
Abstract
Purpose
To explore a new preimplantation genetic testing (PGT) method for de novo mutations (DNMs) combined with chromosomal balanced translocations by whole-genome sequencing (WGS) using the MGISEQ-2000 sequencer.
Methods
Two families, one with maternal Olmsted syndrome caused by DNM (c.1246C>T) in TRPV3 and a paternal Robertsonian translocation and one with paternal Marfan syndrome caused by DNM (c.4952_4955delAATG) in FBN1 and a maternal reciprocal translocation, underwent PGT for monogenetic disease (PGT-M), chromosomal aneuploidy, and structural rearrangement. WGS of embryos and family members were performed. Bioinformatics analysis based on gradient sequencing depth was performed, and parent-embryo haplotyping was conducted for DNM diagnosis. Sanger sequencing, karyotyping, and chromosomal microarray analysis were performed using an amniotic fluid sample to confirm the PGT results.
Results
After 1 PGT cycle, WGS of 2 embryos from the Olmsted syndrome family revealed euploid embryos without DNMs; after 2 cycles, the 11 embryos from the Marfan syndrome family showed only 1 normal embryo without DNM, copy number variations (CNVs), or aneuploidy. Moreover, 1 blastocyst from the Marfan syndrome family was transferred back to the uterus; the amniocentesis test results were confirmed by PGT and a healthy infant was born.
Conclusions
WGS based on parent-embryo haplotypes was an effective strategy for PGT of DNMs combined with a chromosomal balanced translocation. Our results indicate this is a reliable and effective diagnostic method that is useful for clinical application in PGT of patients with DNMs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
De novo mutations (DNMs) are the extreme form of rare genetic variations and are more deleterious than inherited variations because they escape evolutionary selection pressure [1]. These mutations are the primary causes of sporadically occurring genetic diseases with dominant inheritance patterns [1]. In previous studies of patients with DNMs, prenatal diagnosis was the predominant method for identifying genetic risk compared with preimplantation genetic testing (PGT) [2]. This is because, in PGT for monogenic diseases (PGT-M), parental haplotypes are prepared using genomic DNA from nuclear family members or relatives to establish linkages between the mutation and informative markers [3]. However, it is a barrier for DNM patients.
In previous studies, sperms or polar bodies combined with peripheral blood from DNM patients were used to establish linked haplotypes and diagnose embryos [4,5,6]. The analyses required many single sperms to obtain the male haplotype and polar bodies from oocytes retrieved during in vitro fertilization (IVF) treatments to obtain female haploid cells. Recently, single-nucleotide polymorphism (SNP) array and next-generation sequencing (NGS) techniques applied in PGT have enabled more precise diagnosis not only because more genetic markers (such as SNPs) were used instead of the previously used short tandem repeats (STRs), to construct haplotypes, but also because genetic diseases and aneuploidy could be identified concurrently [7, 8]. In addition, other studies reported the direct use of genetic information from parent-linked-embryos to phase haplotypes with preliminary target sequencing, but the diagnosis was customized and unavailable for chromosomes [9].
Whole-genome sequencing (WGS) was performed in PGT to identify mutations and aneuploidy simultaneously in 2015 using the Illumina HiSeq2500 platform at a low sequencing depth; however, this might reduce the resolution for chromosome detection [10]. Moreover, there were no further reports in this area. Herein, we successfully performed PGT for two families with DNMs combined with chromosomal balanced translocations and describe a PGT strategy based on parent-embryo haplotyping and diagnose the embryos using a new WGS platform.
Materials and methods
Ethics approval
The study was approved by the Ethics Committee of Human Study at the Sun Yat-sen Memorial Hospital of Sun Yat-sen University and BGI-Shenzhen, and the principles of the Declaration of Helsinki were followed. All patients underwent genetic counseling and signed a consent form that was approved by the local ethics committee.
Patients and ovarian stimulation
In family 1, the wife was diagnosed with Olmsted syndrome at a different hospital (MIM# 614594), and a de novo pathogenic mutation (c.1246C>T (p.Arg416Trp)) in the TRPV3 gene (MIM* 607066) was detected; her husband was a Robertsonian translocation carrier (Table 1). In family 2, the husband was diagnosed with Marfan syndrome (MIM# 154700) at a different hospital, and a de novo frameshift mutation (c.4952_4955delAATG (p.Glu1651Valfs*30)) was found in the FBN1 gene (MIM* 134797); his wife was a reciprocal translocation carrier (Table 1). These two families underwent controlled ovarian hyperstimulation (COH) for 3 cycles of PGT at our hospital, as shown in Table 1.
Oocyte retrieval, intracytoplasmic sperm injection, polar body, and blastocyst biopsy
Oocyte retrieval was performed using ultrasound-guided puncture, 36 h after human chorionic gonadotropin was administered. Intracytoplasmic sperm injection (ICSI) was performed on metaphase II (MII) oocytes. The oocytes were assessed for fertilization at ~ 18 h post-injection by monitoring for the presence of two pronuclei (2PN) and the second polar body (PB2). Embryos were cultured in G1-plus/G2-plus (Vitrolife, Sweden) sequential media. According to the Gardner grading system on blastocyst assessment [11, 12], blastocysts graded as 3–6, with either of the inner cell mass (ICM) or trophectoderm (TE) graded above C, were considered suitable blastocysts for biopsy. TE biopsy was performed at day 5 or 6 by zona drilling with a laser, and a few biopsied TE cells (5–8 cells) were removed and collected for genetic analysis [13]. In addition, we also biopsied the first polar body (PB1) after ICSI and PB2 at 9 h after ICSI using laser zona drilling to compare these results with those obtained from the TE samples.
Genomic DNA extraction, Sanger sequencing, and WGS
Genomic DNA (gDNA) was isolated from peripheral blood lymphocytes (I:1, I:2, II:1, and II:2) (Fig. 1) using a commercially available genomic DNA extraction kit (Qiagen, Germany). The whole-genome amplification (WGA) of biopsied TE cells and polar bodies was performed using the multiple displacement amplification (MDA) reaction (REPLI-g Single Cell kit, Qiagen, Germany) according to the manufacturer’s protocol.

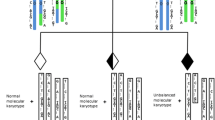
Workflow of preimplantation genetic testing (PGT) for de novo mutations (DNMs) of different origins. a Maternal DNM combined with paternal Robertsonian translocation. (1) pedigree in two generations. Filled symbols represent affected individuals (black indicates patient with the monogenetic disease and gray indicates patient with the chromosomal disease); open symbols represent unaffected individuals. Circles and squares indicate females and males, respectively; (2) mutation (c.1246C>T) confirmed by Sanger sequencing from I:1, I:2, II:1, and II:2; (3) whole-genome sequencing (WGS) performed on all 4 family members to identify the most informative single-nucleotide polymorphism (SNP) markers for PGT; (4) PGT by blastocyst biopsy for combined mutation, and haplotype and copy number variation (CNV) analysis; haplotype construction is based on maternal heterozygous and paternal homozygous sites. In the haplotype, the numbers 1, 2, 3, and 4 indicate different SNP markers, such as A, T, C, and G; the letter W indicates the wild-type allele, and the letter M indicates the mutant allele. b Paternal DNM combined with maternal reciprocal translocation. (1) Pedigree in two generations. Filled symbols represent affected individuals (black indicates patient with the monogenetic disease and gray indicates patient with the chromosomal disease), and open symbols represent unaffected individuals. Circles and squares indicate females and males, respectively; (2) mutation (c.4952_4955delAATG) confirmed by Sanger sequencing from I:1, I:2, II:1, and II:2; (3) WGS performed on all 4 family members to identify the most informative single-nucleotide polymorphism (SNP) markers for PGT; (4) PGT by blastocyst biopsy for combined mutation, and haplotype and CNV analysis; haplotype construction is based on paternal heterozygous and maternal homozygous sites. In the haplotype, the numbers 1, 2, 3, and 4 indicate different SNP markers, such as A, T, C, and G; the letter W indicates the wild-type allele, and the letter M indicates the mutant allele. Mar, Marfan syndrome; Olm, Olmsted syndrome; Rec, reciprocal translocation; Rob, Robertsonian translocation
The TRPV3 and FBN1 sequences were retrieved from the UCSC Genome Browser on Human Dec. 2013 Assembly (hg38) (http://genome.ucsc.edu). The DNMs were detected by Sanger sequencing the polymerase chain reaction (PCR) products of exon 10 in TRPV3 and exon 41 in FBN1, and their flanking intronic regions both in patients and controls, using primers designed using Oligo 6.0 (http://www.oligo.net/downloads.html) (primer sequences are available in Supplemental Table 1). Mutation nomenclature recommended by den Dunnen JT [14] and Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen/) were adopted with + 1 corresponding to the A of the ATG translation initiation codon of the GenBank cDNA sequence NM_001258205 (TRPV3) and NM_000138 (FBN1).
All the MDA products and gDNA from family members (II:1 and II:2) were used for library construction using the MGICare Single Cell Chromosomal Copy Number Variation Detection Kit (MGI, China) according to the manufacturer’s instructions. The libraries were used for WGS in the MGISEQ-2000 sequencer. The sequencing procedure yielded bidirectional sequencing read lengths of 100 bp. Sequencing data were analyzed according to our in-house procedures. Raw image data were processed using Imagine J software. The data was mapped to the reference genome (hg19, GRCh37) using the software BWA (http://bio-bwa.sourceforge.net/). We performed SNP calling using the software Genome Analysis Toolkit (http://www.broadinstitute.org/gatk/). The haplotype was determined according to the informative SNPs. The aneuploidy and copy number variation (CNV) testing using WGS were performed as previously reported [15]. To characterize the relationship between the DNM haplotyping accuracy and WGS sequencing depth, we performed a simulation by subsampling the WGS data from the Marfan syndrome family, which included the couple and their 4 embryos in the first PGT cycle. We found that with > 2× depth of embryo data (Supplemental Table 2) and > 10× depth of family member data (Supplemental Table 3), the haplotype including FBN1 could be determined accurately.
Study design for DNM
As shown in Fig. 1, the PGT strategies used for the two families varied according to the DNM origin, including mutation confirmation, WGS of gDNA, WGS of MDA products, parent-embryo haplotype analysis, CNVs > 4 Mb, and aneuploidy analysis. If any mutant embryo was detected, we aimed to consider this as the proband for DNM haplotyping using WGS. When there was only a wild-type embryo in the homozygote, to avoid the impact of allele dropout (ADO), additional information such as the polar body was used to analyze the linkage. In the family with DNM of maternal origin (Fig. 1a), we focused on the DNM of the TRPV3 gene in the embryos and loci (SNP markers) in which the wife was heterozygous and the husband was homozygous upstream and downstream of TRPV3. That is, using WGS and Sanger sequencing, if one of the embryos was heterozygous at this DNM site, it was hypothesized that the affected embryo inherited the maternal mutation; however, if the embryo was homozygous, this embryo did not inherit the maternal mutation or was in an ADO state. From the WGS results for the embryos and the parents, we could identify the normal and mutant haplotype. In the family with DNM of paternal origin (Fig. 1b), we focused on the DNMs in FBN1 in the embryos, and loci (SNP markers) in which the husband was heterozygous and the wife was homozygous upstream and downstream of FBN1. As with the analysis of the DNMs in TRPV3 in the embryos using sequencing, we identified the normal and mutant haplotypes from the WGS results for the embryos and the parents.
Results
We present the use of WGS for parent-embryo linkage analysis of DNMs to phase the haplotypes for PGT. In each family, informative SNP markers located within a 1 Mb region of the target gene were identified using gDNA prior to the PGT cycle. The amplification efficiencies for MDA were 100%, and the data production including ADO rates are shown in Table 2.
PGT for the maternal DNM in TRPV3 and paternal Robertsonian translocation
In family 1, a total of 4 oocytes were retrieved; all 4 oocytes were in MII when ICSI was performed. Two oocytes were fertilized, and we biopsied and collected PB1 and PB2 of these 2 zygotes. After sequential culture, there were 2 blastocysts suitable for biopsy on day 6 (Table 1). MDA was performed in these 2 TE samples, and 4 PB samples were kept for the next WGS analysis.
From the 578 detected SNPs, there were 130 informative SNPs available for haplotype analysis (Table 2). Based on our PGT strategy, we focused on the DNM of TRPV3 in the 2 biopsied embryos and found no mutations in both embryos using the WGS and Sanger sequencing methods (Fig. 2 and Supplemental Fig. 1). Therefore, we included information from the polar bodies which revealed that both PB1 from the 2 embryos carried the DNM and mutant haplotypes, but both PB2 were homozygous mutant haploids (Fig. 2 and Supplemental Fig. 1). Based on the PB results, we identified that the mutant haplotype was linked to the DNM of TRPV3 from the wife. Both embryos inherited normal maternal and paternal haplotypes and were balanced without CNVs. Hence, both blastocysts were frozen for the next embryo transfer (Fig. 2 and Supplemental Fig. 1).

Preimplantation genetic testing (PGT) for de novo mutations (DNMs) (c.1246C>T) in TRPV3 of maternal Olmsted syndrome (Olm), combined with paternal Robertsonian translocation (Rob). a Results of linkage analyses (partial SNP sites excerpted) of the blastocyst-stage embryos. Eleven SNP markers were selected and linked to the DNM site to identify the mutant haplotype and the normal haplotype in each embryo, PB1, and PB2. Blue bars indicate the paternal normal haplotype, red bars indicate the maternal mutant haplotype, and green bars indicate the maternal normal haplotype. b Copy number variations (CNVs) of the 2 embryos by whole-genome sequencing (WGS) at an average sequencing depth of 30×. No chromosomal abnormality was found. c Outcome of the first PGT cycle. Biopsies of the blastocysts from 2 embryos are subjected to combined haplotype analysis (genotype), and CNV and aneuploidy analysis (karyotype). Chr., chromosome number; F, frozen embryos; ID, reference SNP cluster ID; PB1, the first polar body; PB2, the second polar body; Position, genomic location
PGT for the paternal DNM in FBN1 and maternal reciprocal translocation
In family 2, a total of 38 oocytes were retrieved in 2 PGT cycles, and 11 cleavage embryos became blastocysts which were suitable for biopsy at day 5 or 6 (Table 1). All these TE samples were successful in MDA and WGS.
From the 806 detected SNPs, there were 231 informative SNPs available for haplotype analysis (Table 2). In the first PGT cycle, we identified the mutant haplotype linked with DNM of FBN1 from the husband. Two of four embryos (E02 and E04) did not show mutations or mutant haplotypes but showed pathogenic CNVs (Fig. 3a, c and Supplemental Fig. 2). Therefore, there were no available embryos for freezing or transplantation in this cycle. In the second PGT cycle, only 1 of 7 blastocysts (E03) was normal and did not possess the paternal mutant haplotype, pathogenic CNVs, or aneuploidy (Fig. 3b, c and Supplemental Fig. 2). This blastocyst was implanted to the mother which resulted in pregnancy. The amniocentesis test results revealed that the fetus did not inherit the maternal mutation and carried a chromosomal balanced translocation (Supplemental Fig. 3). A healthy baby boy weighing 2600 g was born at 35 weeks of gestation.

Preimplantation genetic testing (PGT) for de novo mutations (DNMs) (c.4952_4955delAATG) in FBN1 of paternal Marfan syndrome (Mar) combined with maternal reciprocal translocation (Rec). a Genetic results of the first PGT cycle. Left panel: results of linkage analyses (partial SNP sites excerpted) of the blastocyst-stage embryos. Eleven SNP markers were selected and linked to the DNM site to identify the mutant haplotype and the normal haplotype in each embryo. Blue bars indicate the maternal normal haplotype, red bars indicate the paternal mutant haplotype, and green bars indicate the paternal normal haplotype. Right panel: copy number variations (CNVs) of the 4 embryos by whole-genome sequencing (WGS) at an average sequencing depth of 29×. Significant chromosomal abnormalities were identified in embryo E02, E03, and E04 but not embryo E01. b Genetic results of the second PGT cycle. Left panel: results of linkage analyses (partial SNP sites excerpted) of the blastocyst-stage embryos. Eleven SNP markers were identified and linked to the DNM site to identify the mutant haplotype and the normal haplotype in each embryo. Blue bars indicate the maternal normal haplotype, red bars indicate the paternal mutant haplotype, and green bars indicate the paternal normal haplotype. Right panel: CNVs of the 7 embryos by WGS at an average sequencing depth of 8×. Significant chromosomal abnormality was identified in embryos E01, E02, E04, E06, and E07 but not embryos E03 and E05. c Outcome of 2 PGT cycles. Blastocysts biopsied from 11 embryos are subjected to combined haplotype analysis (genotype), and chromosomal CNV and aneuploidy analysis (karyotype). Chr., chromosome number; D, discarded embryos; FET, frozen embryo transferred; ID, reference SNP cluster ID; M, mutant alleles; Position, genomic location; W, wild-type alleles
Discussion
Here, using the new WGS platform, we present a parent-embryo linkage PGT analysis for a maternal DNM in TRPV3 and a paternal DNM in FBN1 with spousal chromosome balanced translocations. This method overcomes the need for family history and affected family members for PGT-M in autosomal or X-linked dominant inheritance diseases. To some extent, in the presence of a mutant embryo, this method can be performed without analyzing many sperms and polar bodies from oocytes; in addition, it allows for the simultaneous detection of CNVs and aneuploidy and specific point mutations. Although a few reports on PGT for DNMs were published [4,5,6], the premise of these studies is haplotype based, using a single-sperm/polar body prior to the PGT-M cycle. Moreover, this method of detection is undoubtedly expensive and time-consuming.
Since most DNMs are dominant, there is a 50% risk that the offspring carries the mutation. Therefore, theoretically in PGT-M, if we have more than two biopsied blastocysts, at least one will carry the DNM from either the mother or father. Therefore, this embryo with the DNM can be considered the proband and we can construct a parent-embryo haplotype linking the mutation with informative genetic markers. Based on the above theory, karyomapping was performed on a de novo deletion in TSC2 for PGT, but this method did not directly reflect the mutation in the embryos [16]. Subsequently, targeted sequencing by NGS was developed, which detected the DNM in FBN1 for PGT-M. However, this required the assistance of a customized probe in the target region and did not detect CNVs and aneuploidy [9]. Moreover, three biopsied blastocysts in this study were considered wild-type homozygotes in the DNM site of FBN1, while the haplotypes were not verified by single-sperm detection and could not exclude ADO in this site which may result in misdiagnoses in the PGT. However, parent-embryo haplotypes were constructed successfully based on the existence of a mutant embryo that was used as the proband. Considering that the ADO rate in MDA is on average 25% (range of 0–60%) [17], but with a rather low error rate (< 0.1%) [18], the normal homozygote in the DNM locus can either be a true normal result or a false negative caused by ADO. However, there is a strong possibility (> 99%) [18] that a heterozygous DNM result is truly accurate. This indicates that there are theoretical accuracy differences between the DNM phasing results deduced from embryos with (> 99% accuracy) and without (average 75% accuracy) heterozygous DNM. From our study, only 2 blastocysts had wild-type homozygotes in family 1, with follow-up analysis of polar bodies performed to avoid misdiagnosis, while 2 of 4 blastocysts had detected DNMs in family 2 in the 1st PGT cycle which might be associated with a good ovarian reserve in the wife (Table 1). This indicated that constructing parent-embryo haplotypes for PGT-M required the consideration of two factors: (i) autosomal/X-linked dominant inheritance and (ii) female with good ovarian reserve. Therefore, the limitation of parent-embryo haplotypes analysis was diminished ovarian reserve which might cause decreased available blastocysts.
In previous studies, advanced WGS using the long fragment read (LFR) technique achieved detection and phasing of embryonic de novo single-nucleotide and short indel mutations [19]. Therefore, it was undoubted that WGS could simultaneously provide effective detection of mutation, CNVs, and aneuploidy on a genome-wide scale. In addition, unlike OnePGT which requires the processing of additional family members due to low sequencing depth [20], our higher sequencing depth could directly identify the mutation site of the embryo. Therefore, in view of our PGT families with DNMs and chromosomal balanced translocation, WGS is a cost-effective, time-saving, and precise method. In our study, the data gradient testing showed that sequencing data at about 10× depth for family members and 2× depth for each embryo could effectively achieve the test results including DNM phasing and detecting CNVs and aneuploidy. Therefore, the samples from the 12 embryos and 4 family members could be used simultaneously for WGS in a single run of the MGISEQ-2000 sequencer.
In conclusion, we present PGT for DNMs combined with chromosomal balanced translocation based on parent-embryo haplotypes using the WGS technique which resulted in successful outcomes. Our study results indicate that this is a reliable and effective diagnostic method that is useful for clinical application in PGT of patients with DNMs.
References
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75.
Mao R, McDonald J, Cantwell M, Tang W, Ward K. The implication of de novo 21-hydroxylase mutation in clinical and prenatal molecular diagnoses. Genet Test. 2005;9:121–5.
Harton GL, De Rycke M, Fiorentino F, Moutou C, SenGupta S, Traeger-Synodinos J, et al. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod. 2011;26:33–40.
Altarescu G, Eldar-Geva T, Varshower I, Brooks B, Haran EZ, Margalioth EJ, et al. Real-time reverse linkage using polar body analysis for preimplantation genetic diagnosis in female carriers of de novo mutations. Hum Reprod. 2009;24:3225–9.
Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. The clinical application of single-sperm-based SNP haplotyping for PGD of osteogenesis imperfecta. Syst Biol Reprod Med. 2019;65:75–80.
Rechitsky S, Pomerantseva E, Pakhalchuk T, Pauling D, Verlinsky O, Kuliev A. First systematic experience of preimplantation genetic diagnosis for de-novo mutations. Reprod BioMed Online. 2011;22:350–61.
Fiorentino F, Bono S, Biricik A, Nuccitelli A, Cotroneo E, Cottone G, et al. Application of next-generation sequencing technology for comprehensive aneuploidy screening of blastocysts in clinical preimplantation genetic screening cycles. Hum Reprod. 2014;29:2802–13.
Handyside AH, Harton GL, Mariani B, Thornhill AR, Affara N, Shaw MA, et al. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J Med Genet. 2010;47:651–8.
Wang S, Niu Z, Wang H, Ma M, Zhang W, Fang Wang S, et al. De novo paternal FBN1 mutation detected in embryos before implantation. Med Sci Monit. 2017;23:3136–46.
Yan L, Huang L, Xu L, Huang J, Ma F, Zhu X, et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc Natl Acad Sci U S A. 2015;112:15964–9.
Gardner DK, Schoolcraft WB. In vitro culture of human blastocysts. In: Jansen R, Mortimer D, editors. Toward reproductive certainty: fertility and genetics beyond 1999. London: Parthenon Publishing; 1999. p. 378–88.
Gardner DK, Schoolcraft WB. Culture and transfer of human blastocysts. Curr Opin Obstet Gynecol. 1999;11:307–11.
Harton GL, Magli MC, Lundin K, Montag M, Lemmen J, Harper JC, et al. ESHRE PGD Consortium/Embryology Special Interest Group--best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS). Hum Reprod. 2011;26:41–6.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9.
Yin X, Tan K, Vajta G, Jiang H, Tan Y, et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol Reprod. 2013;88:69.
Giménez C, Sarasa J, Arjona C, Vilamajó E, Martínez-Pasarell O, Wheeler K, et al. Karyomapping allows preimplantation genetic diagnosis of a de-novo deletion undetectable using conventional PGD technology. Reprod BioMed Online. 2015;31:770–5.
Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I, et al. Whole-genome multiple displacement amplification from single cells. Nat Protoc. 2006;1:1965–70.
Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–85.
Peters BA, Kermani BG, Alferov O, Agarwal MR, McElwain MA, Gulbahce N, et al. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res. 2015;25:426–34.
Masset H, Zamani Esteki M, Dimitriadou E, Dreesen J, Debrock S, Derhaag J, et al. Multi-Centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum Reprod. 2019;34:1608–19.
Acknowledgments
The authors thank all patients and their family members for their participation in this study.
Funding
This work was partially supported by the National Natural Science Foundation of China (No. 81801431), the Chinese Medical Association clinical medical research special fund, Research and development of young physicians in reproductive medicine (No. 18010060735), the Shenzhen Birth Defect Screening Project Lab (JZF No. [2016] 750), and the Shenzhen Municipal Government of China (No. JCYJ20170412152854656).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1
Sanger sequencing results of the Olmsted syndrome family, their 2 embryos, and related the first polar body (PB1) and the second polar body (PB2) for detecting maternal missense mutation (c.1246C > T) in the TRPV3 gene. (PNG 134 kb)
Figure S2
Sanger sequencing results of the Marfan syndrome family and their 11 embryos for detecting paternal frameshift mutation (c.4952_4955delAATG) in the FBN1 gene. (PNG 221 kb)
Fig. S3
Validations of the fetus in the Marfan syndrome family. (a) Sanger sequencing of the amniotic fluid cells confirms that the baby does not have the mutant allele in TRPV3. (b) Chromosomal karyotype analysis reveals that the fetus has the same karyotype as the mother who carries the chromosomal reciprocal translocation (46, XN, t (10; 20) (q26.1; q13.1)). (c) Single nucleotide polymorphism (SNP) array for the detection of copy number variations (CNVs) and aneuploidy shows that the fetus has no chromosomal abnormalities. (PNG 3208 kb)
ESM 4
(DOCX 14 kb)
ESM 5
(DOCX 16 kb)
ESM 6
(DOCX 15 kb)
Rights and permissions
About this article

Cite this article
Yuan, P., Xia, J., Ou, S. et al. A whole-genome sequencing–based novel preimplantation genetic testing method for de novo mutations combined with chromosomal balanced translocations. J Assist Reprod Genet 37, 2525–2533 (2020). https://doi.org/10.1007/s10815-020-01921-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-020-01921-4