
Abstract
This work presents a quantum mechanical model for predicting octanol-water partition coefficients of small protein-kinase inhibitor fragments as part of the SAMPL6 LogP Prediction Challenge. The model calculates solvation free energy differences using the M06-2X functional with SMD implicit solvation and the def2-SVP basis set. This model was identified as dqxk4 in the SAMPL6 Challenge and was the third highest performing model in the physical methods category with 0.49 log Root Mean Squared Error (RMSE) for predicting the 11 compounds in SAMPL6 blind prediction set. We also collaboratively investigated the use of empirical models to address model deficiencies for halogenated compounds at minimal additional computational cost. A mixed model consisting of the dqxk4 physical and hdpuj empirical models found improved performance at 0.34 log RMSE on the SAMPL6 dataset. This collaborative mixed model approach shows how empirical models can be leveraged to expediently improve performance in chemical spaces that are difficult for ab initio methods to simulate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The capability of in silico methodologies to assess lipophilicity is an important research area in computer-aided drug design as this physical property is a key descriptor of molecular interactions between chemical ligands and biological targets. Precise and robust in silico lipophilicity predictions could reduce the cost of generating and optimising a drug candidate through accelerating and improving the results of lead discovery in virtual screening, reducing the need for physical lipophilicity assaying for ligands, and enhancing the assessment of bioavailability and drug toxicity.
To that end, many in silico approaches have been studied with the aim of developing methodologies that can accurately, precisely, and/or expediently predict octanol water partition coefficient (LogP) values for untested ligands comparable to experimental results. They range from molecular modelling explicitly simulating the hydrophilic and hydrophobic intermolecular interactions between the ligand and solvents, to statistical models correlating chemical features derived from previously assayed molecules to their experimental LogP results, thereby predicting LogP values as a function of the chemical features presented from unseen ligands. The partition coefficient can also be derived from the electronic structure of the ligands with the use of quantum mechanical approaches since solvation changes the solute electronic structure [1].
Quantum mechanical approaches may explicitly include solvent molecules in hybrid quantum mechanical/molecular mechanics calculations, or approximate solvent interactions with implicit continuum solvation models. The explicit inclusion of solvent molecules requires the non-trivial computation of solvent-molecule interaction geometries which greatly increases the modelling cost. Implicit solvation models represent solvated ligands within an electrostatic cavity interacting with a dielectric medium that approximates the solvent which is parameterised from physicochemical descriptors such as the dielectric constant and refractive index. Continuum solvation can model this partitioning by calculating the free energy of transfer of the molecule moving from a non-interacting ideal gas phase to the condensed phase of either solvent as shown in Eq. (1). These models are well parameterised for neutral solutes with relatively short computation times compared to methodologies that include explicit solvent molecules [2,3,4].
Investigating modest cost ab initio LogP prediction methodologies
This study will apply the universal solvation model based on solute electron density (SMD) [5] to investigate implicit solvation model performance for determining the partition coefficient from calculated solvation free energies for two datasets. As shown in Eq. (2) and [5], SMD calculates the free energy of solvation \((\Delta G_{S} )\) as the sum of two components consisting of the bulk electrostatic interactions term \((\Delta G_{ENP} )\), calculated from the self-consistent reaction field treatment using the Polarizable Continuum Model (PCM) in Orca [6, 7], and the cavity-dispersion solvent-structure term \((\Delta G_{CDS} )\) that describes solute-solvent interactions in the first solvation shell. This\(( \Delta G_{CDS} )\) term is calculated from descriptors empirically fitted to experiment in Eq. (3) consisting of the atomic surface tension of each atom k\(({\sigma }_{k})\), the molecular surface tension \(({\sigma }^{\left[M\right]})\), and the solvent accessible surface area (SASA), which is dependent on the geometry \((\mathbf{R})\) and the solvent radius \(({r}_{s})\) added to the set of all atomic van der Waals radii \(({R}_{\begin{array}{c}{Z}_{k}\\ \end{array}})\).
The free energy of solvation is calculated with both aqueous and n-octanol solvent parameterisations from a molecular geometry optimised in the gas phase to derive the LogP in Eq. (1). SMD has been extensively studied with high performance for calculating solvation free energies of other ligand families, such as alcohols [2] and organophosphates [1].
The SMD solvation model was highly predictive when used with the hybrid M06-2X Minnesota functional [8] in previous studies [9] and SAMPL challenges [10], however, this method is only recommended to be used for main group chemistry [11]. The elucidation of a physical LogP prediction methodology that is applicable to the wider chemical domain such as organometallics could enable novel applications for LogP prediction models, such as in empirical modelling, where metallic structures are generally discarded as they are incompatible with their calculation methodology. The local M06-L functional [12] omits exact exchange energy calculations compared to the 54% that is used by M06-2X and has been found to perform well in transition metal systems [8]. The M06-L functional also achieved better LogP prediction performance than the M06-2X functional in the toxicological domain [1]. This study will investigate the lipophilicity prediction performance of the M06-L functional through a direct methodological substitution of the M06-2X functional. It is hypothesised the use of the M06-L functional would not substantially reduce predictive performance compared to M06-2X as it has been shown that M06-L can achieve similar performance to M06-2X methods [1].
Addressing SMD model shortcomings with a collaborative approach
The SAMPL6 physical property prediction challenge offers a collaborative opportunity to evaluate and compare distinct approaches for calculating the octanol water partition coefficient through a blind prediction challenge of 11 protein kinase-inhibitor fragments. The current strategy draws upon methodological experience from the empirical modelling field in validating models implementing both Minnesota functionals on a preliminary dataset in the public domain comprised of neutral organic molecules with structural similarity to the SAMPL6 dataset [13] prior to submission to the blind prediction challenge. In doing so, this study will determine the performance differences between the M06-L and M06-2X functionals for calculating the water-octanol partition coefficient with the SMD solvation model. SAMPL6 LogP Prediction Challenge model submissions are divided into several in silico categories based on their methodology. This brings together empirical models that have been constructed with consideration paid to the chemical space of the SAMPL6 challenge molecules. These empirical models feature different characteristics that could be used to address methodological shortcomings of the current physical methodology such as the computationally efficient incorporation of hydrogen bonding properties through empirically modelled descriptors to avoid computationally costly explicit solvation modelling. To that end, this study investigated the octanol water partition coefficient performance using an ensemble model that averages the predictions from an empirical model with our physical model generated in this study.
Methods
Dataset preparation
The SAMPL6 dataset was collated from the SMILES structures of the 11 protein kinase-inhibitor fragments following the chloride salt removal and neutralisation in SM09 and SM12. A preliminary chemical dataset for model prediction validation prior to entry submission was also composed from the public domain OECD Guidelines for Test No. 117: Partition Coefficient (n-octanol/water) [14] (Table 1, Fig. 1). The OECD dataset is a ten-chemical subset of neutral, aromatic molecules resembling the SAMPL6 dataset, chosen from the 60-molecule reference set in SMILES notation. JChem for Office was used to calculate reference JChem LogP (JCLogP) values from these SMILES structures, JChem for Office 19.0.4, 2019, ChemAxon (https://www.chemaxon.com). Initial 3D geometries were generated from isomeric SMILES with a KNIME [15] pipeline consisting of the OpenBabel [16] node for initial 2D structure generation, the RDKit [17] 3D coordinate generation and geometry optimisation nodes. All initial 3D structure geometries were optimised over 50,000 iterations with the Merck Molecular Force Field (MMFF94) [18].

Flowchart from initial SMILES structures to LogP calculated value. The M2 model methodology that would be submitted as dqxk4 is highlighted in grey and omits single point energy calculations to instead use the last energy values from solvated geometry optimisation
Implicit solvation model prediction methodology
The M06-L functional was chosen in this study because it can calculate accurately across the periodic table at a modest computational cost [12] in comparison to M06-2X [11], which was selected due to high performance being found with SMD being parameterised against the M05-2X functional [5]. Orca 4.1.1 [6, 7] was used to implement all Minnesota functional family calculations in this project with the Def2-SVP basis set [19], atom-pairwise dispersion corrections with zero damping (D3Zero) [20], and the Resolution of Identity approximation for both Coulomb and Hartree-Fock Exchange integrals (RIJK) [21]. Since the M06 DFA family is also known to be grid size dependent [22, 23], geometry optimisation calculations were configured with the Lebedev434 angular grid and IntAcc = 5.01 radial grid followed with the Lebedev590 angular grid [24] and IntAcc = 5.34 radial grid for the final energy calculations. The M06-L functional was used for the initial gas phase geometry optimisations with 1.0e-09 au energy change SCF convergence criteria. Two model variants were developed using either the M06-L/SVP (M1) or M06-2X (M2) functionals for calculating the water and octanol free energies of solvation from the gas phase using the SMD solvation model. A Bayesian bootstrap with 10,000 replications was used to quantify the standard error of the mean for each chemical prediction. Model uncertainty was quantified using the root mean squared error (RMSE) for the partition coefficients of a 10-chemical subset from the OECD Guidelines for Test No. 117: Partition Coefficient (n-octanol/water) [14].
The SAMPL6 dataset predictions of the M2 model variant was chosen for submission with the dqxk4 identifier to the SAMPL6 LogP Prediction Challenge because it had the lowest RMSE error for evaluating the OECD dataset.
Octanol water partition coefficient calculation
Following the methodology outlined in [1], and in the SAMPL6 Challenge Overview, we take the difference in free energy of the molecule in both solvated phases:
We then calculate the octanol water partition coefficient by substituting \(\Delta G_{{transfer}}\) into Eq. (1):
where T is temperature in Kelvin and R is the gas constant.
This calculation methodology implements Eq. (1) without needing to calculate single point energies for an optimised structure in the gas phase. This enabled the submitted model (dqxk4) to omit any single point energy calculations to instead use the final single point energy value produced in the solvated phase geometry optimisation to calculate predicted LogP values.
Quantifying the effect of RIJK on LogP prediction performance
Single point energies were calculated for all structures optimised in the solvated phase at the M06-L/SVP or M06-2X/SVP levels of theory with and without RIJK. These single point energy values were used to calculate LogP values resulting in five additional model configurations for each dataset and are named in the format of ‘single point energy method’//’geometry optimisation method’.
Comparing M2 and M06-2X/SVP//M06-2X/SVP LogP prediction performance
The LogP prediction performance between M2, which uses the final energy values from solvated phase geometry optimisation conducted at the M06-2X/SVP level of theory, and M06-2X/SVP//M06-2X/SVP is compared in order to determine if discrete single point energy calculations are redundant.
Model comparison and collaborative composite model development methodology
Following the advice of the SAMPL6 virtual workshop, a collaboration with the empirical modelling entry hdpuj was formed to compare prediction methodologies by incorporating LogP values predicted using the RayLogP multilinear statistical QSPR model for both OECD and SAMPL6 datasets. This model was chosen as it featured practically equivalent performance to the extent of switching places depending on the metric used to rank SAMPL6 models. This greatly simplifies the empirical methodology as we can use model averaging without any further parameterisation to assign weights to each model based on performance since both M2 and RayLogP can be assumed to be equal. Consequently, the DRLogP composite predictive model was developed by averaging the predicted LogP values from the M2 and RayLogP models.
Results and discussion
The geometries of the 21 molecules optimised in the gas phase at the M06-L level of theory for both datasets as shown in Figs. 2 and 3. Further geometry optimisations were conducted with implicit water and octanol SMD solvation at either M06-L or M06-2X levels of theory for the M1 and M2 (dqxk4) physical model variants, respectively. The resulting structures were used for subsequent free energy calculations. The resulting solvation transfer energies were used to calculate the partition coefficients for both OECD and SAMPL6 datasets in Table 2 and quantitatively compared to experimental methods in Table 3. The Bayesian bootstrap quantified the 95% HDI of the primary error metrics used in this project.


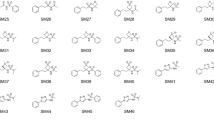
Gas phase 3D optimised structures for 11 SAMPL6 protein kinase inhibitor fragments; see also Table 2
The M2 model based on the M06-2X functional produced better predictive performance with 0.423 log RMSE and 0.363 log MAE compared to 0.453 log RMSE and 0.392 log MAE achieved by the M1 model in the OECD Reference dataset for the Physical methodologies studied in this work (Table 1). This resulted in the selection of M2 to generate blind predictions for submission dqxk4 in the SAMPL6 LogP Challenge. This could be attributed to either the M06-2X functional more precisely calculating the energy, or it may be due to M06-2X functional generating better structures in geometry optimisation compared to M06-L. A series of post hoc geometry optimisations and single point energy calculations were conducted using M06-2X and M06-L combinations as mixing both functionals decouples them and enables investigation of either factor. The use of M06-L for single point energy calculation with M06-2X geometry optimisation featured a higher 0.451 log RMSE compared to, vice versa, using M06-2X for single point energy calculation at 0.441 log RMSE (Table 3). M1 and M06-L/SVP//M06-2X/SVP also supports this finding as M1 has marginally poorer performance without M06-2X geometry optimisation with 0.453 log RMSE compared to 0.451 log RMSE (Table 3, Fig. 4c). This shows more precise energy calculation to have a greater performance impact than improved geometry optimisation for LogP prediction in the OECD Reference dataset. The SAMPL6 dataset finds the M06-2X functional to feature higher error with single point energy calculations, most substantially increasing the error from 0.454 log RMSE to 0.502 log RMSE when using the same M06-L optimised solvated structures in comparison to using M06-L single point energy calculations (Table 3). The M06-L functional performs better in the SAMPL6 dataset than M06-2X.

Correlation plot between experimental and predicted logP values in the OECD dataset with the submitted dqxk4 physical, hdpuj empirical, and DRLogP ensemble models in A. Each gradient shade indicates 0.5 log units error from perfect correlation
We also find the use of the M06-2X/SVP model for both solvated geometry optimisation and single point energy calculations to have equal performance to M2 at 0.423 log RMSE in the OECD dataset and 0.495 log RMSE in the SAMPL6 dataset (Table 3). This indicates the current methodology of using the final energy value from geometry optimisation does not have substantial predictive penalties and is a viable shortcut to skip subsequent single point energy calculations for a M06-2X optimised model.
Lastly, the use of the RIJK approximation in M06-2X models finds equivalent performance to corresponding M06-2X model configurations omitting them in the context of free energy of solvation calculations to predict LogP values (Figs. 4b, d, 5b, d). This finding was replicated across both datasets where the only detectable difference in performance was observed between the M06-2X/SVP//M06-2X/SVP model configuration with and without RIJK at 0.001 log MAE (Table 3), possibly induced via rounding error. The current results agree with those in the literature [25] as the RIJK approximation is known to produce low error and indicate the use of the RIJK integral approximation should be considered to enhance calculation throughput.

Correlation plot between experimental and predicted logP values in the SAMPL6 dataset with the submitted dqxk4 physical, hdpuj empirical, and DRLogP ensemble models in A. Each gradient shade indicates 0.5 log units error from perfect correlation
The M1 model performance does not substantially decrease with 0.454 log RMSE when predicting the SAMPL6 dataset compared to 0.453 log RMSE in the OECD dataset (Table 3). In comparison, all empirical and physical models, except for DRLogP, feature greater RMSE values on the SAMPL6 dataset than the OECD dataset. This resulted in the M06-2X M2 model featuring larger error than previously since in the literature [1] compared to the M06-L M1. This indicates the OECD dataset was not sufficiently representative of the SAMPL6 dataset to precisely rank each method and chemical dataset selection methodology needs improvement to enable better model screening.
High error was seen with OECD molecules 3, 4, and 10 for all models except for JCLogP (Table 1). Molecule SM14 from the SAMPL6 dataset was also overestimated by all models (Table 2). This indicates the current LogP models are not reliably predictive for weakly hydrophobic molecules with less than 2 log units.
In this study, the M2 model submitted as dqxk4 produced 0.494 log RMSE predictive performance on the SAMPL6 dataset in the physical model category, while the DRLogP mixed model found the best performance overall with 0.344 log RMSE. The high accuracy of the DRLogP mixed model could be attributed to the systematic cancelling of errors from averaging both models since M2 generally underestimates LogP while RayLogP generally overestimates LogP. This resulted in values that were more accurate to experimental values with ≤ 0.1 log unit variance for SM16, SM12, and SM02 and ≤ 0.25 log unit variance for SM04 and SM09 (Fig. 5). Averaging M2 and RayLogP also smoothed out model variance on all output predictions. This meant DRLogP had higher error than an individual model alone for molecules that can already well predicted, shown with SM07 and SM13 for M2 and followed with SM08 and SM16 for RayLogP. Conversely, this also enables DRLogP to output more consistent predictions by reducing the frequency of high variance (> 0.5 log unit) predictions to one molecule, SM15, compared to four molecules for RayLogP (SM14, SM16, SM13, SM09) and M2 (SM16, SM15, SM08, SM04). However, the use of the statistical average with no prediction weighting requires both models to have near equal performance otherwise the averaged model could generate lower performance than the higher performing individual model as demonstrated in the OECD dataset results. Here the M2 model had a lower RMSE (0.423 log units) than DRLogP (0.449 log units) due to RayLogP having a comparatively high 0.511 log RMSE. This could be implemented posteriori with a similar experimental design to the current study using a validation dataset that is representative of the blind prediction data to assess model prediction performance.
None of the physical, empirical, or mixed models in this study could precisely predict the lipophilicity trend for all six 4-amino quinazoline compounds. RayLogP featured the best result by ranking the last three compounds correctly (SM12, SM04, SM02) but this was not replicated in the mixed DRLogP model (Table 2).
Both M1 and M2 models underestimate the lipophilicity of 4-amino quinazoline compounds with halogen substituents, namely SM02 and SM12, along with SM04 for M2 (Table 2). In the M2 model, SM04 has the greatest deviation from experimental values with − 0.73 log units in comparison to SM07, which contains a phenyl functional group in place of an aryl halide functional group while SM02 and SM12 have a smaller variance between 0.2–0.3 log units. The M1 model slightly overestimates SM04, however, SM02 was underestimated at 0.3 log units from the experimental value. The cause of this deficiency may be attributed to the use of D3Zero dispersion corrections with the M06-2X functional in M2 which was found to reduce calculation accuracy for hybrid functions due to the complex nature of halogen bonding [26]. This could be further supported by the M2 model finding lower overall variance for the halogenated compounds in the OECD dataset which are smaller than those in the SAMPL6 dataset. These smaller molecules had reduced magnitude D3Zero dispersion corrections resulting in improved performance compared to SAMPL6.
Way forward to improve
The M2 (dqxk4) model performed with a relatively low error in the physical modelling category of the SAMPL6 LogP Prediction Challenge as shown in Fig. 6. However, there are many methodological improvements that future studies could implement to further enhance calculation performance.

Relative position of dqxk4 performance compared to other models in the physical category
Both gas and solvated phase geometry optimisations were calculated until convergence; however, no further analysis was conducted to confirm if the stationery points were at the ground state. Frequency calculations could be conducted to identify local minima but were omitted due to potential imaginary frequencies arising from cavity construction by CPCM instead of from the geometry being analysed in Orca.
This study did not modify the continuum solvation model, which assumes a homogeneous ‘pure’ medium, to reflect the SAMPL6 experimental methodology which used wet octanol. This could be addressed by incorporating aqueous microsolvation in the gas to octanol phase solvation geometry optimisation. Physical models implementing microsolvation have found results close to explicit solvation in modelling reactions [27] so it could improve the existing models by introducing a limited degree of hydrogen bonding to improve the structure of solvated molecules. This could result in a performance improvement when used with implicit solvation models.
The JCLogP empirical model had prediction performance that surpasses most of the current models in both OECD (0.269 log RMSE) and SAMPL6 (0.404 log RMSE) datasets studied in this project. This indicates there is substantial potential for methodological improvement for all modelling categories since the JCLogP model has been extensively modified since the initial parameterisation three decades ago [28].
The LogP prediction error of physical models for halogenated compounds could be reduced through the proposed use of larger basis sets and the omission of dispersion corrections when using density functional theory methodologies [26]. This may not be trivial to implement as larger basis sets greatly increase computational cost and the omission of dispersion corrections could reduce performance for large non-halogenated molecules. Alternatively, empirical corrections could be developed as the DRLogP model demonstrates high predictive accuracy for halogenated SAMPL6 compounds such as SM16, SM02, and SM12, effectively acting as a corrected M2 (dqxk4) model for marginal additional computational cost compared to improving physical methodologies. A permutation feature importance analysis of the RayLogP QSPR model revealed hydrogen bond donor electrotopological indices and electronegativity autocorrelation indices to be important descriptors for its SAMPL6 predictions, which may improve prediction for halogenated compounds. These variables could be implemented at higher levels of theory by physical models; a similar effort has recently been published to efficiently correct non-covalent interactions in density functional theory models using ensemble machine learning methodologies with < 0.22 kcal/mol compared to the gold standard CCSD(T)/CBS results [29].
Conclusions
The use of methods and techniques from the empirical modelling domain with physical methodologies demonstrate potential to efficiently enhance predictive performance. Subsequent analysis of predictions contributing to the combined model can identify strengths and shortcomings of the constitutive empirical and physical models. The current DRLogP model demonstrates enhanced performance due to the capability of the hdpuj empirical model to more precisely predict the LogP for moieties that were underpredicted by the physical dqxk4 model. The use of validation datasets needs to be more than superficially representative of the test dataset to effectively and precisely rank physical models before they are used for blind prediction.
The SAMPL6 LogP Prediction Challenge results have shown there are many SMD implementations with huge performance variation. Our M2 model is the best SMD implementation in the SAMPL6 LogP Prediction Challenge that was only bested by commercial models. This indicates there are many possible pitfalls in implementing a performant SMD model. For the best SAMPL6 LogP prediction performance with the SMD solvation model, use single zeta Karlsruhe basis sets with the M06-L functional, and implement solvated phase geometry optimisations with robust grid and convergence parameters.
References
Vlahovic F et al (2017) Density functional theory calculation of lipophilicity for organophosphate type pesticides. J Serb Chem Soc 82:104–104
Michalík M, Lukeš V (2016) The validation of quantum chemical lipophilicity prediction of alcohols. Acta Chim Slov 9(2):89
Zhang J et al (2017) Comparison of implicit and explicit solvent models for the calculation of solvation free energy in organic solvents. J Chem Theory Comput 13(3):1034–1043
Bayat Z, Movaffagh J (2010) The 1-octanol/water partition coefficient of nucleoside analogs via free energy estimated in quantum chemical calculations. Russ J Phys Chem A 84(13):2293–2299
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396
Neese F (2012) The ORCA program system. Wiley Interdiscip Rev: Computat Mol Sci 2(1):73–78
Neese F (2018) Software update: the ORCA program system, version 4.0. Wiley Interdiscip Rev: Comput Mol Sci 8(1):e1327
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120(1):215–241
Nedyalkova MA et al (2019) Calculating the partition coefficients of organic solvents in octanol/water and octanol/air. J Chem Inf Model 59(5):2257–2263
Jones MR, Brooks BR, Wilson AK (2016) Partition coefficients for the SAMPL5 challenge using transfer free energies. J Comput Aided Mol Des 30(11):1129–1138
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120(1–3):215–241
Zhao Y, Truhlar DG (2006) A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys 125(19):194101
Işık M et al (2019) Octanol-water partition coefficient measurements for the SAMPL6 blind prediction challenge. bioRxiv. https://doi.org/10.1007/s10822-019-00271-3
OECD (2004) Test No. 117: Partition coefficient (n-octanol/water), HPLC method, OECD guidelines for the testing of chemicals, Section 1, OECD Publishing, Paris. https://doi.org/10.1787/9789264069824-en
Berthold MR et al (2008) KNIME: the Konstanz information miner. Springer, Berlin
O'Boyle NM et al (2011) Open babel: an open chemical toolbox. J Cheminform 3(1):33
RDKit: Open-source cheminformatics. http://www.rdkit.org
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17(5-6):490–519
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305
Grimme S et al (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104
Weigend F, Kattannek M, Ahlrichs R (2009) Approximated electron repulsion integrals: Cholesky decomposition versus resolution of the identity methods. J Chem Phys 130(16):164106
Hohenstein EG, Chill ST, Sherrill CD (2008) Assessment of the performance of the M05-2X and M06-2X exchange-correlation functionals for noncovalent interactions in biomolecules. J Chem Theory Comput 4(12):1996–2000
Goerigk L, Grimme S (2011) A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys Chem Chem Phys 13(14):6670–6688
D. Laikov, C. van Wüllen. Lebedev-Laikov-Grids. http://www.ccl.net/cca/software/SOURCES/FORTRAN/Lebedev-Laikov-Grids/index.shtml
Kossmann S, Neese F (2009) Comparison of two efficient approximate Hartee-Fock approaches. Chem Phys Lett 481:240–243
Kozuch S, Martin JML (2013) Halogen bonds: benchmarks and theoretical analysis. J Chem Theory Comput 9(4):1918
Basdogan Y, Keith JA (2018) A paramedic treatment for modeling explicitly solvated chemical reaction mechanisms. Chem Sci 9(24):5341–5346
Viswanadhan VN et al (1989) Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J Chem Inf Comput Sci 29(3):163–172
Li W et al (2019) Efficient corrections for DFT noncovalent interactions based on ensemble learning models. J Chem Inf Model 59(5):1849–1857
Acknowledgements
We acknowledge the National Institutes of Health for supporting the experimental work carried out in the SAMPL6 logP Prediction Challenge.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guan, D., Lui, R. & Matthews, S. LogP prediction performance with the SMD solvation model and the M06 density functional family for SAMPL6 blind prediction challenge molecules. J Comput Aided Mol Des 34, 511–522 (2020). https://doi.org/10.1007/s10822-020-00278-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00278-1