [CVD], upon reasonable request.
Abbreviations
- CL:
-
Cycle length
- EAM:
-
Electroanatomic mapping
- ECG:
-
Electrocardiogram
- EGM:
-
Electrogram
- EP:
-
Electrophysiology
- HPS:
-
His-Purkinje system
- ICD:
-
Implantable cardioverter-defibrillator
- LV:
-
Left ventricle
- PTSD:
-
Post-traumatic stress disorder
- PVC:
-
Premature ventricular contraction
- RI:
-
Regularity index
- RV:
-
Right ventricle
- RVOT:
-
Right ventricular outflow tract
- SCD:
-
Sudden cardiac death
- VF:
-
Ventricular fibrillation
References
Kong MH, Fonarow GC, Peterson ED, et al. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol. 2011;57:794–801.
Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–63.
Fishman GI, Chugh SS, Dimarco JP, et al. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation. 2010;122:2335–48.
Tang PT, Shenasa M, Boyle NG. Ventricular arrhythmias and sudden cardiac death. Card Electrophysiol Clin. 2017;9:693–708.
Haïssaguerre M, Shah DC, Jaïs P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet. 2002;359:677–8.
Haïssaguerre M, Shoda M, Jaïs P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2022;106:962–7.
Tan NY, Christopoulos G, Ladas TP, et al. Regional and temporal variation of ventricular and conduction tissue activity during ventricular fibrillation in canines. Circ Arrhythm Electrophysiol. 2021;14:e010281.
Tri J, Asirvatham R, DeSimone CV, et al. Intramural conduction system gradients and electrogram regularity during ventricular fibrillation. Indian Pacing Electrophysiol J. 2018;18:195–200.
Krummen DE, Hayase J, Vampola SP, et al. Modifying ventricular fibrillation by targeted rotor substrate ablation: proof-of-concept from experimental studies to clinical VF. J Cardiovasc Electrophysiol. 2015;26:1117–26.
Liang X, Evans SM, Sun Y. Insights into cardiac conduction system formation provided by HCN4 expression. Trends Cardiovasc Med. 2015;25:1–9.
Kim YH, Xie F, Yashima M, et al. Role of papillary muscle in the generation and maintenance of reentry during ventricular tachycardia and fibrillation in isolated swine right ventricle. Circulation. 1999;100:1450–9.
Boyden PA, Hirose M, Dun W. Cardiac Purkinje cells. Heart Rhythm. 2010;7:127–35.
Haïssaguerre M, Duchateau J, Dubois R, et al. Idiopathic ventricular fibrillation: role of Purkinje system and microstructural myocardial abnormalities. JACC Clin Electrophysiol. 2020;6:591–608.
Berenfeld O, Jalife J. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3-dimensional model of the ventricles. Circ Res. 1998;82:1063–77.
Haissaguerre M, Cheniti G, Hocini M, et al. Purkinje network and myocardial substrate at the onset of human ventricular fibrillation: implications for catheter ablation. Eur Heart J. 2022;43:1234–47.
Imnadze G, Zerm T. Prevention of ventricular fibrillation through de-networking of the Purkinje system: proof-of-concept paper on the substrate modification of the Purkinje network. Pacing Clin Electrophysiol. 2019;42:1285–90.
Livia C, Sugrue A, Witt T, et al. Elimination of Purkinje fibers by electroporation reduces ventricular fibrillation vulnerability. J Am Heart Assoc. 2018;7:e009070.
Dosdall DJ, Tabereaux PB, Kim JJ, et al. Chemical ablation of the Purkinje system causes early termination and activation rate slowing of long-duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2008;295:H883–9.
DeSimone CV, Asirvatham SJ. Purkinje tissue modification and ventricular fibrillation. Pacing Clin Electrophysiol. 2019;42:1291–3.
Bagdonas AA, Stuckey JH, Piera J. Amer NS, Hoffman BF. Effects of ischemia and hypoxia on the specialized conducting system of the canine heart. Am Heart J. 1961;61:206–18.
Friedman PL, Stewart JR, Fenoglio JJ Jr, Wit AL. Survival of subendocardial Purkinje fibers after extensive myocardial infarction in dogs. Circ Res. 1973;33:597–611.
Marrouche NF, Verma A, Wazni O, et al. Mode of initiation and ablation of ventricular fibrillation storms in patients with ischemic cardiomyopathy. J Am Coll Cardiol. 2004;43:1715–20.
Santoro F, Di Biase L, Hranitzky P, et al. Ventricular fibrillation triggered by PVCs from papillary muscles: clinical features and ablation. J Cardiovasc Electrophysiol. 2014;25:1158–64.
Van Herendael H, Zado ES, Haqqani H, et al. Catheter ablation of ventricular fibrillation: importance of left ventricular outflow tract and papillary muscle triggers. Heart Rhythm. 2014;11:566–73.
Sadek MM, Benhayon D, Sureddi R, et al. Idiopathic ventricular arrhythmias originating from the moderator band: electrocardiographic characteristics and treatment by catheter ablation. Heart Rhythm. 2015;12:67–75.
Haïssaguerre M, Hocini M, Cheniti G, et al. Localized structural alterations underlying a subset of unexplained sudden cardiac death. Circ Arrhythm Electrophysiol. 2018;11:e006120.
Oesterlein T, Frisch D, Loewe A, et al. Basket-type catheters: diagnostic pitfalls caused by deformation and limited coverage. Biomed Res Int. 2016;2016:5340574.
Narayan SM, Krummen DE, Rappel WJ. Clinical mapping approach to diagnose electrical rotors and focal impulse sources for human atrial fibrillation. J Cardiovasc Electrophysiol. 2012;23:447–54.
Acknowledgements
We would like to thank Renee Taubel and the veterinary staff of the Mayo Cardiovascular Innovations Laboratory for their contribution to conducting the animal experiments and Cory Scheuermann and Jamie Bush for their help with Rhythmia mapping.
Funding
This work was supported by the Earl A. Wood Career Development Benefactor Award to Dr. Christopher V. DeSimone, MD, PhD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This study was approved by the Mayo Clinic IACUC committee.
Conflict of interest
CVD, CJM, and SJA report patent filing for intellectual property related to novel tools and methods for ventricular fibrillation mapping and ablation.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ezzeddine, F.M., Ward, R.C., Jiang, Z. et al. Novel insights into the substrate involved in maintenance of ventricular fibrillation: results from continuous multipolar mapping in a canine model. J Interv Card Electrophysiol (2022). https://doi.org/10.1007/s10840-022-01333-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10840-022-01333-7