
Abstract
Microalgae are fundamentally important organisms for global ecosystem functioning with high potential in biotechnology and its applications. The knowledge of their nuclear DNA content has become a prerequisite for many areas of microalgal research. Due to common presence of various pigments, secondary metabolites and complex cell walls, the nuclear DNA content estimation using flow cytometry (FCM) is, however, often laborious or even impossible with the currently used protocols. In this study the performance of six nuclei isolation protocols was compared on various problematic microalgae using FCM. The nuclei isolation methods involved osmotic bursting of cells, razor blade chopping of fresh biomass and two newly introduced protocols, razor blade chopping of desiccated biomass and bead beating. These techniques also involved the use of two different nuclei isolation solutions, Otto I + II solutions, and LB01 buffer. Performance of the particular protocols differed greatly, depending on the used nuclei isolation solution and microalgal group. The most successful method was a newly adopted chopping of desiccated biomass in LB01 buffer. This method seems more appropriate for nuclei isolation in filamentous microalgae; on the other hand, bead beating appears to be more suitable for nuclei isolation in solitarily living algae. Using the optimal protocol for a given species, their nuclear DNA content was estimated, resulting in first DNA content estimates for four investigated taxa (Chlamydomonas noctigama, Gonyostomum semen, Microglena sp. and Stigeoclonium sp.). The estimated DNA content spanned from 0.15 to 32.52 pg.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microalgae are photosynthetic microorganisms that occur across a wide range of habitats from freshwater lakes to desert soils. Due to their polyphyletic origin across the tree of life, they are a remarkably diverse group of organisms. Moreover, microalgae play a key role in the global ecosystem as primary producers and major source of oxygen. Recently, considerable attention has been paid to microalgae as the potential source of next generation biofuels or usable metabolites (Brennan and Owende 2010; Hyka et al. 2013; Milano et al. 2016; Khan et al. 2018). This has led to a need for microalgal DNA content data due to a number of reasons. First, this knowledge enables us to select lineages with potentially higher secondary metabolite production given that an increase in DNA content is often coupled with an increase in gene dosage (e.g. due to aneuploidisation or polyploidisation; Mason 2016; Priyadarshan 2019; Qin et al. 2019). Second, the recent attention drawn to microalgae accelerated the whole-genome sequencing effort, and the DNA amount is the key to designing an optimal sequencing strategy. Further, the nuclear DNA content directly influences the cost of a sequencing project; hence, the low DNA content has become a major criterion in selection of appropriate algal strains (Waaland et al. 2004; Peters et al. 2004; Lin 2006). The combination of DNA content knowledge and high-level phylogeny also opens the ways to determine evolutionary trends in DNA content variation. Such innovative studies brought new insights into microalgal nutrition modes or cell-size changes (Poulíčková et al. 2014; Olefeld et al. 2018). Further, the nuclear DNA content, at least in relative units, is essential for cell cycle determination (Lemaire et al. 1999; Reinecke et al. 2018).
The most suitable method for precise and rapid nuclear DNA content estimation is flow cytometry (FCM). Using FCM, we are able to detect fluorescent-stained particles (e.g. cells, isolated nuclei) in a stream of fluid (Doležel et al. 2007). While FCM has found a broad spectrum of applications in genomic surveys on plants and animals (e.g. Dionisio Pires et al. 2004; Kron et al. 2007; Galbraith 2012; Chang et al. 2018; Sadílek et al. 2019), it has been only rarely applied in algal studies (but see Figueroa et al. 2010; Hyka et al. 2013).
There are several reasons causing the gap of nuclear DNA content estimates in microalgae. First, it is almost always necessary to cultivate microalgal strains from a single cell/filament to obtain sufficient amounts of biomass for FCM analysis. However, this is very time-consuming and for some species even hard to accomplish. Because of the great diversity of microalgae, there is also a wide range of pigments and metabolites that frequently interfere with fluorescent stain and/or create pronounced background noise, prominent especially when whole intact cells are analysed (Simon et al. 1994; Veldhuis et al. 1997; Mazalová et al. 2011). Although the pronounced cytoplasmic autofluorescence as well as nonspecific background fluorescence can be lowered by chemical fixation, such approach is far from optimal due to reduced quality of FCM analyses. Instead, protoplast extraction and preparation of nuclear suspension are much more suitable (Doležel and Bartoš 2005). To achieve this, several methods of cell wall disruption can be implemented in a sample preparation protocol for FCM analysis. However, currently used protocols for microalgae often do not work for FCM. Commonly used nuclei isolation method is chopping the biomass by a razor blade combined with various enzymatic treatments (Mazalová et al. 2011; Weiss et al. 2011; Poulíčková et al. 2014). In many cases the enzymatic treatment was applied to chemically dissolve the cell walls without the need for any further mechanical disruption (Mazalová et al. 2011; Poulíčková et al. 2014). The application of enzymatic treatment on algal samples was originally adopted from plant or fungal studies (Jazwinski 1990; Doležel et al. 2007) and the predominantly used enzymes for microalgal species are cellulase, macerozyme and lyticase (Mazalová et al. 2011; Weiss et al. 2011; Poulíčková et al. 2014). In Mazalová et al. (2011) the authors introduced an enzymatic treatment that was subsequently tested on a broad variety of microalgal species. The enzymatic mixture was primarily developed for streptophyte algae (e.g. the genus Zygnema), but also worked with some Chlorophyta (Chloroidium ellipsoideum, Tetraselmis subcordiformis) and Ochrophyta (Tribonema vulgare). Despite this, the introduced protocol did not work for nearly half of the tested microalgae, among others, for the green algae Trentepohlia sp. or Chlamydomonas noctigama (referred there as C. geitleri).
Unfortunately, the utilization of enzymatic treatment is methodologically demanding as well as time-consuming. Moreover, due to the great algal diversity, enzymatic treatment often requires additional modifications for specific algal groups (Mazalová et al. 2011; Weiss et al. 2011; Potter et al. 2016). However, use of the enzymatic treatment predominates as a protoplast isolation technique in microalgal studies despite these disadvantages. To resolve the situation, new methods of nuclei isolation for FCM analysis need to be established for microalgae. For example, the most common way of nuclei isolation in plants or seaweeds is simple chopping tissue using a razor blade (Galbraith et al. 1983; Asensi et al. 2001; Doležel et al. 2007). Further, beat beating by zirconium or silica beads has been previously used to isolate nuclei of bacteria (Gryp et al. 2020), fungi (Griffin et al. 2002), plants (Roberts 2007) and animals (Harmon et al. 2006). Interestingly, despite its easy and rapid use, neither bead beating nor chopping by a razor blade alone was ever successfully applied to isolate microalgal nuclei for FCM.
The aim of this study is to develop new protocols of microalgal nuclei isolation and test them on a diverse set of species that were referred as problematic in the past (Mazalová et al. 2011; personal observation).
Materials and methods
Origin, cultivation and harvesting of investigated strains
Monoclonal cultures used in this study were obtained from Culture Collection of Algae of Charles University in Prague (CAUP), Culture Collection of Cryophilic Algae (CCCryo), Norwegian Culture Collection of Algae (NORCCA) and from collaborators (Table 1). The algal taxa chosen for this study were selected based on the previous difficulties with their nuclei extraction and/or FCM analysis (Mazalová et al. 2011; author’s personal observation in pilot FCM analyses). A special focus is paid to Zygnema strains as this genus is the model organism in recent studies in our working group (e.g. Pichrtová et al. 2018; Trumhová et al. 2019).
The strains were cultivated either in 50 mm Petri dishes filled with Bold’s Basal medium (BBM; Bischoff and Bold 1963) solidified with 1.5% agar or in 50-mL Erlenmeyer flasks filled with liquid BBM or modified WC medium (MWC; Guillard and Lorenzen 1972). The majority of cultures were maintained at 17 °C with constant light conditions under the illumination of 30–50 μmol photons m-2 s-1. The Chlamydomonas noctigama and Microglena sp. strains were cultivated at 23 °C with 14 h light and 10 h dark conditions under the illumination of 100 μmol photons m-2 s-1. Origin details and cultivation media for particular algal strains are listed in Table 1. The cultures were transferred into a fresh medium 2 to 5 weeks before the planned FCM analyses and their biomass growth regularly checked. Afterwards, the culture biomass was harvested in their exponential phase of growth. Approximately 15-30 mg bulk of biomass were collected from cultures growing on solidified medium (BBM-agar) using an inoculation needle with a bent tip. Similarly, 2 mL of strains cultivated in liquid medium (BBM or MWC) were transferred into an Eppendorf tube, centrifuged (5500 rpm for 5 min) and superfluous medium removed by pipetting.
Nuclei isolation and staining
In total, six nuclei isolation protocols were subsequently tested on the studied algal strains. In each protocol, either LB01 buffer (15 mM Tris, 2 mM Na2EDTA, 0.5 mM spermine tetrahydrochloride, 80 mM KCl, 20 mM NaCl, 0.1 % (v/v) Triton X-100; pH = 8.0; Doležel et al. 1989) or a two-step Otto protocol (Otto I solution consisting of 0.1 M citric acid, 0.5% Tween 20 with pH = 2.0–3.0 and Otto II solution consisting of 0.4 M Na2HPO4·12H2O with pH = 8.0–9.0; Otto 1990) was used.
Protocol 1
Single-celled algal strains (C. noctigama, Microglena sp. and Gonyostomum semen) were prepared for the FCM analysis without any protoplast extraction, i.e. whole cells of each strain were mixed with 550 μL of ice-cold LB01 lysis buffer or Otto I solution, to attempt a release of nuclei by osmotic bursting of cells. The suspension was thoroughly mixed and filtered through a 42-μm nylon mesh into a special 3.5-mL cuvette for direct use with the flow cytometer. Following a 20 min incubationFootnote 1 at room temperature, staining solution consisting of either 1 mL Otto II solution or 550 μL LB01 lysis buffer, of 50 μg mL−1 propidium iodide, of 50 μg mL−1 RNase IIA and of 2 μ mL−1 β-mercaptoethanol was added to the sample.
Protocol 2
Harvested biomass was transferred to a plastic Petri dish and chopped by a razor blade in 550 μL of ice-cold Otto I solution. The resulting suspension was thoroughly mixed and filtered through a 42 μm nylon mesh into a special 3.5-mL cuvette for direct use with the flow cytometer. Following a 20-min incubation1 at room temperature, staining solution consisting of 1 mL of Otto II solution, of 50 μg mL−1 propidium iodide, of 50 μg mL−1 RNase IIA and of 2 μL mL−1 β-mercaptoethanol was added to the sample.
Protocol 3
Harvested biomass was transferred into a plastic Petri dish and chopped by a razor blade in 550 μL of ice-cold lysis buffer LB01. The resulting suspension was thoroughly mixed and filtered through a 42 μm nylon mesh into a special 3.5-mL cuvette for direct use with the flow cytometer. Following a 20 min incubation1 at room temperature, staining solution consisting of 550 μL of LB01 lysis buffer, of 50 μg mL−1 propidium iodide, of 50 μg mL−1 RNase IIA and of 2 μL mL−1 β-mercaptoethanol was added to the sample.
Protocol 4
Harvested biomass was desiccated by transferring into 2-mL Eppendorf tube and placed with an open lid into a zip-lock bag filled with silica gel for 2 to 5 days. The dry algal biomass was then transferred in a plastic Petri dish and chopped by a razor blade in 550 μL of ice-cold lysis buffer LB01. The sample preparation was further completed according to Protocol no. 3.
Protocol 5
Approximately 10 glass beads of 1.5-mm diameter (Sigma-Aldrich) were added into 2-mL Eppendorf tube containing 550 μL of ice-cold Otto I solution and a biomass pellet. The cells were disrupted for 3 min at 25 Hz using Retsch MM200 mixer mill (Retsch, Inc., Germany). The nuclei suspension was then filtered through a 42 μm nylon mesh into a special 3.5-mL cuvette for direct use with the flow cytometer. Following a 20 min incubation1 at room temperature, staining solution consisting of 1 mL Otto II solution, 50 μg mL−1 propidium iodide, 50 μg mL−1 RNase IIA and 2 μL mL−1 β-mercaptoethanol was added to the sample.
Protocol 6
Approximately 10 glass beads of 1.5 mm diameter (Sigma-Aldrich) were added into 2-mL Eppendorf tube containing 550 μL of ice-cold lysis buffer LB01 and the pellet of biomass. The cells were disrupted for 3 min at 25 Hz using Retsch MM200 mixer mill. The sample was filtered through a 42 μm nylon mesh into a special 3.5-mL cuvette for direct use with the flow cytometer. Following a 20 min incubation1 at room temperature, staining solution consisting of 550 μL of LB01 lysis buffer, of 50 μg mL−1 propidium iodide, of 50 μg mL−1 RNase IIA and of 2 μL mL−1 β-mercaptoethanol was added to the sample.
Standardization
Initially, nuclei suspensions for FCM analysis were prepared without a standard. When a suitable nuclei extraction protocol was found for a given species, an internal standard was included into following analyses. Four different plants were used as standards in this study—wild clone of Carex acutiformis (2C = 0.82 pg; Veselý et al. 2012), commercial clone of Solanum pseudocapsicum (2C = 2.59 pg; Temsch et al. 2010), wild clone of Bellis perennis (2C = 3.38 pg; Schönswetter et al. 2007) and Vicia faba cv. Inovec (2C = 26.90 pg; Doležel et al. 1992). To release the standard nuclei, ca. 20 mg piece of fresh leaf tissue was chopped with a razor blade either together with an algal sample (Protocols 2, 3 and 4) or separately, in a fraction of used nuclei isolation solution and later mixed with the protoplast suspension containing the remaining solution (Protocols 5 and 6). When razor chopping was used to isolate nuclei of both algal sample and plant standard, the algal biomass was chopped slightly less than the plant standard. The resulting nuclei suspension was filtered and stained as described in Protocols 1–6.
DNA content estimation
The stained samples were immediately analysed using a Partec CyFlow SL cytometer (Partec GmbH, Germany) equipped with a green solid-state laser (Cobolt Samba, 532 nm, 100 mW), and aside from PI fluorescence intensity, optical parameters forward scatter (FSC) and side scatter (SSC) were recorded. Each sample measurement was taken for up to 5000 particles. The success rate of particular protocol was evaluated as follows: (1) no peak, sample peak undistinguishable from the background noise or not detected; (2) poor analysis, sample peak visible but its position hardly recognizable from the background noise (yet apparent on a relative fluorescence vs. side scatter plots); and (3) good result, sample peak clearly visible with reduced background noise.
To properly analyse DNA content of the studied algal strains, at least three measurements were done on separate days to obtain precise value and to minimize the effect of random instrumental shift. The resulting FCM histograms were analysed using FloMax ver. 2.4d (Partec). The lowest fluorescence intensity sample peaks were identified as G1 (vegetative cells) and additional peaks with double fluorescence intensity (if observed) as G2. Gating of sample nuclei in fluorescence vs. side scatter plots was necessary to remove the background noise connected to the populations of interest in order to obtain more accurate results (with an exception of G. semen). The absolute nuclear DNA content was calculated as the sample G1 peak mean fluorescence/standard G1 peak mean fluorescence × standard 2C DNA content (according to Doležel and Bartoš 2005). Since the ploidy level or life cycle stage of studied organisms is generally unknown, the DNA content results are given in pg cell-1, i.e. the absolute nuclear DNA content measured per cell (1 pg ≈ 978 Mbp; Doležel et al. 2003). The quality and accuracy of resulting DNA content estimates was expressed by averaged coefficient of variation (CV) for individual sample peaks and standard deviation (SD) for measurements error averaged from the three independent measurements.
Results
Comparison of isolation protocols
Altogether, six nuclei isolation protocols were tested and compared on a set of ten problematic algal taxa (Table 1). Protocol success rate was evaluated using a three quantitative scale (see Materials and methods). The results differed greatly according to the used protocol and algal sample tested (Table 2).
Except analysing cells only mixed with nuclei isolation solution (Protocol 1), the broadly used technique of razor blade sample chopping (Protocols 2 and 3) was the least successful method in this study. When Otto I + II solutions were used (Protocol 2), none of the tested strains resulted in a visible sample peak. The razor blade chopping technique was successful only in combination with LB01 isolation buffer (Protocol 3), resulting in clearly visible and well separated peaks for Stigeoclonium sp. and Tribonema vulgare. A partial success of Protocol 3 was also achieved for all Zygnema strains, however, still leading to a high background noise and often hardly distinguishable sample peak (Fig. 1a,d).

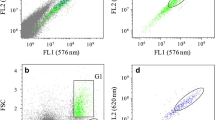
Flow cytometric fluorescence histograms (a-c, g-i) and fluorescence vs. side scatter plots (d-f, j-l) summarizing the results of poor quality (a-f) and suitable (g-l) nuclei isolation protocols of Zygnema sp. OS2, Trentepohlia sp. and G. semen strains with internal reference standards. The nuclei of Zygnema sp. OS2 isolated by a razor blade chopping of fresh biomass in LB01 buffer (Protocol 3) resulted in visible sample and standard peaks with pronounced background noise (a, d), while using razor blade chopping of desiccated biomass (Protocol 4) led to prominent sample and standard peaks (g, j). Isolation of Trentepohlia sp. nuclei using Protocol 3 did not result in visible sample peak (b, e), contrary to nuclei isolation with the Protocol 4, where the sample peak is clearly visible and well separated (h, k). Note that in fluorescence vs. side scatter plot, the peak of presumed haploid zoospores can be identified (k, indicated by the arrow). Protocol 4 did not result in any sample peak for G. semen (c, f); however, when nuclei were isolated by cell bead beating in LB01 buffer (Protocol 6), it led to a clear sample peak with nearly no background noise (i, l)
In contrast, Protocol 4, combining sample desiccation with razor blade chopping in LB01 isolation buffer, was the most successful of all the tested methods. This protocol resulted in clearly visible and well separated peaks for Spirogyra sp., Trentepohlia sp. (Fig. 1h,k) and all three analysed strains of Zygnema sp. (Fig. 1g,j). Interestingly, the same quality of analysis with Trentepohlia was observed when Otto I + II solutions were used instead of LB01 buffer; however, this was not examined for any other microalgal strain. Protocol 4 was further partially successful for the species Stigeoclonium sp. and T. vulgare, however, leading to a more pronounced background noise compared to the same method without the desiccation step (Protocol 3). On the other hand, this method failed to result in any sample peaks for Chlamydomonas noctigama, Microglena sp. and Gonyostomum semen (Fig. 1c,f). The only successful method for analysing these microalgal species was Protocol 6. In this protocol, the nuclei were extracted by bead beating cells in LB01 isolation buffer. This method was particularly suitable for G. semen, where it led to a high-quality analysis with nearly no visible background noise (Fig. 1i,l). Contrarily, the analyses of C. noctigama and Microglena sp. were of very low quality (pronounced background noise and poor peak delimitation). However, Protocol 6 was the only protocol leading to any sample peak for these species. Interestingly, the same method of nuclei isolation by bead beating successful for C. noctigama, Microglena sp. and G. semen did not work when Otto I + II solutions were used (Protocol 5) instead of LB01 isolation buffer (Protocol 6). Therefore, Protocol 5 was not further examined for the remaining strains.
Nuclear DNA content estimation
When the most suitable protocol for particular species was found, their absolute nuclear DNA content per cell was thoroughly investigated (Table 3). The nuclear DNA content of studied microalgal strains is given in pg of absolute nuclear DNA per cell with equivalent values in Gbp (1 pg ≈ 0.978 Gbp; Doležel et al. 2003). The DNA content differed greatly, spanning from 0.15 (0.14) to 32.52 pg (31.81 Gbp). The smallest DNA content belonged to the representatives of the class Chlorophyceae with 0.15 pg (0.14 Gbp) for Stigeoclonium sp., 0.33 pg (0.33 Gbp) for C. noctigama and 0.44 pg (0.43 Gbp) for Microglena sp. and to the representative of the class Xanthophyceae with 0.34 pg (0.34 Gbp) for T. vulgare. In contrast, the largest measured DNA content of 32.52 pg (31.81 Gbp) belonged to G. semen from the class Raphidophyceae. The three analysed strains of the genus Zygnema varied in their DNA content (1.11–2.86 pg ≈ 1.09–2.73 Gbp). The highest quality of DNA content estimates was observed within G. semen and one of Zygnema strains, with coefficients of variation (CVs) < 2% (1.14 and 1.75%, respectively). On the other hand, the lowest quality of DNA content estimates was documented in Stigeoclonium sp., Microglena sp. and Spirogyra, exceeding 13% (13.51, 13.54 and 13.65%, respectively).
Discussion
Nuclei isolation protocols
In this study, two new nuclei isolation protocols for FCM are proposed and applied on various samples of microalgae. The newly introduced methods involve either sample desiccation before razor blade chopping or bead beating of the sample biomass. Both methods are easy to use and bring satisfactory results of DNA content estimation in microalgae, even for problematic taxa. These new methods were compared with more broadly used techniques for microalgae, i.e. analysis of osmotic bursting of cells (applied on unicellular algae) and razor blade chopping of fresh biomass (all tested algae).
Moreover, these new techniques allowed for the first time DNA content estimation in C. noctigama, G. semen, Microglena sp. and Stigeoclonium sp. Further, identical strains of C. noctigama (strain CAUP G224) and Stigeoclonium sp. (strain CAUP J603) were already examined in the study Mazalová et al. (2011) using enzymatic mixture for protoplast extraction, however, without any success. In this work, C. noctigama was successfully analysed by applying bead beating of the biomass in LB01 isolation buffer (Protocol 6). Interestingly, a suitable method for analysing Stigeoclonium sp. was simple razor blade chopping of the biomass in LB01 isolation buffer (Protocol 3), not a protoplast extraction using enzymatic mixture (Mazalová et al. 2011) or any other method used in this study (except Protocol 4, see later). Moreover, Protocol 3 was also the best method to analyse T. vulgare. Although this taxon was already successfully analysed with the use of enzymatic mixture in the study Mazalová et al. (2011), the enzymatic treatment is methodologically demanding as well as time-consuming. In contrast, razor blade chopping of a fresh sample is very simple and rapid method and sometimes, as seen on the example of Stigeoclonium sp. and T. vulgare, also the optimal method for FCM without the need for further optimization. Therefore, this simple method is still worth a try when conducting pilot FCM measurements on other microalgal species. Both Stigeoclonium sp. and T. vulgare were also successfully analysed using desiccation step followed by razor blade chopping (Protocol 4), however, resulting in a reduced quality of the FCM analysis.
In general, razor blade chopping of biomass in LB01 isolation buffer either preceded by the desiccation step (Protocol 4) or without it (Protocol 3) appears to be a more efficient way of nuclei isolation in filamentous microalgae. The success of the desiccation using silica gel is especially interesting since this led to a decrease of quality in FCM analysis of vascular plants (Kolář et al. 2012). However, desiccating the biomass of microalgae might have reduced the negative effect of secondary metabolites interfering with DNA staining. For example, high amounts of secondary metabolites such as phenols have been documented in Trentepohlia sp., Spirogyra sp. and Zygnema spp. (Simić et al. 2012; Pichrtová et al. 2013; Mridha et al. 2017). Phenolic compounds can significantly decrease the quality of FCM analyses (Loureiro et al. 2006a), and the desiccation might reduce their negative effect (along with possibly other metabolites) on FCM analysis. On the other hand, optimal algal material for FCM analysis are young cultures approximately 3 to 5 weeks after their inoculation into a fresh medium, yet young Zygnema cells are known to contain high amounts of phenolic compounds (Holzinger et al. 2018), contradicting the benefits of using young cultures. However, analysis of Zygnema spp. cultures older than 5 weeks resulted only in a background noise (data not shown). Another explanation could be the putative role of desiccation in disturbing layers of polysaccharide present on Zygnema and Spirogyra filaments (Palacio-López et al. 2019), facilitating the release of their nuclei.
Bead beating of biomass in LB01 buffer (Protocol 6) seems to be more suitable for solitarily living algae. Even though the cell disruption by bead beating was previously used to isolate DNA of algae (e.g. Countway and Caron 2006), to my knowledge, it has never been used as a method for nuclei isolation in algal FCM. This technique was particularly suitable for G. semen, where it resulted in clear FCM histograms with very limited background noise (Fig. 1l,i). Bead beating of cells in LB01 buffer (Protocol 6) is also the only method that gained any DNA content estimates for C. noctigama and Microglena sp. not only in this study but also including unsuccessful attempts in Mazalová et al. (2011). However, the outcomes were of very poor quality, and further optimization is needed to obtain more precise results.
It is worth emphasizing the importance of selecting optimal nuclei isolation solution when employing FCM on algal samples. In this study, only two nuclei isolation solutions were used (LB01 buffer and Otto I + II solutions); however, their performance was completely different. When using Otto I + II solutions (Protocol 1, Protocol 2 and Protocol 5), the analyses led to no visible sample peaks (with the exception of Trentepohlia sp. with equally good results under the use of both buffers). Vast majority of the successful analyses were done using LB01 isolation buffer. The differences between LB01 buffer and Otto I + II solutions are in their different chemical composition but also in strikingly distinct pH level (2–3 and 8, respectively; Loureiro et al. 2006b). This stresses the importance of selecting an optimal isolation solution and comparing to others might be a next step in further optimization.
DNA content estimates of the studied algae
The nuclear DNA content of four algal taxa was successfully estimated with the smallest measured DNA content represented by Stigeoclonium sp. with 0.15 pg (0.14 Gbp). To my knowledge this also represents the first DNA content estimate for the whole order Chaetophorales. On the other hand, the largest DNA content measured in this study belongs to the raphidophyte G. semen with 32.52 pg (31.81 Gbp). The only representative of the class Raphidophyceae that has been analysed for DNA content so far was marine Heterosigma carterae possessing a genome five times smaller (5.43–6.12 pg/5.31–5.98 Gbp; Veldhuis et al. 1997). In contrast, more DNA content data are available for the genus Chlamydomonas with estimates ranging from 0.08 to 0.40 pg (0.08–0.39 Gbp; Chiang and Sueoka 1967; Kates et al. 1968; Cattolico and Gibbs 1975; Spring et al. 1978; Veldhuis et al. 1997; Merchant et al. 2007; Reinecke et al. 2018; Nelson et al. 2019). However, only a few of these estimates were acquired using FCM. The DNA content of C. noctigama estimated in this study (0.33 pg/Gbp) is rather large but still falling within the previously published range.
The identical strain of T. vulgare (CAUP D 501) was previously analysed by Mazalová et al. (2011), leading to a slightly different result of 0.41 pg (0.40 Gbp) compared to 0.34 pg/Gbp estimated in this study. This variance might be induced by use of a different FCM standard. In this study, the plant Carex acutiformis was used in opposite to Raphanus sativus cv. Saxa used in the study by Mazalová et al. (2011). However, the latter FCM standard displays many difficulties like high CVs, polyploidy, higher presence of secondary metabolites and reported genome size of different values; therefore, its use was repeatedly discouraged (Doležel et al. 1992; Praça-Fontes et al. 2011; Park et al. 2016; Šmarda et al. 2019). The only available DNA content data for the genera Trentepohlia and Spirogyra originate from DAPI microdensitometry (Kapraun 2005, 2007; López-Bautista et al. 2006). However, DAPI fluorescent stain binds to adenine-thymine-rich regions and therefore may bring the erroneous estimates of AT:GC ratio of the sample and the reference standard (Doležel et al. 1992). For the both genera, the DNA content estimates in this study were the first ones acquired using FCM. The previously published estimates for Trentepohlia sp. span from 1.08 to 4.01 pg (1.10–4.10 Gbp; López-Bautista et al. 2006; Kapraun 2007), and thus the estimate measured in this study (1.17 pg/1.14 Gbp) falls within the published range. Interestingly, in relative fluorescence vs. side scatter plots of some Trentepohlia sp. FCM analyses, it is possible to identify three sample peaks that differ in their ploidy level (Fig. 1 k). An abundant population of nuclei belonging to the intermediate ploidy was identified as nuclei of vegetative filament (G1). The peak of highest Trentepohlia sp. ploidy was determined as dividing nuclei of the vegetative filament (G2), unfortunately partially overlapping with the standard nuclei. The peak of the lowest ploidy with smallest population of nuclei may represent haploid zoospores. The presence of sporangia containing zoospores was subsequently confirmed by observation using light microscopy. The previous DNA content estimates of Spirogyra sp. (3.91–4.01 pg/4.00–4.10 Gbp; Kapraun 2005) were four times higher than in this study (1.03 pg/1.00 Gbp). Despite the fact that the measurements were conducted on different Spirogyra strains and by different techniques, these results probably reflect high DNA content variability within the Spirogyra genus. Similarly, DNA content variability within the genus Zygnema will likely be much higher than documented to date. The known DNA content range is from 0.49 to 1.5 pg (0.50–1.54 Gbp; Kapraun 2005; Mazalová et al. 2011). However, analyses of three Zygnema strains displayed DNA content between 1.11 and 2.86 pg (1.09–2.80 Gbp), and thus the previous DNA content range for the genus was nearly doubled.
I believe that the presented new nuclei isolation protocols will provide alternative ways of microalgal FCM and apply to a broad range of various species of microalgae. Hopefully, the newly introduced protocols will help to extend yet very limited DNA content data of microalgae, and these data will subsequently serve to various microalgae applications.
Data Availability
All data are provided within the manuscript.
Notes
If visible sediment was present after 20 min incubation, an upper layer of nuclei suspension was transferred into a new cuvette and used as a material for analysis.
References
Asensi A, Gall EA, Marie D, Billot C, Dion P, Kloareg B (2001) Clonal propagation of Laminaria digitata (Phaeophyceae) sporophytes through a diploid cell-filament suspension. J Phycol 37:411–417
Bischoff H, Bold H (1963) Phycological studies IV. Some soil algae from Enchanted Rock and related algal species. Phycological Studies, University of Texas IV:1-95
Brennan L, Owende P (2010) Biofuels from microalgae—a review of technologies for production, processing, and extractions of biofuels and co-products. Renew Sustain Energy Rev 14:557–577
Cattolico RA, Gibbs SP (1975) Rapid filter method for the microfluorometric analysis of DNA. Anal Biochem 69:572–582
Chang P, Tseng Y-F, Chen P-Y, Wang C-JR (2018) Using flow cytometry to isolate maize meiocytes for next generation sequencing: a time and labor efficient method. Curr Protoc Plant Biol 3:e20068
Chiang KS, Sueoka N (1967) Replication of chloroplast DNA in Chlamydomonas reinhardi during vegetative cell cycle: its mode and regulation. Proc Natl Acad Sci 57:1506–1513
Countway PD, Caron DA (2006) Abundance and distribution of Ostreococcus sp. in the San Pedro Channel, California, as revealed by quantitative PCR. Appl Environ Microbiol 72:2496–2506
Dionisio Pires LM, Jonker RR, Van Donk E, Laanbroek HJ (2004) Selective grazing by adults and larvae of the zebra mussel (Dreissena polymorpha): application of flow cytometry to natural seston. Freshw Biol 49:116–126
Doležel J, Bartoš J (2005) Plant DNA flow cytometry and estimation of nuclear genome size. Ann Bot 95:99–110
Doležel J, Binarová P, Lucretti S (1989) Analysis of Nuclear DNA content in plant cells by flow cytometry. Biol Plant 31:113–120
Doležel J, Sgorbati S, Lucretti S (1992) Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol Plant 85:625–631
Doležel J, Bartoš J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytom Part A 51A:127–128
Doležel J, Greilhuber J, Suda J (2007) Flow cytometry with plant cells. Wiley-VCH, Weinheim
Figueroa RI, Garcés E, Bravo I (2010) The use of flow cytometry for species identification and life-cycle studies in dinoflagellates. Deep Sea Res II 57:301–307
Galbraith DW (2012) Flow cytometry and fluorescence-activated cell sorting in plants: the past, present, and future. Biomédica 30:65
Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E (1983) Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220:1049–1051
Griffin DW, Kellogg CA, Peak KK, Shinn EA (2002) A rapid and efficient assay for extracting DNA from fungi. Lett Appl Microbiol 34:210–214
Gryp T, Glorieux G, Joossens M, Vaneechoutte M (2020) Comparison of five assays for DNA extraction from bacterial cells in human faecal samples. J Appl Microbiol 129:378–388
Guillard RRL, Lorenzen CJ (1972) Yellow-green algae with chlorophyllide C. J Phycol 8:10–14
Harmon AF, Zarlenga DS, Hildreth MB (2006) Improved methods for isolating DNA from Ostertagia ostertagi eggs in cattle feces. Vet Parasitol 135:297–302
Holzinger A, Albert A, Aigner S, Uhl J, Schmitt-Kopplin P, Trumhová K, Pichrtová M (2018) Arctic, Antarctic, and temperate green algae Zygnema spp. under UV-B stress: vegetative cells perform better than pre-akinetes. Protoplasma 255:1239–1252
Hyka P, Lickova S, Přibyl P, Melzoch K, Kovar K (2013) Flow cytometry for the development of biotechnological processes with microalgae. Biotechnol Adv 31:2–16
Jazwinski MS (1990) Preparation of extracts from yeast. Meth Enzymol 182:154–174
Kapraun DF (2005) Nuclear DNA content estimates in multicellular green, red and brown algae: phylogenetic considerations. Ann Bot 95:7–44
Kapraun DF (2007) Nuclear DNA content estimates in green algal lineages: Chlorophyta and Streptophyta. Ann Bot 99:677–701
Kates JR, Chiang KS, Jones RF (1968) Studies on DNA replication during synchronized vegetative growth and gametic differentiation in Chlamydomonas reinhardtii. Exp Cell Res 49:121–135
Khan MI, Shin JH, Kim JD (2018) The promising future of microalgae: current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb Cell Fact 17:36
Kolář F, Lučanová M, Tešitel J, Loureiro J, Suda J (2012) Glycerol-treated nuclear suspensions–an efficient preservation method for flow cytometric analysis of plant samples. Chromosom Res 20:303–315
Kron P, Suda J, Husband BC (2007) Applications of flow cytometry to evolutionary and population biology. Annu Rev Ecol Evol Syst 38:847–876
Lemaire S, Hours M, Gerard-Hirne C, Trouabal A, Roche O, Jacquot JP (1999) Analysis of light/dark synchronization of cell-wall-less Chlamydomonas reinhardtii (Chlorophyta) cells by flow cytometry. Eur J Phycol 34:279–286
Lin S (2006) The smallest dinoflagellate genome is yet to be found: a comment on LaJeunesse et al. “Symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates”. J Phycol 42:746–748
López-Bautista JM, Kapraun DF, Chapman RL (2006) Nuclear DNA content estimates in the Trentepohliales (Chlorophyta): phylogenetic considerations. Algol Stud 120:41–50
Loureiro J, Rodriguez E, Doležel J, Santos C (2006a) Flow cytometric and microscopic analysis of the effect of tannic acid on plant nuclei and estimation of DNA content. Ann Bot 98:515–527
Loureiro J, Rodriguez E, Doležel J, Santos C (2006b) Comparison of four nuclear isolation buffers for plant DNA flow cytometry. Ann Bot 98:679–689
Mason AS (2016) Polyploidy and hybridization for crop improvement. CRC Press, Boca Raton
Mazalová P, Šarhanová P, Ondřej V, Poulíčková A (2011) Quantification of DNA content in freshwater microalgae using flow cytometry: a modified protocol for selected green microalgae. Fottea 11:317–328
Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Marechal-Drouard L, Marshall WF, Qu LH, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q, Ferris P, Lindquist E, Shapiro H, Lucas SM, Grimwood J, Schmutz J, Cardol P, Cerutti H, Chanfreau G, Chen CL, Cognat V, Croft MT, Dent R, Dutcher S, Fernandez E, Fukuzawa H, Gonzalez-Ballester D, Gonzalez-Halphen D, Hallmann A, Hanikenne M, Hippler M, Inwood W, Jabbari K, Kalanon M, Kuras R, Lefebvre PA, Lemaire SD, Lobanov AV, Lohr M, Manuell A, Meier I, Mets L, Mittag M, Mittelmeier T, Moroney JV, Moseley J, Napoli C, Nedelcu AM, Niyogi K, Novoselov SV, Paulsen IT, Pazour G, Purton S, Ral JP, Riano-Pachon DM, Riekhof W, Rymarquis L, Schroda M, Stern D, Umen J, Willows R, Wilson N, Zimmer SL, Allmer J, Balk J, Bisova K, Chen CJ, Elias M, Gendler K, Hauser C, Lamb MR, Ledford H, Long JC, Minagawa J, Page MD, Pan J, Pootakham W, Roje S, Rose A, Stahlberg E, Terauchi AM, Yang P, Ball S, Bowler C, Dieckmann CL, Gladyshev VN, Green P, Jorgensen R, Mayfield S, Mueller-Roeber B, Rajamani S, Sayre RT, Brokstein P, Dubchak I, Goodstein D, Hornick L, Huang YW, Jhaveri J, Luo Y, Martinez D, Ngau WCA, Otillar B, Poliakov A, Porter A, Szajkowski L, Werner G, Zhou K, Grigoriev IV, Rokhsar DS, Grossman AR (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318:245–250
Milano J, Ong HC, Masjuki HH, Chong WT, Lam MK, Loh PK, Vellayan V (2016) Microalgae biofuels as an alternative to fossil fuel for power generation. Renew Sustain Energy Rev 58:180–197
Mridha A, Nandi C, Pal R, Paul S (2017) Studies on few fresh water green algal species reveals Spirogyra triplicata as the repository of high phenolic and flavonoid content exhibiting enhanced anti-oxidant property. J Pharmacogn Phytochem 6:1291–1297
Nelson DR, Chaiboonchoe A, Fu W, Hazzouri KM, Huang Z, Jaiswal A, Daakour S, Mystikou A, Arnoux M, Sultana M, Salehi-Ashtiani K (2019) Potential for heightened sulfur-metabolic capacity in coastal subtropical microalgae. iScience 11:450–465.
Olefeld JL, Majda S, Albach DC, Marks S, Boenigk J (2018) Genome size of chrysophytes varies with cell size and nutritional mode. Org Divers Evol 18:163–173
Otto F (1990) DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. Meth Cell Biol 33:105–110
Palacio-López K, Tinaz B, Holzinger A, Domozych DS (2019) Arabinogalactan proteins and the extracellular matrix of charophytes: a sticky business. Front Plant Sci 10:447
Park CH, Baskar TB, Park SY, Kim SJ, Arasu MV, Al-Dhabi NA, Kim JK, Park SU, Xiao J (2016) Metabolic profiling and antioxidant assay of metabolites from three radish cultivars (Raphanus sativus). Molecules 21:157
Peters AF, Marie D, Scornet D, Kloareg B, Mark Cock J (2004) Proposal of Ectocarpus siliculosus (Ectocarpales, Phaeophyceae) as a model organism for brown algal genetics and genomics. J Phycol 40:1079–1088
Pichrtová M, Remias D, Lewis LA, Holzinger A (2013) Changes in phenolic compounds and cellular ultrastructure of Arctic and Antarctic strains of Zygnema (Zygnematophyceae, Streptophyta) after exposure to experimentally enhanced UV to PAR ratio. Microb Ecol 65:68–83
Pichrtová M, Holzinger A, Kulichová J, Ryšánek D, Šoljaková T, Trumhová K, Nemcova Y (2018) Molecular and morphological diversity of Zygnema and Zygnemopsis (Zygnematophyceae, Streptophyta) from Svalbard (High Arctic). Eur J Phycol 53:492–508
Potter EE, Thornber CS, Swanson JD, McFarland M (2016) Ploidy distribution of the harmful bloom forming macroalgae Ulva spp. in Narragansett Bay, Rhode Island, USA, using flow cytometry methods. PLoS One 11:e0149182
Poulíčková A, Mazalová P, Vašut RJ, Šarhanová P, Neustupa J, Škaloud P (2014) DNA content variation and its significance in the evolution of the genus Micrasterias (Desmidiales, Streptophyta). PLoS One 9:e86247
Praça-Fontes MM, Carvalho CR, Clarindo WR, Cruz CD (2011) Revisiting the DNA C-values of the genome size-standards used in plant flow cytometry to choose the “best primary standards”. Plant Cell Rep 30:1183–1191
Priyadarshan PM (2019) Induced mutations and polyploidy breeding. In: Priyadarshan PM (ed) Plant breeding: Classical to Modern. Springer, Singapore, pp 329–370
Qin Y, Zhang Y, Mo R, Zhang Y, Li J, Zhou Y, Ma H, Xiao S, Yu Z (2019) Influence of ploidy and environment on grow-out traits of diploid and triploid Hong Kong oysters Crassostrea hongkongensis in southern China. Aquaculture 507:108–118
Reinecke DL, Castillo-Flores A, Boussiba S, Zarka A (2018) Polyploid polynuclear consecutive cell-cycle enables large genome-size in Haematococcus pluvialis. Algal Res 33:456–461
Roberts AV (2007) The use of bead beating to prepare suspensions of nuclei for flow cytometry from fresh leaves, herbarium leaves, petals and pollen. Cytom Part A 71A:1039–1044
Sadílek D, Urfus T, Vilímová J (2019) Genome size and sex chromosome variability of bed bugs feeding on animal hosts compared to Cimex lectularius parasitizing human (Heteroptera: Cimicidae). Cytom Part A 95:1158–1166
Schönswetter P, Suda J, Popp M, Weiss-Schneeweiss H, Brochmann C (2007) Circumpolar phylogeography of Juncus biglumis (Juncaceae) inferred from AFLP fingerprints, cpDNA sequences, nuclear DNA content and chromosome numbers. Mol Phylogenet Evol 42:92–103
Simić S, Kosanić M, Ranković B (2012) Evaluation of in vitro antioxidant and antimicrobial activities of green microalgae Trentepohlia umbrina. Not Bot Horti Agrobot Cluj-Napoca 40:86–91
Simon N, Barlow RG, Marie D, Partensky F, Vaulot D (1994) Characterization of oceanic photosynthetic picoeukaryotes by flow cytometry. J Phycol 30:922–935
Šmarda P, Knápek O, Březinová A, Horová L, Grulich V, Danihelka J, Veselý P, Šmerda J, Rotreklová O, Bureš P (2019) Genome sizes and genomic guanine+cytosine (GC) contents of the Czech vascular flora with new estimates for 1700 species. Preslia 91:117–142
Spring H, Grierson D, Hemleben V, Stöhr M, Krohne G, Stadler J, Franke WW (1978) DNA contents and numbers of nucleoli and pre-rRNA-genes in nuclei of gametes and vegetative cells of Acetabularia mediterranea. Exp Cell Res 114:203–215
Temsch EM, Greilhuber J, Krisai R (2010) Genome size in liverworts. Preslia 82:63–80
Trumhová K, Holzinger A, Obwegeser S, Neuner G, Pichrtová M (2019) The conjugating green alga Zygnema sp. (Zygnematophyceae) from the Arctic shows high frost tolerance in mature cells (pre-akinetes). Protoplasma 256:1681–1694
Veldhuis MJW, Cucci TL, Sieracki ME (1997) Cellular DNA content of marine phytoplankton using two new fluorochromes: taxonomic and ecological implications. J Phycol 33:527–541
Veselý P, Bureš P, Šmarda P, Pavlíček T (2012) Genome size and DNA base composition of geophytes: the mirror of phenology and ecology? Ann Bot 109:65–75
Waaland JR, Stiller JW, Cheney DP (2004) Macroalgal candidates for genomics. J Phycol 40:26–33
Weiss TL, Johnston JS, Fujisawa K, Okada S, Devarenne TP (2011) Genome size and phylogenetic analysis of the A and L races of Botryococcus braunii. J Appl Phycol 23:833–839
Acknowledgements
I thank Martina Pichrtová (Department of Botany, Charles University) and Karin Rengefors (Department of Biology, Lund University) for providing several algal cultures used in this study. I also thank Paul Kron (Department of Integrative Biology, Guelph University) for his suggestions at the initial stage of this study.
Author information
Authors and Affiliations
Contributions
The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results and manuscript preparation.
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Čertnerová, D. Nuclei isolation protocols for flow cytometry allowing nuclear DNA content estimation in problematic microalgal groups. J Appl Phycol 33, 2057–2067 (2021). https://doi.org/10.1007/s10811-021-02433-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-021-02433-z