
Abstract
Nonadditivity (NA) in Structure-Activity and Structure-Property Relationship (SAR) data is a rare but very information rich phenomenon. It can indicate conformational flexibility, structural rearrangements, and errors in assay results and structural assignment. While purely ligand-based conformational causes of NA are rather well understood and mundane, other factors are less so and cause surprising NA that has a huge influence on SAR analysis and ML model performance. We here report a systematic analysis across a wide range of properties (20 on-target biological activities and 4 physicochemical ADME-related properties) to understand the frequency of various different phenomena that may lead to NA. A set of novel descriptors were developed to characterize double transformation cycles and identify trends in NA. Double transformation cycles were classified into “surprising” and “mundane” categories, with the majority being classed as mundane. We also examined commonalities among surprising cycles, finding LogP differences to have the most significant impact on NA. A distinct behavior of NA for on-target sets compared to ADME sets was observed. Finally, we show that machine learning models struggle with highly nonadditive data, indicating that a better understanding of NA is an important future research direction.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
SAR exploration builds strongly on the additivity hypothesis: SAR is typically explored in a cross shape within an R1 by R2 SAR matrix, where one substituent on one R-group vector is varied and all other substituents are kept fixed. With the best substituent identified, the next R-group is explored, keeping the current best substituent in the first vector fixed (Fig. 1). While an efficient way of establishing SAR, this process carries an implicit assumption of additivity across the substituents - if we cannot assume additivity, we would have to synthesize many more R-group combinations which would exhaust any reasonable technical capacity. Therefore, it is important to understand where the additivity hypothesis holds and where it does not.

Schematic R group analysis representation of SAR exploration. Initially, six distinct R1 groups are inspected, while maintaining a constant R2 group across all compounds (i.e. horizontal line). Following this, the R1 group of the most active compound is kept constant, while different R2 groups are investigated (i.e. vertical line). The optimal combination of R1 and R2 substituents is represented by a green dot. Compounds with low activity are denoted by yellow dots, the red dot indicates an inactive compound and a light green dot indicates a compound with moderate activity
In a recent publication, Gogishvili et al. showed that up to 10% of all compounds show nonadditivity (NA) [1], which raises concerns about the effectiveness of the standard, efficient approach to SAR exploration. They also showed that machine learning (ML) models struggle to predict activities of compounds that frequently are part of highly nonadditive local SAR. While a couple of physical phenomena that lead to strong NA have been described, we still lack a quantitative understanding of how often particular events occur, and when to expect them. The instances that have so far been described include structural changes of both protein and ligand, reorientation of the whole ligand, and differences in water networks [2,3,4]. More mundane reasons for strong NA include experimental errors, errors in the assignment of stereochemistry, and substituent interactions.
Technically, NA is calculated from sets of four compounds linked by two identical transformations, e.g. double transformation cycles (DTCs, hereafter referred to as ‘cycles’) [5]. Those transformations match medicinal chemistry thinking in the sense that each transformation tests the variation of a structural element, while keeping all other elements fixed. For practical reasons, in computed NA analyses across whole SAR datasets we typically use matched molecular pair (MMP) transformations. It is the two transformations, plus the four compounds with their measured activities and/or properties, that characterize a cycle. If there is an outlier among the compounds or measurements, we will see the whole cycle as having a high NA. Since individual compounds typically occur in several cycles, there is an amplifying effect where the measurement error of a single entry can propagate, leading to an overestimation of total NA. Yet, it would be too simple to assume that the majority of NA is caused by experimental errors. In in-house data, we occasionally identify a single mismeasurement as cause for NA (results not published), but we here will show that NA is correlated with certain properties of the local SAR, which should not be the case if the majority of NA was caused by random errors.
In this contribution, our overall goal is to better characterize surprising NA, identify drivers behind it, and illustrate its importance for ML models. We are not focusing on the causes of NA in individual instances, as this has been previously explored, but instead, we are interested in examining NA trends across a dataset. To get there, we first report a set of novel descriptors to characterize cycles. We use these descriptors to subgroup cycles and search for trends in NA: If all NA was caused by random errors, we would expect to see NA evenly split among subgroups. Conversely if we see systematically higher NA following identifiable trends in subgroups, we reason that there are common properties that lead to NA, and they should be taken into account when planning SAR exploration. We analyze physicochemical, ADME, and on-target data. We use in-house datasets where we can control the quality and have a sufficient dataset size. Since we look for trends in groups of compounds, we use robust statistics to characterize the NA distribution on subsets. We will show that we can identify features of cycles that lead to very clear increases in NA, and we will discuss the impact of the findings on the practice of SAR exploration.
Methods
Datasets
We collected SAR data of twenty in-house on-target datasets and four ADME datasets. We performed NA analysis and analyzed the distributions of DTCs and ratios of cycles to compounds across the datasets. The on-target datasets belong to current drug discovery programs or previous programs where individual examples can be shared. Some of the latter group includes targets like AEP [6], ATX [7], BACE1 [8], CB2 [9], DDR1 [10], DPP4 [11], GLYT1 [12], PDE10 [13] and SMN2 [14]. The physicochemical and ADME datasets consist of solubility (kinetic solubility at pH 6.5, phosphate buffered), permeability (parallel artificial membrane permeability assay), lipophilicity (LogD7.4) and intrinsic clearance in human liver microsomes (HLM CLInt). Assay technologies to measure the physicochemical and ADME properties have in parts been published elsewhere [15,16,17].
Each dataset possesses a unique chemical space and SAR, characterized by variations in the number of chemical series, the ratio of cycles to the number of compounds, protein families, and other factors (see Fig. 2). The on-target datasets vary in size, ranging from 1,000 to 9,000 compounds, while the cross-project ADME datasets are significantly larger, with sizes ranging from 66,000 to 89,000 compounds. The average ratio of cycles to compounds is considerably lower for ADME properties than the average ratio for on-target properties, averaging at two cycles per compound. In contrast, the on-target properties exhibit a more diverse cycle-to-compound ratio. CB2 and MAGL display the highest number of cycles per compound (9 and 6 compounds, respectively), whereas DDR1 and DPP4 present a ratio of less than one. It is observed that projects with fewer chemical series tend to have a higher number of cycles due to a more localized SAR exploration.
All the compounds within a dataset were measured in the same assay. The activity values consisted of IC50 measurements, with the exception of two ion channel programs where Ki was selected instead and two other programs where the functional assay (EC50) was selected. The negative logarithm of the activity value is used in all the equations and analysis shown. The ADME endpoints were also transformed in logarithmic scale. The geometric mean was applied for the cases with multiple measurements. Chemical structures in the form of isomeric SMILES were standardized, salts and adducts were removed to obtain parent molecules, and the stereoisomer and tautomer structures as deposited by the project teams were used.

(A) Distribution of the number of compounds and number of DTCs per each on-target dataset. (B) Distribution of the number of compounds and number of DTCs per each ADME dataset. Vertical axes have been truncated
NA calculation
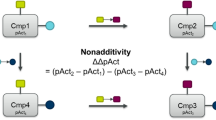
Figure 3A depicts an example of four compounds that are related to each other by two transformations in a cycle. NA is calculated as the activity value (pAct) differences of the four compounds assembling the cycle

(A) DTC with surprising NA for PDE10 dataset. (B) List of NA descriptors with the corresponding values for the depicted cycle. Delta symbol (Δ) represents the difference of the property value between the two compounds of a transformation
Cycle descriptors
A set of descriptors was implemented to categorise cycles and analyze NA. Some descriptors are calculated on the cycle level which implies two transformations and four compounds, like the descriptor Number of atoms between R groups. Other descriptors depend only on the transformation level or on the compound level, such as the Tanimoto similarity or the unassigned stereocenters, respectively. The full list of descriptors is presented in Table 1 and Table S1. Some of the physicochemical descriptors consist of additive properties (i.e. number hydrogen bond donors (HBD), number hydrogen bond acceptors (HBA), topological polar surface area (TPSA)), which allows the calculation on the compound level. By applying this workflow, compound level descriptors and transformation level descriptors which belong to multiple cycles, only need to be calculated once.
The descriptors quantify the variance resulting from the substitutions of the R group. For transformation level descriptors, we always present the biggest effect between the two transformations, and the obtained value is associated with the cycle. For instance, when the chemical similarity of the two transformations is analyzed, the lowest Tanimoto coefficient is reported. For the remaining descriptors, the highest difference between the two transformations is reported.
The 2D structure descriptors were calculated using RDKit, including the atom-based calculation of LogP using Crippen’s approach [18]. According to the level of stereochemical information, cycles are classified as inverted (R groups have opposite chirality), unassigned (the stereocenter of one R group is not defined) and none (the transformation involves chemical structure changes). To determine the shortest topological distance between the two R groups in a cycle, RDKit was used to calculate the number of atoms between R groups.
Compound classification based on NA
NA is a characteristic of a double transformation cycle, therefore it does not make sense to label an individual compound as additive or nonadditive. Yet, we are interested in the impact of NA on predictive quality of ML models. To this end, we categorize compounds into distinct classes based on their embedding in MMPs/cycles and on the average per-compound NA [5] (the per-compound NA for compounds 2 and 4 is ΔΔpAct and for compounds 1 and 3 is (− 1)·ΔΔpAct on the basis of Eq. 1) of all the double transformation cycles they participate in. Each compound is allocated to one of the following groups: compounds with high average per-compound NA, additive compounds (i.e. compounds with low average per-compound NA), MMPs (referring to compounds belonging to matched molecular pairs but not engaged in a double transformation cycle), and the rest, which includes singleton compounds not connected to any matched molecular pair transformation. The set of high NA compounds is further divided into compounds exhibiting surprising NA and those with mundane NA (for the definitions of surprising and mundane see the Results section).
To determine if a compound has high NA or low NA, we compare the average NA across all cycles (µ (ΔΔpAct)Compound) to the NA distribution we would expect from standard experimental uncertainty (σexperiment = 0.2 log units) [5]. The standard deviation of the distribution of all nonadditivities across a dataset, originating from experimental uncertainty only [3], is
If a compound is part of n cycles, we would expect its average NA across cycles, originating from random assay uncertainty, to decrease with √n. So a compound is considered to have high NA at the 2 sigma level (95.4% of the overall population) if
holds. With increasing n, Eq. 3 takes very low numbers, labeling compounds with a low average NA as high NA compounds. Measurements for individual compounds also carry experimental uncertainty. Applying the same 2 sigma level, individual compounds’ average µ(ΔΔpAct)Compound has to be considered normal if it is lower than the amount expected from normal experimental uncertainty [5]
We use Eqs. 3 and 4 together to identify compounds with high NA. In practice, Eq. 3 applies to all compounds that are part of four or less cycles, whereas Eq. 4 applies to all compounds that are part of four or more cycles. Note that the absolute thresholds depend on σexperiment. From in-house experience across many different projects and previous publications [19], we find that an experimental uncertainty of 0.2 log units, estimated by the robust standard deviation, is a good average value (details not shown). If there is a more precise estimate for individual assays, this may be used.
Machine learning details
To prepare the input for the ML models, we generate input files with stratified splits, so that each of the five categories (rest, MMP, additive, mundane NA, surprising NA) is represented according to the distribution in the input data. Random Forest (RF) models were trained using the Scikit-Learn framework [20] and the Graph Neural Network (GNN) models were trained using the Attentive Fingerprint methodology (AttentiveFP) [21] using the Pytorch framework [22]. For the RF models, compounds were encoded using Morgan fingerprints (1024 bits, radius 2, RDKit). Fivefold cross-validation was applied. For the GNNs, molecular graphs were constructed, with atoms being nodes and bonds being edges. The hyperparameters were chosen based on the best internal validation performance. Note that here we are not interested in optimizing the ML models, but rather use a robust baseline approach to understand the impact of NA on prediction quality.
Code
Based on the original Python code to quantify NA [5], we have extended and enhanced the code. Major code refactoring was done and unit/regression testing was added for code stability and maintainability. The code is written in Python making use of the cheminformatics libraries RDKit [23] as well as Pandas and NumPy. The assembly of cycles is based on matched molecular pairs, employing our open-source code mmpdb [24]. The code includes NA classification and corresponding descriptor calculation. The code to perform NA calculations is available from GitHub under https://github.com/Roche/NonadditivityAnalysis.
Results
We have analyzed different factors that lead to NA and investigated how often different reasons for NA occur such as substituent competition, experimental issues or annotation errors. 892,839 cycles were generated: 21% of those are based on on-target properties, 79% are based on ADME properties. A detailed view of the number of cycles per property is shown in Fig. 2.
NA classification
We have developed a set of descriptors to be used to categorize cycles into surprising and mundane based on their structural features. The distinction between surprising and mundane is arbitrary rather than definitive but the intention is to provide a convenient way to enable teams to focus on those high NA cycles that deserve greater attention and further investigation.
Hence, we propose to classify double transformation cycles as mundane when any of the following conditions apply:
-
Number of atoms between R groups ≤ 2 atoms.
-
Tanimoto similarity of the transformation < 0.4.
-
Number of exchanged heavy atoms > 10.
-
Linker exchange transformations.
-
Transformations with unassigned or inverted stereocenters.
Distance between R-groups
Average NA is strongly influenced by the distance between the two R-groups in a cycle. With R-groups being anchored close to each other, it is much more likely that the two groups will interfere and hence influence the activities of the compounds in a nonadditive fashion. Our data shows that the mean absolute NA increases as the topological distance between two R groups, defined as the shortest path in terms of atoms, decreases, see Fig. 4A. This phenomenon occurs in both types of datasets, on-target and ADME. The implemented descriptor calculates the number of atoms between the two R-groups in a cycle. If the number of atoms between R-groups is zero, the two R-groups are connected. A distance of one or two means that the R groups are either attached to the same atom or to neighboring atoms. In all cases, the R groups can have steric as well as electrostatic influence on each other because of the very small spatial and topological distance between the two substituents. With increasing distance between the R-groups, the average NA decreases. As we want to filter out mundane NA and focus on the surprising cases, we use a cutoff of at least three atoms between R-groups to characterize surprising cycles.

(A) Impact of the absolute NA mean values across the distance between R groups, on-target data is colored in red and ADME data is colored in blue. The error bars indicate the standard deviation at 95% confidence. (B) DTC example from the PDE10 dataset where the atom distance between the transformations is zero
Transformation similarity
The more similar the two molecules involved in a transformation are, the less NA is observed. Figure 5 shows a pronounced decrease of NA at minimum Tanimoto coefficients above 0.6. The exceptions to this observation were in the on-target cycles where sets of molecules had a minimum Tanimoto coefficient of 1 and average high NA. Inspection of those cases revealed that all of them were cycles that had a stereochemistry inversion in both transformations. Stereochemical assignment in databases can be unclear or inconsistent, which together with this statistical analysis indicates that an unknown, unassigned or incorrectly assigned stereocenter could lead to NA. This is discussed further below. In the on-target cycle set, very few (2%) had the minimum transformation Tanimoto below 0.4 but in the ADME set there were many more (33%). The majority of these are cycles where the transformed fragments are large (see Fig. 5B) and the compounds largely originate from different projects, sharing a small constant part but exchanging the greater part of their structures. We use a threshold of minimum transformation Tanimoto above 0.4 to characterize surprising cycles.

(A) Influence of the transformation similarity on the absolute NA mean values, data related to on-target properties is represented in red, while data pertaining to ADME properties is depicted in blue. (B) DTC example from CB2 set where the T1 presents the lowest Tanimoto similarity and maximum of heavy atoms exchange is sixteen. (C) Increase of the mean absolute NA according to the maximum number of heavy atoms exchanged
Transformation size
At the beginning of our study, we decided to increase the maximum number of heavy atoms allowed in the transformation to 16 instead of the default value of 10 in the mmpdb code [24] in order to accommodate bigger transformations. As the upper limit for the heavy atom count (HAC) in the transformation is raised, there is a corresponding sharp increase in the number of double transformation cycles observed. Specifically, we cataloged 263,984 such cycles with a maximum of ten HAC within the transformation. This figure increases to 892,839 cycles when the transformation’s maximum HAC is extended to 16. However, expanding the size of the transformation comes with a greater chance of high NA as we have observed in our analysis for both on-target and ADME datasets (Fig. 5C). The mean absolute NA for cycles involving up to 10 HAC is 0.44 for on-target sets and 0.35 for ADME sets. In contrast, for transformations with more than 10 HAC, the mean absolute NA increases to 0.52 for on-target sets and 0.42 for ADME sets, respectively.
Figure 5B shows a cycle with NA of 2.59, where in transformation 1 (T1) 16 heavy atoms are exchanged. In this particular case, the constant part of the cycle is very small, consisting of just a carboxamide linker. Practically the two transformations exchanged almost the entire molecule - in such a case, it is not surprising to find high NA. Generally, when considering a fixed molecular weight range, the constant parts become smaller as the transformed fragments increase in size, which in cases like the one above should not be surprising. The size of the transformation has an effect on the amount of NA seen with both sets, on-target and ADME. Consequently, we propose that a higher number of HAC in the transformation is likely to be associated with instances of mundane NA, and a cutoff needs to be set for the HAC exchanged for surprising NA. While this cutoff may be set specifically for each project based on the individual needs of SAR exploration, for a general analysis such as this one we decided to set it to 10 heavy atoms.
Linker exchange
Exchanging a linker or the core of a molecule can induce substantial conformational changes, depending on the geometry of the linking fragment. To filter out cycles with linker and core exchanges, we have implemented a descriptor based on the number of cut bonds in a transformation. Most of the transformations in our datasets are single cuts. Cycles that include double and triple cuts have a frequency of 34.3% and 6.8% respectively for the on-target set; and 17.3% and 1.5% respectively for the ADME set.

(A) Impact of the absolute NA mean values across the number of fragmentation cuts, data related to on-target properties is represented on the red line, while data pertaining to ADME properties is depicted in blue. (B) DTC example from the CB2 dataset with double cut for the first transformation (T1) and triple cut for the second transformation (T2)
We note that cycles whose transformations involve multiple cuts exhibit slightly higher NA for the on-target datasets (Fig. 6A). The lack of a more pronounced difference could be due to the preservation of geometry and exit vectors across most transformations, thereby avoiding drastic conformational changes. Furthermore, the compounds within cycles have already been developed following a rational design, striving to maintain similar shapes and protein interactions, thus becoming bioisosteric replacements. In practice, project teams explore SAR with a variety of exit vectors and scaffold replacements. However, if the initial one or two examples of each variation do not achieve the desired on-target activity, additional compounds bearing these variations are typically not synthesized, making the generation of cycles containing such compounds less likely.
Stereochemistry
A change in or unclear stereochemistry can cause substantial NA. To account for this, we have incorporated two stereo descriptors that capture stereochemistry at both the transformation and compound levels. At the transformation level, we differentiate cycles whose transformation only implies changes in stereochemistry between R-groups, either due to unassigned stereocenters or inverted ones. Although only 3.1% of the cycles involve stereotransformations, those related to on-target data exhibit significantly higher NA values, particularly for inversion transformations. The average absolute NA for on-target cycles without stereotransformation is 0.49, whereas with inversion stereotransformation it is 0.60. Further investigation into cycles showing high absolute NA with stereoinversion revealed that in many cases the absolute stereochemistry had been assigned arbitrarily, an example is shown in Fig. 7. In other cases, due to incomplete chiral separation, the inactive enantiomer was contaminated with differing proportions of the active enantiomer, leading to an overestimation of the measurement of the potency of the inactive enantiomer for one of the two matched pairs, with a consequent increase in the apparent NA. In contrast, for the ADME data, the average absolute NA values for cycles without stereotransformation and with inversion stereotransformation are 0.40 and 0.22 respectively. The very low value for the inversion stereotransformation cycles can be understood by considering that such cycles consist of two pairs of enantiomers with likely very similar ADME property values within each pair [25]. At the compound level, we highlight cycles containing compounds with unassigned, assigned or no (none) stereocenters, irrespective of the variable part of the MMP transformation. Our data suggests that compounds with unassigned stereochemistry are more likely to exhibit higher NA values, presumably due to the potential failure to incorporate actual stereochemical differences correctly in the cycles. On-target data revealed that cycles where compounds do not have stereocenters have the lowest mean absolute NA (i.e. 0.46), while cycles with fully assigned and partially unassigned stereochemistry have values of 0.52 and 0.53 respectively. In the case of ADME data, cycles with assigned stereochemistry, partially unassigned stereochemistry or without stereo compounds had similar mean absolute NA values (0.38, 0.41 and 0.41 respectively), again consistent with the expected low influence of stereochemistry on ADME SAR. Overall, approximately half (47%) of the cycles contain compounds with some form of stereochemistry. However, in a third of these cycles, the corresponding stereo is unassigned. It is challenging to fully represent structures of compounds containing stereocenters whose absolute configurations are unknown or undetermined, and organizations apply varying structure business rules during compound registration to handle this. In our organization there is a complex system of structure comments which attempt to describe what is known about the stereochemistry of such compounds but these comments are of course not taken into account during the NA analysis. To identify really surprising NA, we propose to ignore cycles where a transformation changes a stereocenter.
Figure 7 illustrates an example cycle, showcasing stereochemistry at both levels. The horizontal transformation involves an inversion of stereochemistry at the transformation level. Despite the vertical transformation altering the R-groups into entirely new chemical structures, the oxazolidinone stereo center is defined for the compounds containing the chlorofluorobenzene group (compounds 1 and 2), but it is undefined for the compounds containing the chloropyridine group (3 and 4). Compounds 1 and 2 were separated from a 1:1 isomeric mixture using chiral chromatography without determining the absolute configuration at the methyl group chiral center; the same applies to compounds 3 and 4. Hence compounds 1 to 4 are all single stereoisomers, but their absolute configurations at the methyl group chiral center were arbitrarily assigned. Taking into account the displayed stereochemistry, this cycle exhibits a strong NA of 3.31. Yet, if the stereochemistries at the methyl group chiral centers of one pair of compounds in the horizontal transformation were assigned inversely (e.g. compound 1 with the stereochemistry and measured value of compound 2, and compound 2 with the stereochemistry and measured value of compound 1), the NA value would drop to 0.13. It is important to note that we can’t confirm stereochemistry assignment through NA analysis, but this instance provides a good example of how NA analysis could propose a correction or at least make it consistent in the dataset.

DTC example from the MAGL dataset where the horizontal transformation consists of stereochemistry inversion transformation and the four compounds have unknown absolute configuration
After categorizing the cycles according to the aforementioned criteria, we identified 40,889 double transformation cycles (21.7%) in the on-target set and 118,902 (16.9%) in the ADME set that qualify as potentially having surprising NA. The majority of the cycles are filtered out (classified as mundane) by these criteria, see Tables S3 and S4. Among all cycles with high NA, this helps to focus on the surprising cases where interesting NA may be present and where further SAR exploration is warranted.
Statistical property analysis on surprising NA cases
After classifying double transformation cycles into potentially surprising and mundane, we here proceed to analyze the commonalities among the surprising cycles. We focus on properties associated with polarity, including LogP, TPSA, number of HBD and HBA. Additionally, we consider structural descriptors related to the steric and shape aspects of the molecules. These encompass the fraction of sp3 hybridized carbons, the number of rotatable bonds, the number of heavy atoms and connectivity indices. All these descriptors are initially calculated at the compound level and then extended to the cycles by taking the largest difference between them. Assigning a descriptor value at the cycle level is crucial, as NA only exists at this level.

NA trends for molecular polarity properties (A) and molecular spatial properties (B) for potentially surprising on-target cycles colored in red and for potentially surprising ADME cycles colored in blue. Error bars represent the standard error at 95% confidence. Each bin on the X-axis, independently of the property, group is composed of at least 20 cycles. The data for the properties ΔLogP, ΔTPSA, and ΔChi1 is categorized into bins, while the remaining properties are represented as integers
Figure 8 summarizes our findings: Lipophilicity (ΔLogP) has the most significant impact on NA in the surprising cycles. If lipophilicity is substantially changed during transformations, the effect on binding might not be additive. To determine the underlying reasons of this observation, a detailed investigation would be necessary for each individual project. Figure 9 illustrates a cycle with a ΔLogP of 4.65. The compounds within this cycle have a broad spectrum of lipophilic character, ranging from the rather hydrophilic compound C1 to the extremely greasy compound C3. The most potent compound C2 has an intermediate lipophilicity arising from the combination of the polar pyrrolidinol and the lipophilic chlorobenzene substituents.
Although the impact is less pronounced, TPSA and number of HBA follow the same trend as lipophilicity across both types of datasets. We note that the difference in number of HBD does not influence the surprising cycles’ NA. This could be due to the lack of significant variability in the number of HBD within a cycle. The median change of number of HBD for the entire dataset is zero.

DTC from the CB2 dataset with strong surprising NA, which is potentially associated with a high difference in lipophilicity
We have not identified a spatial descriptor that strongly correlates with NA. In the case of on-target data, there is a clear trend of increasing NA values as the difference in HAC and the number of rotatable bonds increases, up to a certain point (6 for HAC and 4 for rotatable bonds). For the remaining cycles with higher difference values, which constitute a minority, the effect is unpredictable, as indicated by the error bars. The difference in HAC impacts the size of the molecule, while the difference in the number of rotatable bonds can lead to different spatial configurations. Both descriptors also account for the increase in NA values for the ADME set. While one might expect a correlation between the number of rotatable bonds and the fraction of sp3 hybridized carbons, this does not appear to be the case with the difference of calculated values. We also included the topological Chi indexes [26] as descriptors to capture the connectivity of the molecular graph. We compared the values for order zero (only vertices are represented), order one (for one edge path distance), and order two (for two edges path distance). Lower Chi values indicate a higher number of connectivity subgraphs. Considering that the ΔChi1 ninetieth percentile is 2.2 for on-target and 3.0 for ADME, we can still observe some contribution of connectivity towards NA within this ΔChi1 range.
Property prediction on surprising NA compounds
Gogishvili et al. and Kwapien et al. pointed out that ML models are poor at predicting compounds that are frequently part of high NA cycles [1, 27]. Using the definitions we introduced here to characterize cycles into mundane and surprising, the performance of ML models on predicting compounds belonging to different classes can be evaluated. ML models will assign activity values to compound structures, so a category mapping from the cycle level to the compound level is required.
We choose to assign compounds to one out of five different sets: surprising NA, mundane NA, additive, MMP and rest. If a compound is part of cycles with high NA where the cycles are categorized as both mundane and surprising, it is assigned to the surprising NA set. If a compound is part of cycles with high NA, but the cycles are all categorized as mundane, it becomes part of the mundane set. A compound becomes part of the additive set, if its average per-compound NA falls below the criteria outlined in Eqs. 3 and 4. If a compound is not part of any cycle, but has matched molecular pair neighbors in the dataset, it is assigned to the MMP category. Finally, if a compound does not have any MMPs, it is assigned to the rest category. The distribution of compounds between the five categories is rather consistent across the properties examined for both on-target and ADME properties. The majority of the compounds fall into either the mmp set (with an average of 42.7% and 50.4% of compounds for on-target and ADME sets) or the additive set (averaging 44.6% and 37.5% of compounds for on-target and ADME sets respectively). The remaining categories, namely the rest set, the mundane NA set and the surprising NA set account for 2.1%, 8.5% and 2.1% of compounds in the on-target datasets, and 3.0%, 6.6% and 2.5% respectively for the ADME datasets. We built models using RF as a classical ML algorithm and AttentiveFP as a neural network approach. These are compared to a null model based on the activity mean value of each compound category. Figure 10 illustrates the comparison of performance metrics for two on-target properties (BACE1 and CB2) and two ADME properties (lipophilicity and solubility) as representative sets. The performance for all datasets can be found in Table S5 of the SI.

Performance metrics for two on-target end points (BACE1 and CB2) and for two ADME properties (lipophilicity and solubility) are presented. The red dots represent the RMSE values of RF models, while the blue dots indicate the RMSE values for AttentiveFP models. The gray dots indicate the performance of the null model as a baseline. The RMSE values have the same units as the properties which all are in logarithmic scale. Each model’s values are divided into five categories: mundane NA, surprising NA, additive, mmp, and rest compounds. The percentage values indicate the relative number of compounds in these categories
The AttentiveFP models clearly outperform the RF models when it comes to predicting ADME properties. However, the AttentiveFP models for on-target properties do not surpass the performance of the RF models. While we here only test stratified cross-validation, we expect that other validation setups show a similar result. The compounds which are part of the additive set are typically predicted the best. This is anticipated as a ML model can effectively learn from compounds that exhibit additive behavior. It aligns with findings from Kwapien et al., who asserted that datasets composed solely of additive data points are simpler to predict compared to those containing nonadditive data [27]. On the other hand, the nonadditive sets (i.e. mundane NA and surprising NA sets) generally have a smaller margin between the baseline and prediction, indicating these are the most challenging sets to predict. While the overall model performance varies across all datasets, a clear hierarchy of prediction quality appears. This order, based on RMSE values, is additive < mmp < mundane < surprising ~ rest.
Discussion
NA is a key phenomenon in SAR data that requires attention from drug design teams. Previously, NA has been estimated to affect around 10% of all compounds in a SAR dataset [1]. We here show that the majority of those cases, 70–80%, are mundane kinds of NA that are to be expected due to direct substituent interactions, linker exchanges, stereochemistry assignment issues, and transformations that in combination change the majority of the compounds. Those cases are likely to be expected by experienced design teams. Yet, for the remaining 20–30% of cases, NA comes as a surprise. On a fundamental level, drug design would not be possible without additivity, as otherwise every new combination of substituents would give a very unpredictable result, and many more compounds would have to be synthesized. It would be very useful if we can reduce the amount of surprise. Design teams need to be aware of when to expect additivity and when they cannot in order to steer their synthetic efforts and increase the efficiency of SAR exploration.
Using a set of novel interpretable descriptors that characterize double transformation cycles, we show that transformations whose change in logP add up to a large amount, are likely to cause high NA. Other descriptors that indicate a change in polarity, such as ΔTPSA and ΔHBA, are also correlated with higher NA. For structural descriptors like ΔRotBonds or ΔHAC, on the contrary, we did not find a clear correlation with a change in NA. More work will be required to better understand the individual reasons for NA, in particular understanding this correlation with changing the polarity. To this end, NA has been explained with specific occurrences, such as water networks [2], ligand conformational changes [28], protein side chain flipping [4], or complete ligand rearrangements [3]. However, all of these explanations are retrospective - for drug design teams, it would be very useful if we can derive rules that guide design teams on when to synthesize and test more substituent combinations.
Our findings suggest a distinct behavior of NA for on-target datasets compared to ADME datasets, with on-target datasets exhibiting more NA than ADME datasets (see Table S2). Small molecule drug discovery projects typically aim to identify molecules that interact with proteins in a very specific manner. The primary on-target potency is driven by highly specific protein-ligand interactions, where the compound’s conformation significantly influences and dictates the binding affinity. Therefore factors that impact ligand conformation such as chirality, ortho effects and hydrogen bond formation may heavily impact the SAR by virtue of changes in molecular recognition. The extent of this impact will depend on the characteristics of the pocket and its flexibility. In turn, NA analysis examines the extent to which changes in binding affinity of two substructures of a ligand binding in two distinct regions of the binding pocket are independent of each other. Subtle or more significant changes in ligand conformation may therefore result in high NA. Complex effects such as changes in water networks or ligand binding entropy might also influence NA. In contrast, ADME properties are generally less controlled by specific pharmacophore sites or molecular features and are instead more dependent on the physicochemical properties of the entire molecule. Within the four ADME properties included in our analysis, aqueous solubility showed a significantly higher average absolute NA than lipophilicity, permeability and clearance (see Table S2). Lipophilicity is empirically an additive molecular property while permeability is correlated with lipophilicity and with additive physicochemical descriptors such as the number of hydrogen bonding atoms, polar surface area and rotatable bond count [29, 30]. Microsomal intrinsic clearance might be expected to be affected by molecular recognition by metabolizing enzymes but in practice, at the level of matched molecular pairs, the change in lipophilicity plays a significant role [31]. Solubility is heavily influenced by lipophilicity but also by crystal lattice energy. The molecular recognition event important for solubility is ligand-ligand interactions in the crystal lattice and this is not an additive property. Moreover, measured solubility values are also influenced by sample purity and the nature of counterions which may even lead to different kinetic solubility results for different batches of the same compound. Along with displaying more NA than the other ADME properties, solubility is also the most challenging for the ML models to predict as evidenced by the highest RMSE for AttentiveFP being associated with solubility (see Table S5). Nonetheless, it is interesting that with all four ADME properties we observed similar NA trends for molecular polarity properties as were observed with the on-target datasets (Fig. 8).
Further investigation into the rare examples of high NA within the lipophilicity and permeability datasets revealed that in the majority of cases it could be attributed to an obvious measurement error in one of the four compounds within the cycle. Replacement of the measured value in such cases with the predicted value from an in silico model and subsequent recalculation of the NA reduced the absolute NA of these cycles to within normal experimental error for additive behavior. Inspection of the few remaining high NA cycles showed that the NA could very often be rationalized by a difference in ionization state for one of the four compounds compared to the other three. Ionization has a large effect on lipophilicity and permeability - the ionized fraction partitions largely into the aqueous layer in the LogD7.4 assay and has much lower permeability in the parallel artificial membrane permeability assay (which is run at pH 6.5). An example of such a cycle is shown in Fig. 11. Compound 1 is the only compound that does not have a very high fraction ionized (> 99%) at pH 7.4 and pH 6.5 and therefore has far higher measured lipophilicity and permeability values than the other three compounds.

DTC from the lipophilicity (LogD7.4) and PAMPA permeability (LogPeff) datasets that displays high NA attributed to a difference in fraction ionized for one of the four compounds
The observation that the majority of the observed high NA in the lipophilicity and permeability datasets could be attributed to a large error in the experimental determination of one or more compounds in the cycles leads to a different interpretation of the apparently poor performance of the ML models in predicting the compounds belonging to high NA circles, namely that the model is in reality giving a more accurate estimate of the property value than the experimental result. We have found that when a particular compound is flagged by the NA analysis as showing consistently highly positive or negative per-compound NA [5] and has a large difference between its ML predicted and measured value in the same direction, this is a very strong indication of a mis-measured data point that should be followed up by a new measurement.
This research, for the first time, presents a systematic data analysis on NA across a broad spectrum of on-target assays. We have restricted our analysis to in-house datasets where all experimental values were obtained under the same conditions and repeat measurements were available. This is particularly relevant for NA analysis in order to minimize the experimental uncertainty associated with each assay, and allows us to assume that the noise from each measurement is taken from the same distribution. Given that this analysis aimed to identify the impact of descriptors on NA, it was crucial to be extremely careful in minimizing any additional noise. Experimental uncertainty can have a large impact on SAR analysis. The distribution of NA values is directly tied to an upper limit of experimental uncertainty or experimental noise, which is typically between 0.1 and 0.3 log units [19].
Typically, NA is a property of local SAR, consisting of four compounds. In a few cases, apparent NA is caused by a single error in the database: This could be a wrong measurement, or the wrong compound assigned to a structure. NA analysis will point to compounds that are consistently part of cycles with huge NA. The experiments and structures of those compounds should be checked individually. In our study, we were able to identify inconsistency in the stereochemical assignment in one case, leading to apparent NA. Stereochemistry is tricky, as the exact stereochemistry is often not known, and the relative stereochemistry can be captured in a format that does not translate to the input SMILES strings used for NA analysis. NA analysis will point out such cases and requires corrections to make the assignment consistent on a SMILES level.
NA has a strong influence on the performance of ML models. Gogishvili et al. and Kwapien et al. have shown before that the predictive performance of ML models substantially drops for compounds involved in high NA SAR [1, 27]. Previous work has also shown that ML models have challenges predicting activity cliffs [32, 33]. Activity cliffs are a property for pairs of compounds, whereas NA is a property of four compounds in a cycle and the cycle could contain activity cliffs without necessarily exhibiting NA. We here confirm the finding that ML models have challenges when predicting properties of compounds that show high NA. We further expand this finding by showing that there is a hierarchy of ML models performance, depending on how well compounds are embedded in known local SAR. The hierarchy in order of decreasing performance is additive < mmp < mundane < surprising ~ rest. This is in line with previous research on applicability domain, yet offers a different, arguably more intuitive approach to understand predictivity of ML models. The hierarchy holds for both on-target and ADME datasets. High NA, no matter whether surprising or mundane, leads to compounds being predicted as badly as compounds that have no MMP neighbors, i.e. are chemically very distant to the rest of the dataset. Most likely, the overall performance of ML models will, inter alia, depend on how densely the SAR space is sampled within the training dataset. Will descriptor- or graph-based models even be able to accurately capture and model the reasons for NA? Given the results we show here, we feel that this is an important future research question for ML modeling.
The categories that we introduce here - additive/nonadditive, MMP and rest - offer an alternative to the traditional model applicability domain, as they are able to separate compounds into such that are predicted well and others that are not. Traditionally, applicability domain is evaluated as Tanimoto similarity to the five nearest molecules in the training set [34]. The classification we offer here is arguably more intuitive, as it is based on the molecular transformations, the most fundamental way to analyze SAR datasets. Compounds from the double transformation sets have at least three close neighbors related by substructural transformations, facilitating more accurate predictions in a higher resolved SAR space. The compounds in the rest set are structurally the most dissimilar, followed by those in the MMP set. The three sets in which double transformation exists - additive, surprising and mundane - have comparable median Tanimoto coefficients but still differ substantially when it comes to the accuracy of predictions within them. Applicability domain based only on Tanimoto similarity is intrinsically different from similarity based on substructure/mmp consideration. Therefore we argue that it is crucial to better understand NA to improve ML models.
Although we have not generated 3D conformations of the compound sets or incorporated protein information, the established descriptors are designed to capture 3D information at the ligand level. However, if applicable, project teams would always analyze the SAR results together with 3D modeling. Ideally, the insights derived from NA analysis could stimulate thought processes for highlighting potential NA effects and strategies to circumvent them. Possible outcomes could include examining outliers through repeated measurements or aiding in the assignment of stereochemistry, synthesizing and testing new combinations of R-groups, and monitoring for significant changes in certain properties. We chose to keep the analysis within the cheminformatics realm for rapid computation and immediate NA assessment. Beyond the scope of this study, one could generate docking poses for all four members of a cycle, and evaluate the RMSD between the four compounds. Alternatively, one could perform FEP calculations to determine the binding free energy differences between the four compounds and see whether FEP can recapitulate high NA.
Conclusions
In this contribution, we further developed the understanding of causes of NA in molecular activity by analyzing a dataset of double transformation cycles from various consistent in-house datasets. We identified descriptors that help differentiate between mundane, i.e. explainable, and surprising NA in cycles. Factors such as the proximity of substituent groups, transformation similarity, size of molecular changes, linker exchanges, and changes in stereochemistry were found to substantially increase NA. These descriptors can help project teams filter out cycles with mundane NA and focus on the surprising NA cases that require further investigation for unexpected SAR patterns.
We also investigated the relationship between NA and molecular properties derived from polarity and steric factors. The difference in lipophilicity (ΔLogP) was found to have the most profound effect on NA. Notably, there were distinct NA behaviors observed between on-target and ADME datasets, highlighting the importance of precise spatial location of compound moieties in determining binding activity for on-target properties.
For ML model predictions, we show that the precision differs remarkably between different NA categories, including surprising NA, mundane NA, additive, MMP, and rest. The models performed better on compounds in the additive set, while compounds in the NA sets were more challenging to predict. This categorization of NA introduces a novel way to measure the applicability domain and suggests the use of new descriptors to enhance predictability.
Overall, NA is still a key SAR phenomenon that we are only beginning to understand. Further investigations will be needed to understand and predict NA in SAR datasets, which will in turn be crucial to improve ML models and SAR exploration strategies.
Data availability
The code for performing nonadditivity analysis is available at https://github.com/Roche/NonadditivityAnalysis. While the datasets analyzed in this study are proprietary and not publicly accessible, the PDE10 dataset is provided in the repository to illustrate usage.
Abbreviations
- NA:
-
Nonadditivity
- MMP:
-
Matched molecular pair
- DTC:
-
Double transformation cycle
- SAR:
-
Structure–activity relationship
- ML:
-
Machine learning
- RF:
-
Random forest
- CB2:
-
Cannabinoid type 2
- BACE1:
-
Beta-Secretase 1
- PDE10:
-
Phosphodiesterase type 10
- DPP4:
-
Dipeptidyl peptidase-4
- MAGL:
-
Monoacylglycerol lipase
- DDR1:
-
Discoidin Domain Receptor Tyrosine Kinase 1
- ADME:
-
Absorption, Distribution, Metabolism and Excretion
- GLYT1:
-
Glycine transporter type-1
- ATX:
-
Autotaxin
- SMN2:
-
Survival of motor neuron 2
- AEP:
-
Asparagine endopeptidase
References
Gogishvili D, Nittinger E, Margreitter C, Tyrchan C (2021) Nonadditivity in public and inhouse data: implications for drug design. J Cheminformatics 13:47. https://doi.org/10.1186/s13321-021-00525-z
Biela A, Betz M, Heine A, Klebe G (2012) Water makes the difference: rearrangement of water solvation layer triggers non-additivity of functional group contributions in protein-ligand binding. ChemMedChem 7:1423–1434. https://doi.org/10.1002/cmdc.201200206
Kramer C, Fuchs JE, Liedl KR (2015) Strong nonadditivity as a key structure–activity relationship feature: distinguishing structural changes from assay artifacts. J Chem Inf Model 55:483–494. https://doi.org/10.1021/acs.jcim.5b00018
Gomez L, Xu R, Sinko W et al (2018) Mathematical and Structural characterization of strong nonadditive structure–activity relationship caused by protein conformational changes. J Med Chem 61:7754–7766. https://doi.org/10.1021/acs.jmedchem.8b00713
Kramer C (2019) Nonadditivity Analysis. J Chem Inf Model 59:4034–4042. https://doi.org/10.1021/acs.jcim.9b00631
Krummenacher D, He W, Kuhn B et al (2023) Discovery of orally available and Brain Penetrant AEP inhibitors. J Med Chem 66:17026–17043. https://doi.org/10.1021/acs.jmedchem.3c01804
Hunziker D, Reinehr S, Palmhof M et al (2022) Synthesis, characterization, and in vivo evaluation of a novel potent autotaxin-inhibitor. Front Pharmacol 12
Hilpert H, Guba W, Woltering TJ et al (2013) β-Secretase (BACE1) inhibitors with high in vivo efficacy suitable for clinical evaluation in Alzheimer’s Disease. J Med Chem 56:3980–3995. https://doi.org/10.1021/jm400225m
Nettekoven M, Adam J-M, Bendels S et al (2016) Novel triazolopyrimidine-derived cannabinoid receptor 2 agonists as potential treatment for inflammatory kidney diseases. ChemMedChem 11:179–189. https://doi.org/10.1002/cmdc.201500218
Richter H, Satz AL, Bedoucha M et al (2019) DNA-Encoded Library-Derived DDR1 inhibitor prevents fibrosis and renal function loss in a genetic mouse model of Alport Syndrome. ACS Chem Biol 14:37–49. https://doi.org/10.1021/acschembio.8b00866
Lübbers T, Böhringer M, Gobbi L et al (2007) 1,3-Disubstituted 4-aminopiperidines as useful tools in the optimization of the 2-aminobenzo[a]quinolizine dipeptidyl peptidase IV inhibitors. Bioorg Med Chem Lett 17:2966–2970. https://doi.org/10.1016/j.bmcl.2007.03.072
Pinard E, Alanine A, Alberati D et al (2010) Selective GlyT1 inhibitors: Discovery of [4-(3-Fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a Promising Novel Medicine to treat Schizophrenia. J Med Chem 53:4603–4614. https://doi.org/10.1021/jm100210p
Tosstorff A, Rudolph MG, Cole JC et al (2022) A high quality, industrial data set for binding affinity prediction: performance comparison in different early drug discovery scenarios. J Comput Aided Mol Des 36:753–765. https://doi.org/10.1007/s10822-022-00478-x
Ratni H, Karp GM, Weetall M et al (2016) Specific Correction of Alternative Survival Motor Neuron 2 splicing by small molecules: Discovery of a potential Novel Medicine to treat spinal muscular atrophy. J Med Chem 59:6086–6100. https://doi.org/10.1021/acs.jmedchem.6b00459
Alsenz J, Kansy M (2007) High throughput solubility measurement in drug discovery and development. Adv Drug Deliv Rev 59:546–567. https://doi.org/10.1016/j.addr.2007.05.007
Wagner B, Fischer H, Kansy M et al (2015) Carrier mediated distribution system (CAMDIS): a new approach for the measurement of octanol/water distribution coefficients. Eur J Pharm Sci 68:68–77. https://doi.org/10.1016/j.ejps.2014.12.009
Chen X, Murawski A, Patel K et al (2008) A Novel Design of Artificial membrane for improving the PAMPA Model. Pharm Res 25:1511–1520. https://doi.org/10.1007/s11095-007-9517-8
Wildman SA, Crippen GM (1999) Prediction of Physicochemical parameters by Atomic contributions. J Chem Inf Comput Sci 39:868–873. https://doi.org/10.1021/ci990307l
Kramer C, Dahl G, Tyrchan C, Ulander J (2016) A comprehensive company database analysis of biological assay variability. Drug Discov Today 21:1213–1221. https://doi.org/10.1016/j.drudis.2016.03.015
Pedregosa F, Varoquaux G, Gramfort A et al Scikit-learn: machine learning in Python. Mach Learn PYTHON
Xiong Z, Wang D, Liu X et al (2020) Pushing the boundaries of molecular representation for Drug Discovery with the graph attention mechanism. J Med Chem 63:8749–8760. https://doi.org/10.1021/acs.jmedchem.9b00959
Paszke A, Gross S, Massa F et al (2019) PyTorch: an imperative style, High-Performance Deep Learning Library. Advances in neural information Processing systems. Curran Associates, Inc
RDKit Open-source cheminformatics
Dalke A, Hert J, Kramer C (2018) J Chem Inf Model 58:902–910. https://doi.org/10.1021/acs.jcim.8b00173. mmpdb: An Open-Source Matched Molecular Pair Platform for Large Multiproperty Data Sets
Leach AG, Pilling EA, Rabow AA et al (2012) Enantiomeric pairs reveal that key medicinal chemistry parameters vary more than simple physical property based models can explain. MedChemComm 3:528–540. https://doi.org/10.1039/C2MD20010D
Hall LH, Kier LB (1991) The Molecular Connectivity Chi indexes and Kappa shape indexes in Structure-Property Modeling. Reviews in Computational Chemistry. Wiley, Ltd, pp 367–422
Kwapien K, Nittinger E, He J et al (2022) Implications of Additivity and Nonadditivity for Machine Learning and Deep Learning models in Drug Design. ACS Omega 7:26573–26581. https://doi.org/10.1021/acsomega.2c02738
Kuhn B, Mohr P, Stahl M (2010) Intramolecular Hydrogen Bonding in Medicinal Chemistry. J Med Chem 53:2601–2611. https://doi.org/10.1021/jm100087s
Veber DF, Johnson SR, Cheng H-Y et al (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623. https://doi.org/10.1021/jm020017n
Diukendjieva A, Tsakovska I, Alov P et al (2019) Advances in the prediction of gastrointestinal absorption: quantitative structure-activity relationship (QSAR) modelling of PAMPA permeability. Comput Toxicol 10:51–59. https://doi.org/10.1016/j.comtox.2018.12.008
Dossetter AG (2012) A matched molecular pair analysis of in vitro human microsomal metabolic stability measurements for methylene substitution or replacements – identification of those transforms more likely to have beneficial effects. MedChemComm 3:1518. https://doi.org/10.1039/c2md20226c
van Tilborg D, Alenicheva A, Grisoni F (2022) Exposing the Limitations of Molecular Machine Learning with Activity cliffs. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.2c01073
Tamura S, Miyao T, Bajorath J (2023) Large-scale prediction of activity cliffs using machine and deep learning methods of increasing complexity. J Cheminformatics 15:4. https://doi.org/10.1186/s13321-022-00676-7
Sheridan RP (2015) The relative importance of Domain Applicability Metrics for estimating prediction errors in QSAR Varies with Training Set Diversity. J Chem Inf Model 55:1098–1107. https://doi.org/10.1021/acs.jcim.5b00110
Acknowledgements
We would like to acknowledge each project team at Roche that contributed to the generation of the on-target data sets analyzed in this study, as well as to Björn Wagner and Kenichi Umehara for their contributions to the ADME data sets. We thank Michael Reutlinger for providing valued input and code for ML models.
Author information
Authors and Affiliations
Contributions
L.G. and N.M. are shared first authors. N.M. wrote the code and performed NA analysis. L.G. and C.K. supervised the study and wrote the paper. J.G.C. provided input to the design of the study and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Laura Guasch and Niels Maeder are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guasch, L., Maeder, N., Cumming, J.G. et al. From mundane to surprising nonadditivity: drivers and impact on ML models. J Comput Aided Mol Des 38, 26 (2024). https://doi.org/10.1007/s10822-024-00566-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10822-024-00566-0