
Abstract
Objective
To explore inheritance of the m.3697G > A mitochondrial DNA (mtDNA) mutation and the effectiveness of preimplantation genetic diagnosis (PGD) for the carrier.
Methods
The study encompassed a pedigree of m.3697G > A mtDNA mutation, including one asymptomatic patient who pursued for PGD treatment. Twelve cumulus oocyte complexes (COCs) were collected in the first PGD cycle and 11 COCs in the second cycle. The efficiency of cumulus cells, polar bodies, and trophectoderm (TE) in predicting the m.3697G > A heteroplasmy of embryos was analyzed.
Results
From 23 COCs, 20 oocytes were fertilized successfully. On day 5 and 6 post-fertilization, 15 blastocysts were biopsied. The m.3697G > A mutation load of TE biopsies ranged from 15.2 to 100%. In the first cycle, a blastocyst with mutation load of 31.7% and chromosomal mosaicism was transferred, but failed to yield a clinical pregnancy. In the second cycle, a euploid blastocyst with mutation load of 53.9% was transferred, which gave rise to a clinical pregnancy. However, the pregnancy was terminated due to fetal cleft lip and palate. The mutation loads of different tissues (47.7 ± 1.8%) from the induced fetus were comparable to that of the biopsied TE and amniotic fluid cell (49.7%). The mutation load of neither cumulus cells (R2 = 0.02, p = 0.58) nor polar bodies (R2 = 0.33, p = 0.13) correlated with TE mutation load which was regarded as a gold standard.
Conclusions
The m.3697G > A mutation showed a random pattern of inheritance. PGD could be used to reduce the risk of inheritance of a high mutation load. Cumulus cells are not a suitable predictor of blastocyst mutation load.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria are semi-autonomous organelles located in the cytoplasm, wherein the oxidative phosphorylation system (OXPHOS) generates ATP to supply cellular energy. Mitochondria contain their own genetic material, mitochondrial DNA (mtDNA), which is a closed circular molecule of 16,519 bp. Pathogenic mutations in mtDNA are able to contribute to multi-systemic mitochondrial diseases, such as Leigh’s syndrome (LS). LS is a mitochondrial encephalopathy caused by either nuclear or mitochondrial encoded gene mutation, which is involved in oxidative phosphorylation function. Affected children almost always develop disease within the first year of life, with rapid progression and even death [1, 2]. Unlike nuclear DNA, mtDNA exists as multiple copies, and mutated copies can exist alongside wild-type copies in any cell, a state known as heteroplasmy [3].
This state of heteroplasmy can occur in oocytes and is inherited by offspring, but inheritance of mtDNA mutations is further complicated by the existence of a genetic bottleneck in the female germline. Although an oocyte contains a large mtDNA copy number, only a small amount is inherited by the next generation of primordial germ cells in early development. The subsequent amplification of this small number of founders gives rise to unpredictable shifts in heteroplasmy across individual oocytes [4]. If this mutation load exceeds on onset threshold, it may lead to disabling or fatal maternally inherited mitochondrial diseases. There is currently no effective treatment for mitochondrial disease [5]. It has been reported that the incidence of mtDNA genetic disease is about 1/5000 in a UK population [6, 7], so there is great demand for techniques to prevent the inheritance of mtDNA diseases. Thus, the development of techniques to enable mtDNA disease patients to produce healthy children has become a pressing issue in reproductive research.
The variable mutation load between oocytes provides the opportunity to adopt established reproductive genetic techniques at the zygote/embryo stage. Following in vitro fertilization (IVF), preimplantation genetic diagnosis (PGD) can genotype embryos and allow selection of healthy embryos for transfer, preventing inheritance of genetic diseases. Subsequently, attempts were made to additionally measure mutation load of pathogenic mtDNA mutations during the routine PGD, and select embryos with lower mutation load to bypass inheritance of mtDNA disease. A consensus has emerged that a TE biopsy is more representative of the mutation load of the wider blastocyst than a PB [8]. Both cleavage stage and TE biopsy have successfully been used to produce live offspring with low mutation load of various mtDNA mutations [9,10,11,12]. However, given the unpredictable patterns of inheritance and presentation of various mtDNA diseases, there is no agreed upon threshold for a healthy heteroplasmic embryo, and this will likely vary by mutation. The long-term effect of these methods on the health of offspring remains to be seen.
Biopsy of the TE is a relatively difficult method. An uninvestigated alternative could be the cumulus cells of the oocyte, which play an important supportive role in oogenesis and maturation [13]. As cumulus cells are closely related to oocytes only before IVF, they may have potential as a simpler predictor of embryo mtDNA mutation load, without the biopsy of any embryo biomass. It would also open the possibility of a more ethical PGD approach, as oocytes would not have to be fertilized only to later be discarded.
We present data on PGD cycles of a carrier of the LS causing single-nucleotide polymorphism (SNP) m.3697G > A. In addition, we collected cumulus cells of oocytes prior to intracytoplasmic sperm injection (ICSI) and correlated the mutation load of cumulus cells to that of a later TE biopsy or whole embryo. This data will enable a greater understanding of m.3697G > A inheritance and how PGD can circumvent it, as well as aid optimization of PGD.
Case report
Case description
A 30-year-old, asymptomatic female carrier of m.3697G > A with 52.3% mutation load in blood requested IVF with PGD to prevent inheritance of LS in her offspring. The patient was informed of her carrier status after a LS diagnosis was made for her symptomatic 2-year-old daughter. Whole-exome sequencing and copy number variation analysis showed no abnormalities in the daughter. The daughter started walking at 1 year and 3 months of age but by the age of 2 was losing walking ability concomitant with hypophrenia. Brain MRI showed symmetrical damage of bilateral basal ganglia. Lactic acid and pyruvate concentration in blood were elevated (4.02 mmol/L and 174 µmol/L) relative to reference ranges.
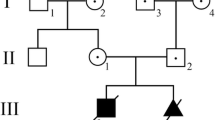
The LS causing m.3697G > A mutation was detected in the blood of all family members, ranging from 52.3 to 99.0%. The patient carried 52.3% heteroplasmy, while the symptomatic daughter carried 99%. A wide range of additional SNPS were detected (Table 1). A threshold of > 1%/ < 98% was applied in the identification of legitimate heteroplasmic mutations, leaving only the m.3697G > A mutation. The pedigree is visualized in Fig. 1, and their mtDNA in Fig. 2.

The pedigree

The predicted structure of mtDNA, the ND1 gene and the coded protein
Ethical approval
The entire mtDNA from blood samples of the family was amplified and sequenced. This study was approved by the Ethics Committee of the Anhui Medical University (Hefei, China; Approval No. 20150038). All methods described in the present study were performed with patient consent.
Methods
A GnRH agonist long protocol was performed in the patient’s first cycle, while an antagonist regimen was used in the second cycle. The COCs were transferred from follicular fluid into gamete medium (COOK, Australia) for preliminary washing, then individual COCs washed five times with fertilization medium (COOK, Australia). Cumulus cells surrounding oocytes were removed via pipette and collected in PCR tubes containing 2 µL PBS. Remaining oocytes were moved into hyaluronidase solution (Thermo Fisher, USA) in the incubator at 37 °C, and observed while blowing and sucking. The COCs were further blown and sucked by 150 μm denuding pipette for several times until cumulus cells almost fell off completely. Mature (MII) oocytes were then transferred into a 7.5 µL micromanipulation droplet of Gamete medium covered with sterile mineral oil. Oocytes were fertilized by ICSI using fresh sperm, via inverted microscope (Olympus IX71, Japan) equipped with a stage warmer (http://www.tokaihit.com) and micromanipulators (Narishige, Japan). Following ICSI, oocytes were cultured in cleavage medium (COOK, Australia). Fertilization was confirmed approximately 18 h after ICSI by the presence of two pronuclei and second polar body extrusion. Degenerate oocytes were collected in 2 µL PBS PCR tubes for subsequent analysis.
Zygotes were cultured in cleavage medium for 3 days then transferred to blastocyst medium (COOK, Australia) until day 5 or 6. Arrested embryos in the first cycle were collected in PCR tubes containing 2 µl PBS. High quality blastocysts underwent laser-assisted drilling (Hamilton, USA) and TE biopsy performed by laser-assisted micromanipulation. Biopsies were collected in PCR tubes containing 2 µl PBS, and the remaining blastocyst cryopreserved. All samples in PCR tubes were stored at − 80°. During the second PGD cycle, polar body biopsy was conducted. After ICSI, oocytes underwent laser-assisted drilling, and polar body biopsy was performed using 15 µm polar body pipette (COOK, Australia). Polar bodies were collected individually in PCR tubes containing 2 µl PBS.
Embryos in developmental arrest underwent laser-assisted drilling, and blastomeres were collected individually using an aspiration pipette with an inner diameter of 20 µm (COOK, Australia). Blastomeres were collected individually in PCR tubes containing 2 µl PBS.
Whole mtDNA amplification from all samples was performed using REPLI-g Single-Cell Kit (Qiagen, Germany). For all samples, whole mtDNA was amplified in a single fragment via PCR using high-fidelity DNA polymerase (primer sequence is shown in Table 2). Amplified mtDNA was separated by agarose gel electrophoresis and purified using gel extraction kit. Amplicons were sheared to around 200 bp fragments by ultrasonification (Cavoris, USA), and repaired using DNA end repairing agent (NGS Fast DNA Library Prep Set for Illumina, Joy Orient, China). Adapters were ligated to blunt-end products via T4 DNA ligase (NGS Fast DNA Library Prep Set for Illumina, Joy Orient, China). DNA products were amplified by 4–6 rounds of ligation-mediated PCR; then, a magnetic bead method was used to purify PCR products (NGS Fast DNA Library Prep Set for Illumina, Joy Orient, China). The T2100 Bioanalyzer (Agilent, USA) was used to detect the length of ligated fragments and q-PCR was used to quantify the effective concentration. Libraries were sequenced via Novaseq6000 (Illumina, USA) in paired-end, 150 bp reads. Quality control was conducted and low-quality data removed.
Sequence data was aligned to rCRS (NC_012920) using BWA. Samtools and Pindel were used to call single-nucleotide polymorphisms and insertion/deletions respectively. Read depth and quality were examined to screen for reliable variants. Variants were referenced against the MITOMAP human mitochondrial genome database, then MitoTIP to screen for “confirmed pathogenic” and “likely pathogenic” variants.
Results
Embryo development following ICSI
In the first PGD cycle, a total of 12 oocytes were initially retrieved, encompassing 9 MII, 1 MI, and 2 GV oocytes. Following ICSI of MII oocytes, 7 zygotes were identified, yielding 3 blastocysts on day 5, a further 2 blastocysts on day 6, and 2 arrested embryos. The 3 immature oocytes were all mature after 24 h in vitro culture and underwent ICSI, yielding 2 blastocysts at day 5, and one arrested embryo (Table 3 and Supplementary Table 1).
In the second cycle, 10 MII oocytes were retrieved and 1 degenerate oocyte. After ICSI, all MII oocytes were successfully fertilized. Eight developed into blastocysts at day 5 post-fertilization, while 2 arrested (Table 3).
Mutation load of m.3697G > A in biopsies, arrested embryos, and cumulus cells
The heteroplasmy of TE, arrested embryos, blastomeres, polar bodies, the degenerate oocyte, and corresponding cumulus cells are shown in Table 3 and Fig. 3. The mutation load of TE biopsies ranged from 15.2 to 100%. The range in cumulus cells was 44.2 to 98.8%. Arrested embryos of the first cycle carried a mutation load range of 23.5 to 99.3%. In the second PGD cycle, 2 arrested embryos (2.3 and 2.9) were divided into individual blastomeres. A total of 6 available blastomeres were obtained and analyzed from each embryo and the blastomeres saw a high degree of concordance in each embryo (Table 3). Large differences in mutation load were apparent between cumulus cells and associated TE biopsies/embryos. The maximal difference between a cumulus cell sample and associated TE biopsy/arrested embryo was 83.6%, with a minimum of 3.8% and average difference of 33.3%. Likewise, the maximal, minimal, and average difference between a polar body sample and associated TE biopsy/arrested embryo were 38%, 0, and 20.7% (Fig. 3). The correlation of heteroplasmy between cumulus cells and TE/arrested embryos is visualized in Fig. 4 (R2 = 0.02, p = 0.58), so is the correlation between polar bodies and TE/arrested embryos (R2 = 0.33, p = 0.13).

Heteroplasmy of TE/arrested embryos and their corresponding cumulus cells/polar bodies

The correlation of heteroplasmy between cumulus cells or polar bodies and TE/arrested embryos
Outcome of implantation following PGD
In the first cycle, after appropriate consultation and signing informed consent, the couple decided to transfer a blastocyst (1.2) with a mutation load of 31.7% and a ploidy of mosaic chromosomes 1, 17, and 18. Serum β-hCG was positive 14 days after single-embryo transfer. However, vaginal ultrasound failed to detect clinical pregnancy 30 days after transfer.
In the second PGD cycle, the chromosomal copy number in the second cycle showed more euploidies. The patients opted to transfer a male euploid blastocyst (2.1) with a mutation load of 53.9%. This transfer yielded a successful clinical pregnancy detected the vaginal ultrasound scanning 30 days post-blastocyst transfer. The amniotic fluid puncture result showed a mutation load of 49.7% of m.3697A > G when the patient was at the 20nd week of pregnancy. Unfortunately, the fetal color Doppler ultrasonography indicated a severe cleft lip and palate (Fig. 5) and the couple decided to terminate the pregnancy. We detected the mutation load of m.3697A > G in different tissues and organs of the induced fetus and its appendages. A uniform heteroplasmy of m.3697A > G in these organs (47.7 ± 1.8%) was showed in Table 4. The karyotype of amniotic fluid cell was 46; XY and the amniotic fluid cells did not detect chromosomal aneuploidy or clearly pathogenic genomic copy number variations of more than 100 kb (Fig. 6a). A total of 4 confirmed pathogenic and 12 likely pathogenic gene mutations were screened by whole-exome sequencing (Novogene, China) of the terminated fetus (Supplementary Table 2). We found a likely pathogenic gene, ERCC2, which is responsible for nucleotide excision repair, has been contributed to the cleft lip and palate in previous study [14]. Sanger sequencing verified that the ERCC2 heterozygous mutation carried by the fetus was inherited from the father (Fig. 6b).

The echo in the fetal palate is interrupted and the plow bone is seen. a The hard palate echo is interrupted by about 6 mm. b The echo of soft palate is interrupted by about 2.9 mm. c The upper alveolar echo is interrupted. The right side width is 3.4 mm and the left side width is 2.5 mm. d Upper lip echo is interrupted. The right side is 2.6 mm in width and the left side is 2.5 mm. The fetal alar is collapsed

a The amniotic fluid cells did not detect chromosomal aneuploidy or clearly pathogenic genomic copy number variations of more than 100 kb. b Sanger sequencing verified that the ERCC2 heterozygous mutation carried by the fetus was inherited from the father
Discussion
The blood of all family members carried m.3243A > G heteroplasmy at varying mutation loads, with the daughter being the highest. The m.3697G > A mutation leads to substitution of the 131st amino acid from glycine to serine (Fig. 2). This change likely affects the normal synthesis and function of subunit 1 of NADH dehydrogenase, which plays an important role in electron transport of the respiratory chain [15, 16]. Previous studies have reported the pathogenic onset threshold of the m.3697G > A mutation might be extremely high [15, 16], explaining why the daughter was first to present with symptoms.
In the female carrier of m.3243A > G (52.3% in blood) undergoing PGD, the m.3697G > A heteroplasmy of embryos ranged from 15.2 to 100%, indicating a random drift pattern of inheritance for this mutation. It has been shown that the inheritance of different pathogenic mtDNA mutations can follow different trends and sometimes shift towards homoplasmy [11, 17], but previous analysis has not included m.3697G > A. The high threshold for symptomatic onset of m.3697G > A may explain why this mutation exhibits a random pattern of inheritance without any evidence of purifying selection. A lack of biochemical dysfunction at even moderately high heteroplasmy may allow it to escape any selection against dysfunctional mitochondria in the germline. Since the high onset threshold of m.3697G > A for LS symptoms which further confirmed by this pedigree, several embryos were deemed sufficiently low in mutation load, suggesting PGD is an appropriate technique for m.3697G > A carriers.
Considering the exclusive maternal inheritance of mtDNA mutations, a male blastocyst with mutation load of 31.7% and a karyotype of mosaic chromosomes 1, 17, and 18 was selected for transfer in the first PGD cycle. However, a successful clinical pregnancy did not occur. Mosaicism of multiple chromosomes may have compromised the viability of this blastocyst [18]. In the second PGD cycle, a male euploid blastocyst with relatively low mtDNA mutation load (53.9%) was selected to transfer. This yielded a successful clinical pregnancy and the amniotic fluid puncture result showed a mtDNA heteroplasmy similar to the TE biopsy result at the 22nd week of pregnancy. Unfortunately, the fetal was identified with severe cleft lip and palate via color Doppler ultrasonography. Although the exact relationship between this deformity and mitochondrial genetic mutations has not been verified, the comparable heteroplasmy of m.3697G > A in different tissues from the induced fetus and TE biopsy/amniocentesis confirmed the efficiency of PGD for this kind of carriers. On the other hand, due to the normal phenotype of the PGD patient and her brother and her mother, who have parallel mutation load of m.3697G > A compared with the fetus, we speculate that the fetal malformation was not related to heteroplasmy and karyotype. A previous work on PGD for mtDNA disease has reported the successful birth of children with low mutation load at birth [9,10,11,12]. They assessed only the mtDNA mutant load and no karyotype data was analyzed. Our data suggest this should be integrated into PGD.
Given the maternal inheritance of mtDNA genetic diseases and the close relationship between oocytes and cumulus cells [19], we speculated that measurement of the mutation load of the cumulus cells may reflect that of the corresponding blastocysts. However, obvious discrepancies and poor correlations were observed between heteroplasmy of cumulus cells and that of subsequently developing embryos. Thus, the mutation load of a blastocyst or embryo cannot be accurately predicted from that cumulus cells. Unlike an oocyte which is maintained from early development throughout a long quiescent period [20], cumulus cells undergo more typical metabolism and cell divisions which may give rise to divergences in heteroplasmy relative to the oocyte. The heteroplasmy of a polar body biopsy was low correlated with that of a TE biopsy or whole embryo. However, the examination of 12 blastomeres from the 2 arrested cleavage stage embryos revealed a uniform distribution of heteroplasmy across day 3 cleavage embryos. This suggests a cleavage stage biopsy can accurately predict an embryos wider mutation load, corroborating work on other mtDNA mutations [10, 12]. Together, this suggests that measuring heteroplasmy of cleavage stage blastomeres is a useful tool in PGD, and more predictive of blastocyst heteroplasmy than that of a cumulus cell or polar body biopsy.
Our results provide new insights on the inheritance of the m.3697G > A mutation. To the best of our knowledge, this study is the first to perform PGD for the m.3697G > A mutation. Due to random drift in the inheritance of m.3697G > A, a wide spectrum of mutation loads were measured across oocytes and embryos. This allows a transfer of a blastocyst, but it has low posibility to produce embryos with low mutation load. Depending on successful birth and future health of the child, it may be better for LS patients seeking to have children to undergo mitochondrial replacement therapy [21], but such procedures are tightly regulated. While our data corroborated previous reports suggesting a cleavage embryo biopsy is useful in PGD, the extent to which heteroplasmy of this biopsy correlates with that of the inner cell mass in human blastocyst from which a fetus develops [22] or even the epiblast specifically should be confirmed.
References
Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–3.
Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39(3):343–51.
Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26(6):711–23.
Zhang H, Burr SP, Chinnery PF. The mitochondrial DNA genetic bottleneck: Inheritance and beyond. Essays Biochem. 2018;62(3):225–34.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080.
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77(5):753–9.
Thompson K, Collier JJ, Glasgow R, Robertson FM, Pyle A, Blakely EL, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis. 2020;43(1):36–50.
Neupane J, Ghimire S, Vandewoestyne M, Lu Y, Gerris J, Van Coster R, et al. Cellular heterogeneity in the level of mtDNA heteroplasmy in mouse embryonic stem cells. Cell Rep. 2015;13(7):1304–9.
Heindryckx B, Neupane J, Vandewoestyne M, Christodoulou C, Jackers Y, Gerris J, et al. Mutation-free baby born from a mitochondrial encephalopathy, lactic acidosis and stroke-like syndrome carrier after blastocyst trophectoderm preimplantation genetic diagnosis. Mitochondrion. 2014;18:12–7.
Sallevelt SC, Dreesen JC, Drusedau M, Hellebrekers DM, Paulussen AD, Coonen E, et al. PGD for the m.14487 T>C mitochondrial DNA mutation resulted in the birth of a healthy boy. Hum Reprod. 2017;32(3):698–703.
Steffann J, Frydman N, Gigarel N, Burlet P, Ray PF, Fanchin R, et al. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43(3):244–7.
Treff NR, Campos J, Tao X, Levy B, Ferry KM, Scott RJ. Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertil Steril. 2012;98(5):1236–40.
Assou S, Haouzi D, De Vos J, Hamamah S. Human cumulus cells as biomarkers for embryo and pregnancy outcomes. Mol Hum Reprod. 2010;16(8):531–8.
Olshan AF, Shaw GM, Millikan RC, Laurent C, Finnell RH. Polymorphisms in DNA repair genes as risk factors for spina bifida and orofacial clefts. Am J Med Genet A. 2005;135(3):268–73.
Negishi Y, Hattori A, Takeshita E, Sakai C, Ando N, Ito T, et al. Homoplasmy of a mitochondrial 3697G>a mutation causes Leigh syndrome. J Hum Genet. 2014;59(7):405–7.
Spangenberg L, Grana M, Greif G, Suarez-Rivero JM, Krysztal K, Tapie A, et al. 3697G>a in MT-ND1 is a causative mutation in mitochondrial disease. Mitochondrion. 2016;28:54–9.
Otten A, Sallevelt S, Carling PJ, Dreesen J, Drusedau M, Spierts S, et al. Mutation-specific effects in germline transmission of pathogenic mtDNA variants. Hum Reprod. 2018;33(7):1331–41.
Munne S, Blazek J, Large M, Martinez-Ortiz PA, Nisson H, Liu E, et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of high-resolution next-generation sequencing. Fertil Steril. 2017;108(1):62–71.
Uyar A, Torrealday S, Seli E. Cumulus and granulosa cell markers of oocyte and embryo quality. Fertil Steril. 2013;99(4):979–97.
Pan B, Li J. The art of oocyte meiotic arrest regulation. Reprod Biol Endocrinol. 2019;17(1):8.
Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, et al. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157(7):1591–604.
Taylor TH, Gitlin SA, Patrick JL, Crain JL, Wilson JM, Griffin DK. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update. 2014;20(4):571–81.
Funding
This study was supported by the National Natural Science Foundation of China (81871216, 81971455, U20A20350).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ji, D., Li, X., Pan, J. et al. Preimplantation genetic diagnosis for a carrier with m.3697G > A mitochondrial DNA mutation. J Assist Reprod Genet 38, 3251–3260 (2021). https://doi.org/10.1007/s10815-021-02354-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-021-02354-3