
Abstract
Generative approaches to molecular design are an area of intense study in recent years as a method to generate new pharmaceuticals with desired properties. Often though, these types of efforts are constrained by limited experimental activity data, resulting in either models that generate molecules with poor performance or models that are overfit and produce close analogs of known molecules. In this paper, we reduce this data dependency for the generation of new chemotypes by incorporating docking scores of known and de novo molecules to expand the applicability domain of the reward function and diversify the compounds generated during reinforcement learning. Our approach employs a deep generative model initially trained using a combination of limited known drug activity and an approximate docking score provided by a second machine learned Bayes regression model, with final evaluation of high scoring compounds by a full docking simulation. This strategy results in molecules with docking scores improved by 10–20% compared to molecules of similar size, while being 130 × faster than a docking only approach on a typical GPU workstation. We also show that the increased docking scores correlate with (1) docking poses with interactions similar to known inhibitors and (2) result in higher MM-GBSA binding energies comparable to the energies of known DDR1 inhibitors, demonstrating that the Bayesian model contains sufficient information for the network to learn to efficiently interact with the binding pocket during reinforcement learning. This outcome shows that the combination of the learned latent molecular representation along with the feature-based docking regression is sufficient for reinforcement learning to infer the relationship between the molecules and the receptor binding site, which suggest that our method can be a powerful tool for the discovery of new chemotypes with potential therapeutic applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Identifying a new compound active against a desired target within the immensity of chemical space requires screening libraries of drug-like molecules, currently at sizes of billions of compounds [1], for new leads that occur with a best case frequency of approximately 1 in 1000 (1 in 30,000) for 1 μM (100 nM) compounds [2]. Both traditional computer aided drug design (CADD) and new machine learning (ML) drug design approaches are concerned with increasing the rate of new active identification and the quality of the actives identified. Using CADD this goal is often accomplished through virtual screening, in which known compounds from a database such as ZINC [3] or REAL [4] are rapidly evaluated either by docking against a known protein structure [5], or by evaluating their similarity to known active compounds [6]. Conversely, machine learning approaches often attempt to accelerate this process by eliminating the library and instead incorporating generative models that are capable of designing new active molecules directly [7].
The efficacy of the traditional CADD approach was recently illustrated in one of the largest ever reported virtual screening and synthetic campaigns [2]; in this work, Lyu et al. screened 138 million make on demand molecules against the D4 receptor using DOCK3.7 [8] and selected for testing 549 compounds with different scaffolds across a range of docking scores. In general, when considering known active compounds, docking scores typically show greater correlation with geometric root mean squared deviation from the known binding pose than with activity [9]. This was also true of the assayed results reported by Lyu et al., the docking scores showed little correlation with individual compound activity, however the probability of a compound being active was shown to correlated strongly, with the top scoring compounds having upwards of 25% hit rates compared to below 1% at lower scores. A comparable machine learning driven effort is that of Zhavoronkov et al. [10] who used a variational auto encoder (VAE) combined with a tensor train encoded gaussian mixture model (GMM) to localize sampling to regions of high activity within the VAE latent space. Using this approach, Zhavoronkov et al. generated 30,000 new molecules that were then prioritized using traditional CADD techniques, and ultimately identified a new Ponatinib [11] derivative with 9 nM activity. Subsequently, a number of other generative methods have contributed significantly to the identification of new preclinical lead candidates [12, 13].
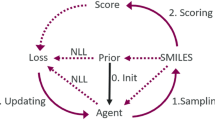
Based on the success of the above campaigns, we set out to combine the generative model described by Zhavoronkov et al. [10] with a reward function that could rapidly and accurately approximate the docking affinity of the generated compounds. The goal of this combination is to overcome the known limitation of generative molecular models which tend toward high similarity with the active molecules used to condition the generative process [14] as well as lessening the amount of initial data needed for such a model to be applicable to a new drug target [15]. To approximate the docking scores, we investigated several techniques and show Bayesian Ridge regression based on Morgan radius 2 finger prints (MFP2, equivalent to ECFP4) [16] to have the best combination of accuracy, speed, and extensibility beyond the training set. An overview of the training process is depicted in Fig. 1A.

Pipeline Overview A Training of the Reinforcement Learner: Training of our pipeline occurs in three distinct stages. In Stage (1) known active molecules are combined with reference molecules and used to train the GMM-VAE. This yields a probability distribution that models the location of the known actives within the latent space and a decoder model that converts the latent space coordinate to a SMILEs string. In Stage (2) random molecules are generated based on the initial probability distribution and docked using SMINA. These docking scores along with those of the known actives are used to train a regression model to predict docking score based on molecular feature. In Stage (3) the regression model is converted to an exponential reward function, and used to update the probability distribution through reinforcement learning. After training, new compounds are generated and high scoring compounds are validated by docking. B T-SNE Embedding of the Reinforcement Learning Trajectory: This plot was generated from the MFP2 features of known and generated molecules and shows the progress of the reinforcement learner over 2000 training epochs. At each indicated epoch, 500 molecules were sampled and included in the embedding. Known DDR1 inhibitors are shown in red, and inhibitors of other kinases in khaki. As seen in the embedding, learning occurs primarily in two modes, iteration on the scaffold of known actives resulting in random exploration of the chemical space locally around training compounds (see the circled region), and population drift starting from the center of the distribution guided by the reinforcement gradient toward high scoring scaffolds (As indicated by the arrows). C Sample Generated Molecules Shown are 6 molecules that demonstrate docking scores well above the average of known inhibitors (− 9.5), low ChEMBL similarity, and high weight normalized docking score. Poses of these molecules are available in Supplemental Fig. 2. Depictions of the latent space are a 3D T-SNE embedding of the latent space descriptors of the training molecules. Coloring is based on the training sets
We are not the first to consider combining traditional CADD approaches with machine learning. Other researchers have directly incorporated docking programs as scoring functions within larger machine learned models designed to iteratively optimize the molecular docking scores [17,18,19]. Another popular approach is using machine learning as a fast prescreening step [20,21,22] to identify molecules from a library that are likely to have high docking scores prior to the more time consuming step of running the docking simulation. Most similar to this work, Choi and Lee presented V-Dock [20] a multilayer perceptron (MLP) model for predicting docking scores, QED, and Tanimoto similarity of molecules to known targets. This was combined with MolFinder [23], a SMILEs based evolutionary optimization algorithm, to develop diverse molecules with high docking scores and QED. In another similar recent work, Ciepliński et al. [24] developed a docking-based benchmark from 1200–10,000 docked compounds for 4 different targets. For each target, an MLP was trained that could predict the docking score of compounds toward that protein, with the MLP output then being used as an optimization goal for three generative networks (CVAE [25], GVAE [26], and REINVENT [27]) resulting in molecules with docking scores considerably above the 50th percentile of random ZINC molecules, though below the 10th percentile. Our work expands upon these previous efforts in several ways. First, our GMM-VAE model is considerably more complex than previously explored generative models primarily due to the GMM mapping of the latent space generated during the initial training. This allows us to employ a transfer learning approach wherein the GMM is initially trained on known actives, but approximate docking scores are used during reinforcement learning, enabling a greater diversity of generated compounds, while still incorporating known activity data within the GMM. Finally, we show that Bayes regression based on MFPs compares comparably to the MLP approaches in the previous works, and is independent of similarity to known active compounds.
As in the case of Zhavoronkov et al. [10], we elected to investigate the discoidin domain receptor 1 (DDR1), a receptor tyrosine kinase implicated in fibrosis and solid tumor formation [28,29,30]. We show that our approach results in a significant increase in average docking score as compared to a similarity only reinforcement across all ranges of similarity to known molecules and all molecular weights. Further, by comparison to randomly selected molecules from the ZINC database using scores normalized by molecular weight, we calculate an approximately 670-fold enrichment in the best scoring (< − 0.03/amu) compounds across all molecular weights. When accounting for the time necessary to train the encoder and conduct reinforcement learning, this corresponds to a roughly 130-fold speed up in top scoring compound identification. Finally, we show that these increased docking scores correlate with docking poses with interactions similar to known inhibitors and result in high MM-GBSA binding energies comparable to known DDR1 inhibitors, demonstrating that the Bayesian model contains sufficient information for the network to learn to efficiently interact with the binding pocket during reinforcement learning.
Results and discussion
Training the GMM-VAE
The GMM-VAE was trained in batches of 1000 molecules composed of 25%/25%/10%/40% of DDR1 kinase inhibitors (1.5 k)/other kinase inhibitor (10 k)/non-kinase inhibitors (10 k)/uninvestigated druglike molecules (1.6 M). Training was conducted over 50 epochs where each epoch consisted of complete encoding of the uninvestigated druglike molecules with each other set randomly sampled to fill the batch. Molecules were represented as simplified molecular input line entry system (SMILES) strings that were converted to vectors using an integer embedding to represent the atom and bond characters. Stereochemistry was omitted from the encoding. The GMM model was conditioned using a binary activity flag that indicated whether a molecule was active against any kinase with an IC50 of less than 10uM. During training, the loss function (see Zhavoronkov et al. [10]) decreased rapidly during the first 5 epochs as the autoencoder reached upwards of 99% accuracy in reconstruction of the SMILES strings. Over the remaining 45 epochs, the GMM model continued to converge, though improvements were minimal in the final epochs. After training, the GMM was used to direct sampling from the regions of the latent space predicted to have high likelihood of containing DDR1 inhibitors. Prior to reinforcement learning, approximately 25% of samples yielded valid SMILES strings. Visualization of the sampling and training molecules using a t-distributed stochastic nearest neighbor (T-SNE) [31] embedding of the MFP2 features showed samples to be mostly normally distributed within the T-SNE space with regions of high concentration near the DDR1 inhibitors, and scattered samples among the other kinase inhibitors (Fig. 1B, Supplemental Fig. 1). Given the significant difference between the latent representation and the T-SNE, this represents a well-trained and diversely featured model.
Developing and training the docking regressor
In developing the docking regressor, we had several desired characteristics for the constructed model. First it must be accurate enough to enable optimization of the docking score during reinforcement learning. Second, it must be fast enough in execution to be included in the reinforcement learning, without meaningfully increasing the training time. Third, the loss of accuracy outside the training domain must be gradual; the generation of new molecules necessarily means some extrapolation outside the training data must occur, meaning any model that loses accuracy rapidly will be unsuitable for new chemotype development. To ensure our training data spanned the entirety of the chemical space considered we used SMINA [32] to dock all collected DDR1 and other kinase inhibitors, as these define the breadth of the chemical space considered as seen in the T-SNE embeddings. We supplemented this with an additional 2500 molecules sampled from the GMM-VAE, to account for any chemical groups enriched in that model but absent in the set of kinase inhibitors.
Using this docking data, we trained 16 common regression models (See Table 1) available in Scikit-Learn [33] to predict the docking scores based on the molecules MFP2 features. To generate an initial set of new molecules from the GMM-VAE, we initially conducted reinforcement learning in a manner similar to Zhavoronkov et al. [10] using two self-organized maps (SOMs) [34] that return the probability of a molecule being either a DDR1 kinase inhibitor, or a kinase inhibitor of any type. After reinforcement, new molecules were generated and docked, and these generated molecules were used as a blind validation for each regressor model. Pearson’s r, R2, mean absolute error(MAE), and root mean squared error (RMSE) were used as the regressor evaluation criteria. For each model, ten fold cross validation was used to measure the uncertainty in these parameters. Execution time was also measured for each model. Based on these criteria Bayesian ridge regression narrowly outperformed stochastic gradient descent, with the remaining regressors showing considerably worse performance. Additionally, the Bayesian Ridge regressor takes only 2 ms per epoch to estimate the docking score, a negligible amount of time compared to the 2.5 s needed to conduct the sampling and reinforcement, and a considerable speed up compared to the 0.4 h needed to locally conduct the docking scoring for the same number of molecules. Of note, Bayesian regressors have been used extensively by the Liotta group at Emory University for molecular design and virtual synthesis showing them to be accurate and versatile with little training data [35] and useful in the development of inhibitors against a range of targets [36,37,38,39].
Reinforcement learning
To conduct reinforcement learning, we converted the output of the Bayesian Ridge regressor to a reward function based on the average of the known inhibitors. We tried several forms for this function and found an exponential function to result in the fastest reinforcement learning process. Our sampling strategy was a combination of direct sampling of the GMM model and an exploratory sampling based on the average and standard deviation of latent space values sampled. We settled on a 90/10 ratio of these two modes based on the empirical result that either 90/10 or 10/90 ratios resulted in the best performing molecules, with all ratios between exhibiting lower performance. At each epoch in the reinforcement process, 500 molecules were sampled and the average reward was calculated and baselined using the previous averages. Models were saved at various points during the training up to 3000 epochs, and for each saved model, 500 molecules were sampled, and docking conducted. Based on a combination of docking scores, generation of valid SMILES strings, and the percentage of unique SMILES strings, 1000 epochs was selected as the optimal stopping point (Supplemental Fig. 3). While continued improvement in docking scores occurred after this point, it was primarily due to tighter convergence that resulted in fewer unique molecules being generated.
We also used T-SNE embeddings to visualize the convergence of the reinforcement model (Fig. 1B, Supplemental Fig. 1A). From this view it is clear that the reinforcement learning occurs primarily by two separate processes. First is optimization local to the known inhibitors by modification of those scaffolds with new substituents that improve the docking score, but do not correspond to large movements in the chemical space. Second is a gradient descent away from the center of the chemical space in the direction of the highest docking compounds identified.
Performance of generated compounds
To evaluate the performance of the compounds generated by the reinforcement learning process, we first generated and docked a total of 30,000 compounds. As controls we used four separate sets: (1) 30,000 molecules provided by Zhavoronkov et al. (generated using the same network architecture, but a similarity-based SOM reward function) [10], (2) 30,000 molecules sampled from ZINC with a molecular weight and LogP Distribution matching the DDR1 inhibitors, (3) 2,500 molecules sampled from the GMM-VAE prior to reinforcement learning, and (4) the known DDR1 inhibitors. Of these sets, the compounds generated using our method had the highest average docking score, though a sub-population of known DDR1 inhibitors clearly outperforms any generated compound (Fig. 2A). Surprisingly, the randomly sampled ZINC molecules also perform quite well, though we expect this would decrease if the LogP and molecular weight constraints were removed (as would be the case against a target with fewer known inhibitors).

Evaluation of Compounds Generated by Bayesian Guided Reinforcement Learning Molecular sets considered are DDR1—the known DDR1 inhibitors, ZINC—randomly sampled from a set with molecular weight and LogP matching DDR1, Zhavoronkov—molecules provided as supplemental information and generated based only on molecular similarity, GMM-VAE—molecules generated in this work prior to reinforcement learning, RL—molecules generated in this work based on Bayesian reinforcement learning A Distribution of docking scores B Distribution of molecular weights, C Distribution of docking efficiency (score/MW), D docking score plotted versus molecular weight, E distribution of docking scores in each molecular weight range, F Distribution of docking efficiency in bins defined by the maximum Tanimoto similarity to any known CHEMBL molecule. In E, F numbers below each distribution indicate the percentage of that set represented by the distribution
Accounting for the molecular weight distribution of the sets helps to explain these observations. The generated compounds tend to be lower in molecular weight than most of the considered molecules (Fig. 2B) ranging primarily between 280 and 380 amu, roughly corresponding to the size of molecules considered “lead-like” (typically 250–350) [40]. This may be partially due to the relatively low molecular weight (< 340 amu) of the MOSES molecules used as training data, however, a decrease in average molecular weight during reinforcement learning also suggests identifying better scoring, high molecular weight compounds to be more difficult within this framework. To allow more even comparison across these groups, we also considered the docking efficiency of each compound (docking score / molecular weight), a common metric that corrects for the tendency of large molecules to be higher scoring on the basis of surface area alone [41]. This metric reveals significant optimization within the generated set compared to all others with an average docking efficiency of − 0.03 amu−1 compared to approximately − 0.02 amu−1 for the ZINC, GMM-VAE, and known DDR1 inhibitors (Fig. 2C). An alternate view is provided in Fig. 2D which shows the docking score plotted against molecular weight for each molecular set, with the generated compounds being at the top of the combined distribution for all molecular weights at which they are present. A version of the same plot binned by molecular weight (Fig. 2E) shows the distribution of scores of each molecular set, in each bin.
Finally, because generative models tend to perform best closest to their training data, we considered how the docking efficiency of our compounds was related to their similarity to known inhibitors. To do so, for each molecule we queried the CHEMBL API for the Tanimoto similarity [42] of the most similar molecule in the database. These similarities were used to bin the molecules, and the distribution of docking efficiency in each bin is shown in Fig. 2F. In each bin the generated compounds have the highest efficiency, and while all sets show decreasing efficiency with decreasing similarity to known inhibitors, the efficiency drop off is markedly lower for the compounds generated here.
Time comparison versus docking only
Finally, we consider the relative time needed to identify 10,000 high docking efficiency (< − 0.03amu−1). In our ZINC set, these occurred at a rate of approximately 0.07%, meaning approximately 14 million compounds would need to be docked. At a rate of approximately 1000 molecules per hour on our server, this would correspond to a wall time of over 18 months. By contrast, training of the GMM-VAE takes roughly 48 h, an additional 20 h were required for docking the molecules used to train the reward function, reinforcement learning took 6 h, and docking of the sampled compounds takes another 30 h, approximately 4.3 days total, or a speed up of 134 times compared to docking alone.
Comparison of docked protein–ligand interactions
When designing our training approach, we were most interested in creating a model in which docking scores could supplement known activity data to allow generative modelling to be deployed against developing targets. High docking scores on their own however are not meaningful indicators of activity. Docking a simple alkane such as dodecane will often give high scores due to high van der Waals contacts from the flexible backbone while being devoid of potential pharmaceutical value. In contrast, a successful docking campaign aims to identify important interactions between the ligand and protein that are responsible for activity. While our training approach did not explicitly account for any specific interactions, we were interested in the consistency of the interactions of the generated molecules, and how these interactions compared to those of the known inhibitors. Essentially, we sought to determine whether this reinforcement learning strategy can develop something akin to a pharmacophore model within the network weights.
To attempt to answer this question, we generated ligand interaction fingerprints (LIFPs) for the highest scoring docking pose of each known inhibitor, the randomly selected ZINC molecules, and each generated molecule using the ProLIF package [43]. ProLIF creates a one-hot encoded vector of ligand interactions where each feature represents a residue and type of interaction (e.g. vdW, pi-pi, H-bond, etc.) resulting in a detailed representation of the ligand pose. We compared the ProLIF fingerprints of each batch of molecules using several metrics. To qualitatively analyze the trends during reinforcement learning, we created a t-SNE embedding of the LIFPs of molecules generated during reinforcement learning and compared them to the randomly selected ZINC molecules and the known inhibitors (Fig. 3). In this representation, the ZINC molecules are spread most widely within the t-SNE space, along with the molecules from early training epoch samples. With more training epochs, however, the generated molecules are found more concentrated in the areas of highest known inhibitor concentration, demonstrating that despite the low chemical similarity to the known inhibitors the binding interactions are similar. This suggests that the model has some internal understanding of isosteric substitutions. The increased similarity with known inhibitor binding modes was further shown by the average maximum Tanimoto similarity of the LIFP of each generated compound to a known inhibitor; this metric increased from approximately 0.75 at the beginning of reinforcement learning to 0.85 after 1000 epochs of training, demonstrating that the increased docking scores are also correlated with binding modes that are increasingly similar to the known inhibitors. Note that these similarities are high comparable to the MFP Tanimoto scores due to the much smaller size of the LIFP (132) compared to MFP (2048). Interestingly, the randomly selected Zinc molecules also displayed high Tanimoto similarities in this metric, averaging 0.78, though the docking scores were consistently lower.

tSNE Embedding of Ligand Interaction Fingerprints during reinforcement learning. As training proceeds, the generated molecules localize near high concentrations of known inhibitors
Comparison of MM-GBSA binding energies
As a final evaluation, we also considered MM-GBSA binding energies of the generated molecules as a comparison to both the compounds generated using the SOM reward function, DDR1 inhibitors that had entered clinical trials, and the six molecules selected by Zhavoronkov et al. [10] For each molecule, we conducted a 30 ns molecular dynamics simulation in OpenMM [44] using the top scoring docked pose as the starting geometry, and calculated the MM-GBSA binding affinity using the last 15 ns of each trajectory. Rankings generated by this methodology are more reliable than docking due to the more comprehensive forcefield used for scoring, the consideration of multiple nearby conformations, and a better representation of solvent effects [45]. However, due to the greater computational demands the number of molecules studied is necessarily reduced. To create a representative sample from both the Bayes and SOM reward function generated molecules, we clustered the highest-scoring compounds using a 0.6 Tanimoto similarity cut-off for cluster inclusion. We then analyzed the top 21 cluster centers which corresponded to roughly the top 200 molecules. As seen in Fig. 4, the MM-GBSA energies of the top scoring molecules generated using the Bayes reward function are significantly higher than those generated using the SOM only approach, and are slightly above both the seven DDR1 inhibitors that have advanced to clinical trials, including four approved drugs. The six molecules selected by Zhavoronkov also perform well here, comparable to the clinical inhibitors. These, however, were selected from 30,000 compounds after considerable additional screening using docking and pharmacophore analysis and are of comparatively high similarity to the known inhibitors. Our training approach more directly yields compounds with MM-GBSA properties comparable to known inhibitors with higher novelty suggesting that incorporation in future development pipelines can enrich the generated molecules in high-binding energy compounds.

Comparison of the distribution of MM-GBSA calculated Gibbs binding energies of the molecules in each set. Use of the estimated docking score (Bayes) significantly improves predicted binding energies over the similarity only based approach (SOM) to parity with clinical DDR1 inhibitors. The molecules included in each set can be seen in Supplemental Fig. 5
Conclusions
We have demonstrated a new training strategy for the generation of new lead-like molecules using deep learning and a GMM-VAE generative model. The key to this approach is the implementation of a Bayesian ridge regression model to predict the docking score of generated compounds during reinforcement learning. We have shown that Bayesian ridge regression outperforms other regression approaches for predicting the docking score of generated molecules. The molecules generated using our approach have higher docking scores in all ranges of molecular weights generated, though future work is needed to increase this range. Additionally, unlike the comparison set, docking efficiency was consistently high even as the molecular similarity to known molecules decreased. Further, an analysis of the protein–ligand interaction fingerprints of the docked molecules shows that this reinforcement approach yields molecules that converge toward the binding pattern of known inhibitors, demonstrating the ability of the model to both generate and retain an internal model of pharmacophore interactions based on the docking trends during reinforcement learning. Finally, MM-GBSA analysis of the generated compounds showed the top-scoring compounds from the Bayesian reward function to have average binding affinities comparable to the set of DDR1 inhibitors that have proceeded to clinical trial, and significantly higher than the molecules generated from the similarity-based SOM reward function. The computational resources needed to conduct this work are modest, and the inclusion of the docking data does not substantially increase the model training time with data collection to lead compound generation occurring in as little as two weeks. We anticipate the work demonstrated here will allow researchers to identify new lead molecules more efficiently than traditional docking studies that require high levels of parallelism to screen molecular databases in a time efficient manner. Finally, we believe this result is significant in creating a foundation to extend generative models to more data limited regimes such as one-shot [46] and few-shot [15] learning against newly identified targets.
Methods
Computational resources
All machine learning calculations were conducted on a dual GPU server with 2 × 32 GB Geforce3090TI GPUs with dual AMD EPYC 7302 16-Core Processors, and 128 GB RAM.
Other than for benchmarking purposes, docking was conducted using resources provided by the Nanjing University Scientific Computing Center.
Collection of training data
Training data is divided into several groups, each of which was collected using slightly different methods based on the importance of that set to the model goal. DRR1 Inhibitors: These molecules were collected from ChEMBL [47], the CDDI database, and the patent literature by way of SciFinder [48]. Other Kinase Inhibitors: These molecules were collected solely from ChEMBL. Non-Kinase Inhibitors: These molecules were collected solely from ChEMBL. Lead and Druglike Molecules: These molecules were assembled from the MOSES benchmark set [49] and the ZINC database [50]. ZINC molecules were selected from the drug-like set with clean reactivity. These were further sub-selected to match the molecular weight and LogP distribution of the DDR1 Inhibitors. This was accomplished by dividing the DDR1 Inhibitors into two dimensional bins defined by strides of 10 in molecular weight and 0.2 in LogP. The percent of all DDR1 molecules in each bin was then used to bootstrap a training set of ZINC molecules within the same property range. Detailed information for the origin of the training data is available in the Supplementary Information.
Molecular docking
Molecular docking was performed using SMINA [32] a version of the popular Autodock Vina [51] software modified for higher throughput. The structure of DDR1 was obtained from the Protein Data Bank, accession number 6FEX, a 1.29 Å resolution crystal structure containing CHEMBL4635278 a 29 nM IC50 DDR1 inhibitor [52]. CHEMBL4635278 was used to define the docking site with the autobox_ligand parameters. The receptor was prepared using the built in Dock Prep and Minimization routines of UCSF Chimera [53] to add protons and alleviate any steric clashes that resulted. The GMM-VAE is encoded without stereochemical information, but generates molecules with stereocenters; therefore, for each generated smiles, we generate every stereoisomer using RDKit [54] prior to docking. Also in RDKit, an initial minimum energy 3D conformation was generated for each molecule by enumerating 100 structures and minimizing using the UFF forcefield and retaining the lowest energy conformation. Atomic charges were then assigned using the mmff94 forcefield [55] within Open Babel [56]. Docking was then conducted using these prepared structures with seed 0 and exhaustiveness 8. After docking, for the purposes of reward function generation, molecules were again processed using RDKit, stereocenters removed, and SMILES deduplicated, keeping the highest docking score. SMINA Docking scores are derived from known physical interactions (a van der Waals-like potential (defined by a combination of a repulsion term and two attractive Gaussians), a nondirectional hydrogen-bond term, a hydrophobic term, and a conformational entropy penalty), and are nominally in kcal/mol. However, given the significant modifications made to these parameters toward the goal of pose replication over binding affinity, they are at best a pseudo-energy. As such we have referred to them throughout in a unitless notation to avoid confusion with experimental affinity values.
Generation of the reward function
All regression models were generated using Scikit Learn with the default parameters. Molecular features were non-stereoisomeric MFP2 features generated from RDKit. The SOM pseudo-regression was built using MiniSOM [57]. The number of neurons in the SOM was calculated as 5√N where N is the number of molecules in the training set. For each neuron the average docking score of the contained molecules was returned for any new molecules that activated that neuron. Docking scores were then converted to a reward function using several functional forms, the most successful of which was a bounded exponential reward:
where R is the reward function, mfp is the morgan fingerprint for each molecule, \(\widehat{DS}\) is the regression function used for docking score prediction. Threshold is the minimum docking score that we expect generative molecules to achieve after reinforcement, set as -9. A is a scaling factor for numerical stability, such that predicted scores of 12 give rewards of 1. B controls the aggressiveness of the reward above and below the target minimum value. Values were typically 0.5 and 0.1.
GMM-VAE
The GMM VAE used is a modified version of the Generative Tensorial Reinforcement Learning (GENTRL) model provided by Zhavoronkov et al. [10] The most major change is the replacement of the self-organized map (SOM) reward function with a reward derived from a regression model of docking scores derived from a test data set. The remainder of the network structure was left intact, though several hyperparameters were adjusted for improved performance. First the initial learning rates were increased in the GMM-VAE (1e-4 to 1e-3, and a 5% learning rate decay per epoch was added) and the reinforcement learning (RL, 2e-5 to 1e-5) training. The training penalty of − 5 for invalid SMILES was reduced to 0. Batch size in reinforcement learning was increased from 200 to 500. These changes collectively reduced overfitting of the model and increased the diversity of molecules generated. The generative model was rewarded when the structures of generated molecules got excessive scores in reward function:
where \(\overleftarrow {R}_{Batch}\) is the average reward function values of molecules in current batch.
Data availability
All necessary data and code to evaluate or reproduce this work are publicly available through the cited sources, or included as supplemental files with this submission. Data: Training data as curated by our team is available as a supplemental file. The sources of the molecules are indicated in Supplementary Table 1. Software: Smina can be obtained from https://sourceforge.net/projects/smina/. The GENTRL source code can be obtained from https://github.com/insilicomedicine/GENTRL. Information for how to obtain scikit-learn is available at https://scikit-learn.org/.
References
Lyu J, Irwin JJ, Shoichet BK (2023) Modeling the expansion of virtual screening libraries. Nat Chem Biol 19:712–718. https://doi.org/10.1038/s41589-022-01234-w
Lyu J, Wang S, Balius TE et al (2019) Ultra-large library docking for discovering new chemotypes. Nature 566:224–229. https://doi.org/10.1038/s41586-019-0917-9
Irwin JJ, Tang KG, Young J et al (2020) ZINC20—a free ultralarge-scale chemical database for ligand discovery. J Chem Inf Model 60:6065–6073
Shivanyuk AN, Ryabukhin SV, Tolmachev A et al (2007) Enamine real database: making chemical diversity real. Chemistry today 25:58–59
Varela-Rial A, Majewski M, De Fabritiis G (2022) Structure based virtual screening: Fast and slow. WIREs Comput Mol Sci 12:e1544. https://doi.org/10.1002/wcms.1544
Bragina ME, Daina A, Perez MA et al (2022) The SwissSimilarity 2021 web tool: novel chemical libraries and additional methods for an enhanced ligand-based virtual screening experience. Int J Mol Sci 23:811
Martinelli DD (2022) Generative machine learning for de novo drug discovery: a systematic review. Comput Biol Med 145:105403. https://doi.org/10.1016/j.compbiomed.2022.105403
Coleman RG, Carchia M, Sterling T et al (2013) Ligand pose and orientational sampling in molecular docking. PLoS ONE 8:e75992. https://doi.org/10.1371/journal.pone.0075992
Xu W, Lucke AJ, Fairlie DP (2015) Comparing sixteen scoring functions for predicting biological activities of ligands for protein targets. J Mol Graph Model 57:76–88. https://doi.org/10.1016/j.jmgm.2015.01.009
Zhavoronkov A, Ivanenkov YA, Aliper A et al (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 37:1038–1040. https://doi.org/10.1038/s41587-019-0224-x
Gainor JF, Chabner BA (2015) Ponatinib: accelerated disapproval. Oncologist 20:847–848. https://doi.org/10.1634/theoncologist.2015-0253
Zeng X, Wang F, Luo Y et al (2022) Deep generative molecular design reshapes drug discovery. Cell Rep Med. https://doi.org/10.1016/j.xcrm.2022.100794
Li Y, Zhang L, Wang Y et al (2022) Generative deep learning enables the discovery of a potent and selective RIPK1 inhibitor. Nat Commun 13:6891. https://doi.org/10.1038/s41467-022-34692-w
Grant LL, Sit CS (2021) De novo molecular drug design benchmarking. RSC Med Chem 12:1273–1280. https://doi.org/10.1039/D1MD00074H
Vella D, Ebejer J-P (2022) Few-shot learning for low-data drug discovery. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.2c00779
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742–754
Jeon W, Kim D (2020) Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci Rep 10:22104. https://doi.org/10.1038/s41598-020-78537-2
Thomas M, Smith RT, O’Boyle NM et al (2021) Comparison of structure- and ligand-based scoring functions for deep generative models: a GPCR case study. J Cheminform 13:39. https://doi.org/10.1186/s13321-021-00516-0
Sadybekov AA, Sadybekov AV, Liu Y et al (2022) Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601:452–459. https://doi.org/10.1038/s41586-021-04220-9
Gentile F, Yaacoub JC, Gleave J et al (2022) Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat Protoc 17:672–697
Berenger F, Kumar A, Zhang KYJ, Yamanishi Y (2021) Lean-docking: exploiting ligands’ predicted docking scores to accelerate molecular docking. J Chem Inf Model 61:2341–2352. https://doi.org/10.1021/acs.jcim.0c01452
Bucinsky L, Bortňák D, Gall M et al (2022) Machine learning prediction of 3CL SARS-CoV-2 docking scores. Comput Biol Chem 98:107656. https://doi.org/10.1016/j.compbiolchem.2022.107656
MolFinder: an evolutionary algorithm for the global optimization of molecular properties and the extensive exploration of chemical space using SMILES | Journal of Cheminformatics | Full Text. https://jcheminf.biomedcentral.com/articles/https://doi.org/10.1186/s13321-021-00501-7. Accessed 21 Jun 2023
Ciepliński T, Danel T, Podlewska S, Jastrzȩbski S (2023) Generative models should at least be able to design molecules that dock well: a new benchmark. J Chem Inf Model 63:3238–3247. https://doi.org/10.1021/acs.jcim.2c01355
Gómez-Bombarelli R, Wei JN, Duvenaud D et al (2018) Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci 4:268–276
Kusner MJ, Paige B, Hernández-Lobato JM (2017) Grammar variational autoencoder. In: International conference on machine learning. PMLR, pp 1945–1954
Olivecrona M, Blaschke T, Engkvist O, Chen H (2017) Molecular de-novo design through deep reinforcement learning. J Cheminform 9:48. https://doi.org/10.1186/s13321-017-0235-x
Gao Y, Zhou J, Li J (2021) Discoidin domain receptors orchestrate cancer progression: a focus on cancer therapies. Cancer Sci 112:962–969. https://doi.org/10.1111/cas.14789
Moll S, Desmoulière A, Moeller MJ et al (2019) DDR1 role in fibrosis and its pharmacological targeting. Biochimica et Biophysica Acta (BBA) - Mol Cell Res 1866:118474. https://doi.org/10.1016/j.bbamcr.2019.04.004
Tian Y, Bai F, Zhang D (2022) New target DDR1: A “double-edged sword” in solid tumors. Biochimica et Biophysica Acta (BBA) -Rev Cancer 1878:188829
Hinton GE, Roweis S (2002) Stochastic neighbor embedding. Advances in neural information processing systems 15. https://proceedings.neurips.cc/paper_files/paper/2002/hash/6150ccc6069bea6b5716254057a194ef-Abstract.html
Koes DR, Baumgartner MP, Camacho CJ (2013) Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model 53:1893–1904
Pedregosa F, Varoquaux G, Gramfort A et al (2011) Scikit-learn: machine learning in python. J Machine Learn Res 12:2825–2830
Kohonen T (1990) The self-organizing map. Proc IEEE 78:1464–1480
Kaiser TM, Burger PB, Butch CJ et al (2018) A machine learning approach for predicting HIV reverse transcriptase mutation susceptibility of biologically active compounds. J Chem Inf Model 58:1544–1552
Kaiser TM, Dentmon ZW, Dalloul CE et al (2020) Accelerated discovery of novel ponatinib analogs with improved properties for the treatment of parkinson’s disease. ACS Med Chem Lett 11:491–496
Pribut N, Kaiser TM, Wilson RJ et al (2020) Accelerated discovery of potent fusion inhibitors for respiratory syncytial virus. ACS Infect Dis 6:922–929
Cox BD, Prosser AR, Sun Y et al (2015) Pyrazolo-piperidines exhibit dual inhibition of CCR5/CXCR4 HIV entry and reverse transcriptase. ACS Med Chem Lett 6:753–757
Shi Q, Kaiser TM, Dentmon ZW et al (2015) Design and validation of FRESH, a drug discovery paradigm resting on robust chemical synthesis. ACS Med Chem Lett 6:518–522
Lipinski CA (2004) Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1:337–341
Pan Y, Huang N, Cho S, MacKerell AD (2003) Consideration of molecular weight during compound selection in virtual target-based database screening. J Chem Inf Comput Sci 43:267–272
Bajusz D, Rácz A, Héberger K (2015) Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Chem 7:1–13
Bouysset C, Fiorucci S (2021) ProLIF: a library to encode molecular interactions as fingerprints. J Cheminform 13:72. https://doi.org/10.1186/s13321-021-00548-6
Eastman P, Swails J, Chodera JD et al (2017) OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 13:e1005659
Tuccinardi T (2021) What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin Drug Discov 16:1233–1237. https://doi.org/10.1080/17460441.2021.1942836
Altae-Tran H, Ramsundar B, Pappu AS, Pande V (2017) Low data drug discovery with one-shot learning. ACS Cent Sci 3:283–293. https://doi.org/10.1021/acscentsci.6b00367
Mendez D, Gaulton A, Bento AP et al (2019) ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 47:D930–D940
Gabrielson SW (2018) SciFinder. J Med Libr Assoc: JMLA 106:588
Polykovskiy D, Zhebrak A, Sanchez-Lengeling B et al (2020) Molecular sets (MOSES): a benchmarking platform for molecular generation models. Front Pharmacol 11:565644
Sterling T, Irwin JJ (2015) ZINC 15–ligand discovery for everyone. J Chem Inf Model 55:2324–2337
Trott O, Olson AJ (2009) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem NA-NA. https://doi.org/10.1002/jcc.21334
Richter H, Satz AL, Bedoucha M et al (2018) DNA-encoded library-derived DDR1 inhibitor prevents fibrosis and renal function loss in a genetic mouse model of Alport syndrome. ACS Chem Biol 14:37–49
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Bento AP, Hersey A, Félix E et al (2020) An open source chemical structure curation pipeline using RDKit. J Cheminform 12:1–16
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519
O’Boyle NM, Banck M, James CA et al (2011) Open Babel: an open chemical toolbox. J Cheminform 3:1–14
Vettigli G (2022) MiniSom
Acknowledgements
Support for this work was provided by start up funds issued by Nanjing University and by Ice-Kredit.
Funding
Support for this work was provided by startup funds issued by Nanjing University and by Ice-Kredit.
Author information
Authors and Affiliations
Contributions
YWs numbered as in the author order. YX YW1 YW2 PY CB and JW collected and analyzed data and created figures. YW3 LG and CB developed and guided the research. CB wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
LG is the CEO and shareholder of IceKredit.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiong, Y., Wang, Y., Wang, Y. et al. Improving drug discovery with a hybrid deep generative model using reinforcement learning trained on a Bayesian docking approximation. J Comput Aided Mol Des 37, 507–517 (2023). https://doi.org/10.1007/s10822-023-00523-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-023-00523-3