
Abstract
We have used the first-principle calculations of density functional theory within full-potential linearized augmented plane-wave method to investigate the electronic and ferromagnetic properties of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) alloys. The electronic structures of \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\) and \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}\) exhibit a half-metallic ferromagnetic character with spin polarization of 100 %. The total magnetic moment per V atom for each compound is integral Bohr magneton of 2 \(\mu _{\mathrm{B}}\), confirming the half-metallic feature of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\). Therefore, these materials are half-metallic ferromagnets useful for possible spintronics applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Spintronics is an emerging discipline of microelectronics, which explores the spin degree freedom of electrons in information processing to improve the performance of novel information storage and logic devices [1–4]. Diluted magnetic semiconductors (DMSs) have received increasing interest and are attractive materials for spintronics because they combine both magnetic and semiconductor properties. Among the half-metallic ferromagnetic materials, DMS-based III–V semiconductors are considered as the main candidates for the development of practical spintronics devices due to their important properties such as the ferromagnetic state stability at temperatures higher than the room temperature [5, 6] and the half-metallic behavior [7, 8]. In the recent years, several scientific studies have been devoted to explore the half-metallic ferromagnetism in new DMS-based III–V and II–VI semiconductors for spintronics applications [9–23].
Aluminum antimonide (AlSb) is a binary semiconductor belonging to the III–V group with wide indirect bandgap [24], and it is often used as a part of III–V compounds grown on GaSb substrates [25]. There are limited works on the electronic and magnetic properties of DMS-based AlSb; however, the half-metallic ferromagnetic property has been theoretically performed in \(\hbox {Al}_{1-x}\hbox {Mn}_{x}\hbox {Sb}\) [26] and \(\hbox {Al}_{1-x}\hbox {Cr}_{x}\hbox {Sb}\) [27]. Besides, AlSb is considered as a possible candidate DMS material according to the theoretical study of (Al, Mn)Sb [28, 29] and to the experimental work of (\(\hbox {Al}_{1-x},\hbox {Fe}_{x})\hbox {Sb}\) [30]. Katayama-Yoshida, Sato [28, 29] have predicted the stability of ferromagnetism in AlSb doped with 5 % Mn. Recently, Anh et al., [30] have experimentally studied the crystal structure, transport, and magnetic properties of (\(\hbox {Al}_{1-x}, \hbox {Fe}_{x})\hbox {Sb}\) thin films, and found an intrinsic ferromagnetism in insulating (Al, Fe)Sb which paves the way to realize a spin-filtering tunnel barrier that is compatible with well-established III–V semiconductor devices.
To the best of our knowledge, there are no experimental and theoretical studies of electronic and magnetic properties of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) alloys. The aim of this study is to predict the electronic and ferromagnetic properties of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) at concentrations of \(x = 0.25, 0.5\), and 0.75 in the zinc blende structure, using first-principle calculations of density functional theory (DFT) [31, 32].
2 Method of calculations
The theoretical calculations were performed by means of the framework of the DFT [31, 32] within the first-principle full-potential linearized, augmented plane-wave (FP-LAPW) method as implemented in WIEN2K package [33]. The generalized gradient approximation functional of Wu and Cohen (GGA-WC) was used for exchange correlation potential [34], which is more accurate for solids than for any existing GGA and meta-GGA forms [35].
The averages of non-overlapping muffin–tin radii (\(R_{\mathrm{MT}})\) of Al, Sb and V have been chosen in such a way that the muffin–tin spheres do not overlap. The wave functions are expanded in the interstitial region to plane waves with a cutoff of \(K_{\max } = 8.0/R_{\mathrm{MT}}\) (where \(K_{\max }\) is the magnitude of the largest K vector in the plane wave, and \(R_{\mathrm{MT}}\) is the average radius of the muffin–tin spheres). The maximum value for partial waves inside the atomic sphere was \(l_{\max } = 10\), while the charge density was Fourier expanded up to \(G_{\max } = 12\) (a.u.)\(^{-1}\), where \(G_{\max }\) is the largest vector in the Fourier expansion. For the sampling of the Brillouin zone, we used the Monkhorst–Pack mesh [36, 37] of (\(4 \times 4 \times 4\)) k-points for AlSb, \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}\) and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\); and (\(4 \times 4 \times 3\)) k-points for \(\hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\). The self-consistent convergence of the total energy was set at 0.1 mRy.
3 Results and discussions
3.1 Structural properties
The AlSb semiconductor has zinc blende (B3) structure with space group of \(F\bar{4}3m\) No. 216, where the Al is situated at (0, 0, 0) and Sb atom at (0.25, 0.25, 0.25) positions. The \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds with the concentrations of \(x = 0.25, 0.5\), and 0.75 are obtained, respectively, by substituting one, two, and three Al cation sites by V atoms in supercells of 8 atoms of AlSb. We get the \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb} (1 \times 1 \times 1)\) and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb} (1 \times 1 \times 1)\) standard unit cells of 8 atoms, respectively, for the concentrations of \(x = 0.25\) and 0.75 with cubic structure and space group of \(P{4}''\) No. 215. The \(\hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb} (1 \times 1 \times 1)\) supercell of 8 atoms is obtained for the concentration of \(x = 0.5\) with tetragonal structure and space group of \(P\bar{4}m2\) No. 115.
To verify the phase stabilities of our materials in the paramagnetic (PM), ferromagnetic (FM), or anti-ferromagnetic (AFM) state configurations, we have calculated the differences in the total energies \(\Delta E_1 =E_{\mathrm{PM}} -E_{\mathrm{FM}} \) between FM and PM states and \(\Delta E_2 =E_{\mathrm{AFM}} -E_{\mathrm{FM}} \) between FM and AFM states. The (\(\Delta E_1 \) and \(\Delta E_2 )\) are (0.740 and 0.071 eV) for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}\); (1.198 and 0.275 eV) for \(\hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\); and (8.273 and 0.488 eV) for \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\). The total differences between energies \(\Delta E_1 \) and \(\Delta E_2 \) are positive, meaning that the ferromagnetic state arrangements of \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\) are more stable compared with the paramagnetic and anti-ferromagnetic state configurations.
The formation enthalpy is used to describe the phase stability of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) alloys. To determine the thermodynamic stability of the present compounds in the ferromagnetic zinc blende phase, we have calculated the formation enthalpies (\(\Delta \hbox {H}\)) of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) using the following expression:
where the \(E_{\mathrm{total}}(\hbox {Al}_{4-y}\hbox {V}_{y}\hbox {Sb}_{4})\) is the minimum total energy of each alloy; and the E(Al), E(V), and E(Sb) are the minimum total energies per atom of each bulk Al, V, and Sb, respectively, and the \(y = 1, 2\), and 3 are the numbers of the substitute V atoms in supercell. The values of \(\Delta \hbox {H}\) are −7.77, −5.94, and −4.32 eV for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\), respectively. Thermodynamically the negative values of formation enthalpies indicate that these compounds are stable in the ferromagnetic zinc blende phase.
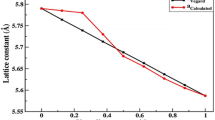
For the stable ferromagnetic state configuration, we have fitted the plots of variations of total energies as a function of equilibrium volumes by the empirical Murnaghan’s equation of state [38] to determine the structural parameters of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds. The computed equilibrium lattice constants (a), bulk modules (B), and their pressure derivatives \((B')\) of AlSb and \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) with other theoretical [39, 40] and experimental [41, 42] data are summarized in Table 1. The optimized lattice constants of binary AlSb such as 6.168 Å maintain good agreement with the experimental value of 6.1355 Å. Also, the calculated lattice parameter of AlSb is better than the theoretical value of 6.228 Å of S. Kacimi et al. [39] with GGA-PBE approximation [43] and than the theoretical lattice constant of 6.188 Å of Caro et al. [40] using the Heyd–Scuseria–Ernzerhof (HSE) hybrid-functional approach [44, 45]. This is due to the better performance of GGA-WC approximation for structural optimization [12, 34, 46–48].

Spin-polarized band structures of majority spin (up) and minority spin (down) for AlSb. The Fermi level is set to zero (horizontal dotted line)

Spin-polarized band structures of majority spin (up) and minority spin (down) for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}\). The Fermi level is set to zero (horizontal dotted line)

Spin-polarized band structures of majority spin (up) and minority spin (down) for \(\hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\). The Fermi level is set to zero (horizontal dotted line)

Spin-polarized band structures of majority spin (up) and minority spin (down) for \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\). The Fermi level is set to zero (horizontal dotted line)
For ternary \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds, the lattice parameter decreases as the concentration (x) of V increases; this is due to the fact that the ionic radius of V may be smaller than that of Al atom. Thus, the bulk modulus increases with the increasing concentration of V, indicating that AlSb is more easily compressible than the \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) systems.
3.2 Electronic and magnetic properties
We have used the theoretical lattice constants to determine the electronic structures and the origin of ferromagnetic state in \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds. The spin-polarized band structures of AlSb, \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\) are displayed in Figs. 1, 2, 3, and 4 respectively. Figure 1 depicts that the majority-spin and minority-spin bands of AlSb have similar band structures with bandgap at Fermi level, confirming the semiconductor feature of this material. Recently, the S. Das et al. [24] have found experimentally that the gaps of AlSb varied between 1.56 and 1.66 eV for indirect transitions and between 2.11 and 2.27 eV for direct transitions. We have found that AlSb has values of 1.25 and 1.45 eV for indirect gap and direct gap, respectively. The obtained indirect bandgap of 1.25 eV of AlSb is in good agreement with the recent theoretical value of 1.227 eV of Kacimi et al. [39], the difference between them not exceeding 1.9 %. For \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) materials, the Figs. 2, 3, and 4 show that the spin-up states are metallic, while the spin-down states are semiconductors. The metallic nature of minority spin is originated from the 3d (V) partially occupied states that occur at Fermi level (\(E_{F})\). In contrast, the minority-spin bands have a direct bandgap, which separates the valence bands maximum (VBM) and the conduction bands minimum (CBM). Thus, the \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds are half-metallic ferromagnets with spin polarization of 100 %.
The minority-spin bands of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) are characterized by two bandgaps: the half-metallic ferromagnetic (HMF) gap (\(E_{\mathrm{HMF}})\), and HM gap (\(G_{\mathrm{HM}})\). The calculated gaps (\(E_{\mathrm{HMF}}\) and \(G_{\mathrm{HM}})\), the VBM, and the CBM of minority-spin bands are given in Table 2. From Figs. 2, 3, and 4, we show that the VBM and the CBM are located at the \(\Gamma \) symmetry point, meaning that minority-spin channels have a direct HMF gap; they are 1.266, 1.153, and 1.24 eV for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\), respectively. The HM gap (flip-gap) determines the minimal energy bandgap for a spin-flip excitation required for generating a hole or an electron in minority spin [20]. For \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\), the minority spin has a HM gap situated between the conduction band minimum and Fermi level (0 eV), which describes the smallest energy (gap) of a spin excitation for generating an electron in the conduction bands. The HM gaps are 0.555, 0.332, and 0.353 eV, respectively, for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\). Thus, the large HM gaps shape the \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds as right half-metallic ferromagnets, and make them potential candidates for practical spintronics applications.

Spin-polarized total and partial DOS of \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}\). The Fermi level is set to zero (vertical dotted line)

Spin-polarized total and partial DOS of \(\hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\). The Fermi level is set to zero (vertical dotted line)

Spin-polarized total and partial DOS of \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\). The Fermi level is set to zero (vertical dotted line)

The double-exchange mechanism in \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) compounds
The calculated spin-polarized total (T) and partial (P) densities of states (DOS) for \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\) are given in Figs. 5, 6, and 7, respectively. They show a similar half-metallic behavior, resulting from the contribution of the spin-up and spin-down states at Fermi level (\(E_{\mathrm{F}})\), where the majority spin behaves with a metallic nature and minority spin has a bandgap at \(E_{\mathrm{F}}\). We have noticed that the upper part of valence bands is formed by the 3d (V) partially occupied states, and the bottom of the conduction bands is dominated by the 3d (V) unoccupied states above \(E_{\mathrm{F}}\). However, the 3d (V) states are divided into two states due to the effect of tetrahedral crystal field created by surrounding Sb ions, leading to the splitting of the degenerate 3d (V) states into twofold low-lying \(e_{g} \left( {d_{z^{2}} ,\hbox { and }d_{x^{2}-y^{2}} } \right) \) and the threefold high-lying \(t_{2g} \quad \left( {d_{xy} ,d_{xz} ,\hbox { and }d_{yz} } \right) \) symmetry states. The \(e_{g}\) are completely filled bonding states that cross a little over the Fermi level (\(E_{\mathrm{F}})\) from lower side, whereas the \(t_{2g}\) are totally empty states located directly above \(E_{\mathrm{F}}\) with anti-bonding character. This confirms that the vanadium atom is located in the tetrahedral environment formed by the neighboring Sb ions.
For the majority spin, the main contribution around the Fermi level is principally formed by the p–d hybridization between 3p (Sb) and 3d (V) states, which suggests that the ferromagnetism is explained by Zener’s double-exchange model [49]. On the other hand, the localized 3d (V) are partially occupied anti-bonding states that occur at Fermi level and stabilize the ferromagnetic state configuration [50, 51] associated with the double-exchange mechanism [52], which is displayed by the schema of Fig. 8. We understand that both double-exchange and p–d exchange mechanisms contribute to the stabilization of the ferromagnetic state arrangement in \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) systems.
The \(e_{g}\) (V) majority-spin \(\hbox {states}\) are totally filled with two electrons, but the \(t_{2g}\) (V) majority spins are empty states. However, the two unpaired electrons of \(e_{g}\) (V) states generate a total magnetic moment of \(2~\mu _{\mathrm{B}}\) per V atom; (\(\mu _{B}\) is the Bohr magneton) for \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\). The local and total magnetic moments of \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\) are given in Table 3. It shows that the total magnetic moments are integral multiples of Bohr magneton of \(2~\mu _{B}\) per V atom, implying the true half-metallic behavior of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\). The main contribution of total magnetic moments is localized at vanadium (V) atom and due to large p–d exchange interaction, the smaller local magnetic moments are induced in the nonmagnetic Al and Sb sites. The ferromagnetic (anti-ferromagnetic) interaction results from the parallel (antiparallel) arrangement of magnetic moments in \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) systems. Then, the positive signs of Al and V magnetic moments describe the ferromagnetic interaction between Al and V atoms, whereas the antiparallel alignment of positive and negative magnetic moments, respectively, for Al and Sb atoms explains the anti-ferromagnetic interaction between V and Sb atoms.
4 Conclusion
First-principles calculations of DFT within the FP-LAPW method with GGA-WC potential have been used to predict the electronic and ferromagnetic properties of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) at concentrations of \(x = 0.25, 0.5\), and 0.75. We have found that \(\hbox {Al}_{0.75}\hbox {V}_{0.25}\hbox {Sb}, \hbox {Al}_{0.5}\hbox {V}_{0.5}\hbox {Sb}\), and \(\hbox {Al}_{0.25}\hbox {V}_{0.75}\hbox {Sb}\) are half-metallic (HM) ferromagnets with 100 % spin polarization. The total magnetic moment per V atom for each concentration is integral Bohr magneton of 2 \(\mu _{B}\), confirming the HM ferromagnetic behavior of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\). In these compounds, the stability of ferromagnetism is explained by the contribution of both p–d and double-exchange mechanisms. Therefore, \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) alloys are predicted to be promising candidates for possible semiconductor spintronics applications.
References
Wolf, S.A., Awschalom, D.D., Buhrman, R.A., Daughton, J.M., von Molnár, S., Roukes, M.L., Chtchelkanova, A.Y., Treger, D.M.: Spintronics: a spin-based electronics vision for the future. Science 294, 1488–1495 (2001)
Žutić, I., Fabian, J., Das Sarma, S.: Spintronics: fundamentals and applications. Rev. Mod. Phys. 76, 323–410 (2004)
Miah, M.I.: Generation and detection of spin current in semiconductors: semiconductor spintronics. Mater. Today 2, 5111–5116 (2015)
Han, W.: Perspectives for spintronics in 2D materials. APL Mater. 4, 032401–032409 (2016). (9pp)
Sato, K., Katayama-Yoshida, H.: Material design of GaN-based ferromagnetic diluted magnetic semiconductors. Jpn. J. Appl. Phys. 40, L485–L487 (2001)
Wu, S.Y., Liu, H.X., Gu, L., Singh, R.K., Budd, L., van Schilfgaarde, M., McCartney, M.R., Smith, D.J., Newman, N.: Synthesis, characterization, and modeling of high quality ferromagnetic Cr-doped AlN thin films. Appl. Phys. Lett. 82, 3047–3049 (2003)
Doumi, B., Tadjer, A., Dahmane, F., Djedid, A., Yakoubi, A., Barkat, Y., Ould Kada, M., Sayede, A., Hamada, L.: First-principles investigation of half-metallic ferromagnetism in V-doped BeS, BeSe, and BeTe. J. Supercond. Nov. Magn. 27, 293–300 (2014)
Mokaddem, A., Doumi, B., Sayede, A., Bensaid, D., Tadjer, A., Boutaleb, M.: Investigations of electronic structure and half-metallic ferromagnets in Cr-doped zinc-blende BeS semiconductor. J. Supercond. Nov. Magn. 28, 157–164 (2015)
Rajamanickam, N., Rajashabala, S., Ramachandran, K.: Effect of Mn-doping on the structural, morphological and optical properties of ZnO nanorods. Superlattices Microstruct. 65, 240–247 (2014)
Rajendar, V., Dayakar, T., Shobhan, K., Srikanth, I., Venkateswara Rao, K.: Systematic approach on the fabrication of Co doped ZnO semiconducting nanoparticles by mixture of fuel approach for Antibacterial applications. Superlattices Microstruct. 75, 551–563 (2014)
Singh, J., Verma, N.K.: Correlation between structure and ferromagnetism in cobalt-doped CdSe nanorods. J. Supercond. Nov. Magn. 27, 2371–2377 (2014)
Doumi, B., Mokaddem, A., Sayede, A., Dahmane, F., Mogulkoc, Y., Tadjer, A.: First-principles investigations on ferromagnetic behaviour of \(\text{ Be }_{1-x}\text{ V }_{x}\text{ Z }\) (Z = S, Se and Te) (\(x = 0.25\)). Superlattices Microstruct. 88, 139–149 (2015)
Kaur, P., Kumar, S., Singh, A., Chen, C.L., Dong, C.L., Chan, T.S., Lee, K.P., Srivastava, C., Rao, S.M., Wu, M.K.: Investigations on doping induced changes in structural, electronic structure and magnetic behavior of spintronic Cr-ZnS nanoparticles. Superlattices Microstruct. 83, 785–795 (2015)
Boutaleb, M., Doumi, B., Sayede, A., Tadjer, A., Mokaddem, A.: Theoretical predictions of electronic structure and half-metallic ferromagnetism in \(\text{ Al }_{1-x}\text{ Mn }_{x}\)P diluted magnetic semiconductors. J. Supercond. Nov. Magn. 28, 143–150 (2015)
Wang, S.F., Chen, L.Y., Zhang, T., Song, Y.L.: Half-metallic ferromagnetism in Cu-doped ZnO nanostructures from first-principle prediction. J. Supercond. Nov. Magn. 28, 2033–2038 (2015)
Doumi, B., Mokaddem, A., Ishak-Boushaki, M., Bensaid, D.: First-principle investigation of magnetic and electronic properties of vanadium- and chromium-doped cubic aluminum phosphide. Sci. Semicond. Process. 32, 166–171 (2015)
Shayesteh, S.F., Nosrati, R.: The structural and magnetic properties of diluted magnetic semiconductor \(\text{ Zn }_{1-x}\text{ Ni }_{x}\)O nanoparticles. J. Supercond. Nov. Magn. 28, 1821–1826 (2015)
Doumi, B., Mokaddem, A., Dahmane, F., Sayede, A., Tadjer, A.: A novel theoretical design of electronic structure and half-metallic ferromagnetism in the 3d (V)-doped rock-salts SrS, SrSe, and SrTe for spintronics. RSC Adv. 112, 92328–92334 (2015)
Saini, H.S., Kashyap, M.K., Kumar, M., Thakur, J., Singh, M., Reshak, A.H., Saini, G.S.S.: Generating magnetic response and half-metallicity in GaP via dilute Ti-doping for spintronic applications. J. Alloy Compd. 649, 184–189 (2015)
Doumi, B., Mokaddem, A., Temimi, L., Beldjoudi, N., Elkeurti, M., Dahmane, F., Sayede, A., Tadjer, A., Ishak-Boushaki, M.: First-principle investigation of half-metallic ferromagnetism in octahedrally bonded Cr-doped rock-salt SrS, SrSe, and SrTe. Eur. Phys. J. B 88, 93–109 (2015). (9pp)
Kervan, S., Kervan, N.: First-principles study on half-metallic ferromagnetism in the diluted magnetic semiconductor (DMS) \(\text{ Al }_{1-x}\text{ Mn }_{x}\)P compounds. J. Magn. Magn. Mater. 382, 63–70 (2015)
Boutaleb, M., Doumi, B., Sayede, A., Tadjer, A.: Half-metallic ferromagnetic properties of Cr- and V-doped AlP semiconductors. J. Magn. Magn. Mater. 397, 132–138 (2016)
Mahmood, Q., Alay-e-Abbas, S.M., Yaseen, M., Mahmood, A., Rashid, M., Noor, N.A.: Theoretical investigation of half-metallic ferromagnetism in \(\text{ Mg }_{0.75}\text{ Ti }_{0.25}\text{ Y }\) (Y = S, Se, Te) alloys by using DFT-mBJ studies. J. Supercond. 29, 1387–1397 (2016)
Das, S., Ghosh, B., Hussain, S., Bhar, R., Pal, A.K.: Pulsed laser deposition: a viable route for the growth of aluminum antimonide film. J. Cryst. Growth 419, 12–19 (2015)
Nilsen, T.A., Patra, S.K., Breivik, M., Fimland, B.O.: Thermal dependence of the lattice constant and the Poisson ratio of AlSb above room temperature. J. Cryst. Growth 336, 29–31 (2011)
Rahman, G., Cho, S., Hong, S.C.: Half metallic ferromagnetism of Mn doped AlSb: a first principles study. Phys. Stat. Sol. (b) 244, 4435–4438 (2007)
Saeed, Y., Shaukat, A., Nazir, S., Ikram, N., Reshak, A.H.: First principles calculations of electronic structure and magnetic properties of Cr-based magnetic semiconductors \(\text{ Al }_{1-x}\text{ Cr }_{x}X\) (\(X = \text{ N, } \text{ P, } \text{ As, } \text{ Sb }\)). J. Solid State Chem. 183, 242–249 (2010)
Katayama-Yoshida, H., Sato, K.: Materials design for semiconductor spintronics by ab initio electronic-structure calculation. Physica B 327, 337–343 (2003)
Katayama-Yoshida, H., Sato, K.: Spin and charge control method of ternary II-VI and III-V magnetic semiconductors for spintronics: theory vs. experiment. J. Phys. Chem. Solids 64, 1447–1452 (2003)
Anh, L.D., Kaneko, D., Hai, P.N., Tanaka, M.: Growth and characterization of insulating ferromagnetic semiconductor (Al, Fe)Sb. Appl. Phys. Lett. 107, 232405 (2015)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136, B864–871 (1964)
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–1138 (1965)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: In: Schwarz, K. (ed.) WIEN 2K, an Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties. Technische Universität Wien, Wien (2001)
Wu, Z., Cohen, R.E.: More accurate generalized gradient approximation for solids. Phys. Rev. B 73, 235116–235126 (2006). (6pp)
Sharma, S., Verma, A.S., Bhandari, R., Kumari, S., Jindal, V.K.: First principles study of the structural, electronic, optical, elastic and thermodynamic properties of \(\text{ CdXAs }_{2}\) (X = Si, Ge and Sn). Mater. Sci. Semicond. Process 27, 79–96 (2014)
Monkhorst, H.J., Pack, J.D.: Special points for Brillonin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976)
Pack, J.D., Monkhorst, H.J.: “Special points for Brillonln-zone integrations”-a reply. Phys. Rev. B 16, 1748–1749 (1977)
Muranghan, F.D.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 30, 244–247 (1944)
Kacimi, S., Mehnane, H., Zaoui, A.: I-II-V and I-III-IV half-Heusler compounds for optoelectronic applications: Comparative ab initio study. J. Alloy Compd. 587, 451–458 (2014)
Caro, M.A., Schulz, S., O’Reilly, E.P.: Origin of nonlinear piezoelectricity in III-V semiconductors: internal strain and bond ionicity from hybrid-functional density functional theory. Phys. Rev. B 91, 075203–075209 (2015)
Adachi, S.: Properties of Group-IV. III-V and II-VI Semiconductors. Wiley, New York (2009)
Madelung, O.: Semiconductors: Data Handbook, 3rd edn. Springer, Berlin (2004)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
Heyd, J., Scuseria, G.E., Ernzerhof, M.: Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003)
Heyd, J., Scuseria, G.E.: Efficient hybrid density functional calculations in solids: assessment of the Heyd–Scuseria–Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 121, 1187–1192 (2004)
Sajjad, M., Manzoor, S., Zhang, H.X., Noor, N.A., Alay-e-Abbas, S.M., Shaukat, A., Khenata, R.: The half-metallic ferromagnetism character in \(\text{ Be }_{1-x}\text{ V }_{x}\text{ Y }\) (Y = Se and Te) alloys: An ab-initio study. J. Magn. Magn. Mater. 379, 63–73 (2015)
Doumi, B., Mokaddem, A., Sayede, A., Boutaleb, M., Tadjer, A., Dahmane, F.: Half-metallic ferromagnetic property related to spintronic applications in 3d (V, Cr, and Mn)-doped GaP DMSs. J. Supercond. Novel Magn. 28, 3163–3172 (2015)
Cherfi, Y., Mokaddem, A., Bensaid, D., Doumi, B., Sayede, A., Dahmane, F., Tadjer, A.: A novel theoretical investigation of electronic structure and half-metallic ferromagnetism in 3d (V)-doped InP for spintronic applications. J. Supercond. Novel Magn. 29, 1813–1819 (2016)
Zener, C.: Interaction between the d-shells in the transition metals. II. Ferromagnetic compounds of manganese with perovskite structure. Phys. Rev. 82, 403–405 (1951)
Sato, K., Dederichs, P.H., Araki, K., Katayama-Yoshida, H.: Ab initio materials design and Curie temperature of GaN-based ferromagnetic semiconductors. Phys. Status Solidi C 7, 2855–2859 (2003)
Sato, K., Katayama-Yoshida, H., Dederichs, P.H.: Curie temperatures of III-V diluted magnetic semiconductors calculated from first-principles in mean field approximation. J. Supercond. 16, 31–35 (2003)
Akai, H.: Ferromagnetism and its stability in the diluted magnetic semiconductor (In, Mn)As. Phys. Rev. Lett. 81, 3002–3005 (1998)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zerouali, A., Mokaddem, A., Doumi, B. et al. First-principle calculations of electronic and ferromagnetic properties of \(\hbox {Al}_{1-x}\hbox {V}_{x}\hbox {Sb}\) . J Comput Electron 15, 1255–1262 (2016). https://doi.org/10.1007/s10825-016-0912-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10825-016-0912-4