
Abstract
Advanced energy storage technologies have been developed to achieve safe, high-capacity, and stable cathode materials for aqueous Zn-ion batteries. Vanadium oxide has lately become a popular cathode material thanks to its advantages of a sufficient operating voltage window and a highly reversible redox reaction. However, vanadium pentoxide has certain drawbacks, such as poor electrical conductivity and slow ion diffusion. To resolve these problems, V2O5 sheets with amorphous carbon were synthesized by an ultrafast (within one minute), lower temperature (180 ℃), one-step and large-scale combustion synthesis method. We demonstrate the introduction of carbon improves the conductivity and increases the content of low-valence vanadium, resulting in higher electrochemical activity and lower polarization of V2O5/C. Moreover, the introduction of C increases the concentration of oxygen defects in V2O5, further enhancing its conductivity and providing higher active sites for electrochemical reactions, thereby increasing its battery capacity. The synthesized V2O5/C sample exhibited a specific capacitance of 220 mAh g−1 (compared to 180 mAh g−1 for V2O5) in aqueous electrolytes at 4 A g−1, demonstrating a high specific capacity of 85.5% after 500 cycles with nearly 100% Coulomb efficiency. These results indicate improved specific capacitance and cyclic stability.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Aqueous zinc-ion batteries (ZIBs) have garnered significant attention for their unique advantage of using zinc as the negative electrode, due to their high theoretical capacity, low redox potential (− 0.76 V relative to the standard hydrogen electrode), high stability, and abundance at a low cost [1]. ZIBs are becoming increasingly attractive due to their excellent safety, high ionic conductivity, low cost, and superior performance over several decades [2, 3]. To date, cathode materials for ZIBs are mainly obtained from manganese-based oxides (α-, γ-phase) [4, 5], vanadium-based materials [6, 7], and Prussian blue analogues [8, 9], such as copper hexacyanoferrate and zinc hexacyanoferrate, which possess analogous crystalline structures [10]. Among these materials, vanadium pentoxide (V2O5) has distinct characteristics that make it an ideal candidate for cathode materials, including a conventional layered structure, an abundant material source, and a high energy density. However, due to its poor electronic conductivity, the performance of V2O5 is compromised under high current densities, leading to substantial polarization [11, 12]. V2O5 also has the limitation of large volume expansion [13] and disintegration of active species, resulting in poor rate capability and capacity diminishing during cycles [14].
To overcome these limitations, high-capacity transition metal oxides are hybridized with or deposited over a broad surface area of carbon to reduce localized stress and enhance electrical conductivity [15]. In response to these chemical variations, V2O5/C composites have been developed. Zhang [16] demonstrated that edge-rich cathodes with vertically oriented graphene (VG) nanosheets serving as templates for V2O5 nanosheets showed a high capacity of 370 mAh g−1 (at a current density of 0.2 A g−1) for ZIBs through hydrothermal synthesis. However, this hydrothermal method requires a long processing time and yields a low production output. Chen [17] reported that the high conductivity of carbon nanotubes (CNTs) is used to improve the rate capability of the V2O5 electrode. After 6000 cycles at 5 A g−1, the capacity retention ratio of the V2O5@CNTs electrode was 72%, which is 36% higher than that of pure V2O5 synthesized through hydrothermal methods. Nonetheless the discharge specific capacity of V2O5@CNTs electrode in 1 M ZnSO4/1 M Na2SO4 mixed electrolyte initial specific capacity is only 129 mA hg−1 at 5 A g−1. Wang [18] presented the impregnation of a porous CNT film with an aqueous V2O5 sol under vacuum, yielding a highly flexible composite film of carbon nanotube film and V2O5 (CNTF@V2O5) with high strength and high conductivity. As a result, this CNTF@V2O5 film delivers a high capacity of 356.6 mAh g−1 at 0.4 A g−1. However, it does not perform well under high current conditions. The initial specific capacity is 98 mAh g−1 at 4 A g−1. Xu [19] successfully synthesized a new type of vanadium pentoxide-carbon fiber cloth (V2O5-CFC) through electrospinning and high-temperature calcination techniques. As a cathode material for aqueous zinc-ion batteries, V2O5-CFC possesses specific capacity of 132 mA h g−1 at 1 A g−1. Corpuz [20] demonstrated binder-free, centimeter long, single-crystal, V2O5 nanofibers (BCS-VONF) on carbon cloth, as the cathode material for AZBs synthesized via a simple one-step hydrothermal. And reached a maximum discharge capacity of 155 mAh g−1 until ~ 30th cycle at 100 mA g−1.
Various methods have been developed to synthesize vanadium oxide, such as hydrothermal techniques, chemical vapor deposition, sol-gel, powder metallurgy, the liquid exfoliation technique, soft template approaches, and supercritical solvothermal reactions [21,22,23,24,25]. However, these methods often involve intricate processes, including long-term aging responses, ethanol immersion processes, long waits, several washings, and heating at various steps to obtain the final product. Therefore, a simple and cost-effective method with good extensibility is needed for practical use.
Solution combustion synthesis (SCS) [26] is an innovative and suitable method for producing V2O5/C for aqueous zinc-ion batteries. SCS offers the following advantages [27,28,29]: (1) low experimental environment requirements; (2) simplicity and low cost of instrumentation; (3) uniform reaction process due to the mixture being promoted to flow during the heating process; (4) fast and efficient reaction process; and (5) easy modification of product compositions by adjusting the combustion settings. By utilizing these advantages and adding C6H12O6 to the combustion feedstock, carbon-containing vanadium pentoxide powder can be obtained quickly and easily.
This paper proposes the use of the SCS strategy to fabricate V2O5/C as the cathode material for ZIBs and systematically analyzes its structure and electrochemical performance. The high-conductivity C is in contact with the V2O5 particles, significantly enhancing the conductivity of the V2O5/C cathode. An appropriate amount of C6H12O6 addition leads to morphological differences. The introduction of carbon increases the content of low-valent vanadium, higher concentration of oxygen defects, and enhances the electrochemical activity, resulting in lower polarization and thereby improving its conductivity.
2 Experimental procedure
All reagents used in the synthesis were of analytical quality and were used as received without additional purification. Ammonium metavanadate (NH4VO3) (0.03 M) was used as the vanadium source, while ammonium nitrate (NH4NO3) (0.4 M) and urea (CO(NH2)2) (0.2 M) were used as oxygen and oxidizing agents, respectively. C6H12O6 was added as the carbon source (0.01 M). The starting solution was prepared by dissolving all reactants in 100 ml of deionized water in a 500 ml glass at room temperature. The resulting mixture turned into a turbid white liquid after being stirred evenly with a magnetic stirrer. The glass was then placed in an electrical furnace heating at 300 ℃ for 20 min, and the majority of the water was evaporated, leaving behind a sticky paste. The solution was then instantly combusted, resulting in a foamy product. The power to the heating furnace is switched off simultaneously instantly at the moment of combustion. The reaction time measured is 1 min, and the reaction temperature is 180 ℃. For comparison purposes, a reaction without the addition of C6H12O6 was also prepared in the same manner (Fig. 1).

Schematic diagram for the preparation of V2O5 and V2O5/C
The powders’ phases were investigated at room temperature using X-ray diffraction (XRD, Rigaku D/max-RB12, Japan). Raman spectra were obtained using a Thermo Scientific DXR2xi Raman microscope with a 514 nm laser excitation source. Fourier transform infrared spectroscopy (FTIR) spectra were collected on a BRUKER ALPHA spectrometer across a range of 4000 to 400 cm−1. Thermogravimetric analysis (TGA) was performed with a TGA instrument in the temperature range 30–600 °C at a heating rate of 10 °C min−1 in air. To examine the chemical valence composition of the product’s surface components, X-ray photoelectron spectroscopy (XPS) was utilized with a PHI-5300 instrument. The powders’ morphology was characterized using both scanning electron microscopy (SEM, Zeiss, ULTRA 55, Germany) and transmission electron microscopy (TEM, FEI, Tecnai G2 F20, USA).
Electrochemical measurements were conducted using CR2032-type coin cells. The active materials were mixed with acetylene black and a binder at a weight ratio of 7:2:1 to produce the functioning electrode (PVDF). PVDF was dissolved in N-methyl pyrrolidone, and the mixture was stirred with an ultrasonic and vibrating stirrer to form a slurry that was evenly applied to the Ti foil. The prepared electrode sheets were then dried at 80 °C in a drying oven for 8 h, with a mass loading of 1 mg cm2 active materials. The CR2032-type coin cells were assembled in the air for electrochemical characterization, with a 3 M ZnSO4 aqueous solution used as the electrolyte. A Zn metal disk served as the counter electrode for electrochemical testing. Galvanostatic charge and discharge tests were carried out using a Neware tester (CT-4008). Cyclic voltammetry (CV) curves were collected using an electrochemistry workstation (CHI618D) in a voltage window of 0.2 to − 1.6 V, and electrochemical impedance spectroscopy (EIS) measurements were performed on the same workstation in the frequency range of 0.01 Hz to 100 kHz.
3 Results and discussion
To clarify how C6H12O6 influenced the formation of the V2O5/C composite, the morphology of the V2O5/C composite was analyzed using SEM characterization. The V2O5 sample (Fig. 2a) showed a glossy structure with regular blocks, and the average sheet size was around 200 nm. In contrast, the V2O5/C product (Fig. 2c) displayed a flake-like structure with a sheet structure and a few small pieces. The layer was smooth, and many fragments formed from sheet packing around 80 nm. With the addition of a small amount of C6H12O6, the crystal development of V2O5 was altered, resulting in a rather rough surface structure of V2O5/C. Additionally, the particle size of V2O5/C was observed to be smaller than that of V2O5. The possible reason for this situation is that adding C6H12O6 may lead to the refinement of V2O5 grains, and there are literature reports that excessive glucose can hinder the formation of intermolecular gaps in V2O5 [30].

a SEM pattern of V2O5; b TEM pattern of V2O5; c SEM pattern of V2O5/C; d HRTEM image of V2O5/C; e SAED pattern of V2O5/C; f TEM pattern of V2O5/C; g–i the elemental mappings about various elements of V2O5/C
The microstructures of V2O5 and V2O5/C were analyzed using TEM (Fig. 2b, f). It was observed that both V2O5 and V2O5/C exhibit a stacked sheet structure. The d-spacing of the lattice fringes is 0.34 nm, consistent with the (110) plane of V2O5/C (Fig. 2d). Additionally, the SAED pattern confirms the single-crystal structure of V2O5/C (Fig. 2e), as (-10, -1, -101, 002) diffraction points can be observed. The EDS mappings reveal a homogeneous distribution of V, O, and C throughout the structure of V2O5/C (Fig. 2g–i), indicating a successful carbon mix of V2O5. From Fig. 2i, it can be observed that there is also some carbon material outside the particles. This is due to the fact that during TEM testing, the sample is supported by a carbon film substrate, resulting in carbon film signals appearing in the outer region of the sample.

a XRD patterns of V2O5 and V2O5/C samples after calcination; b Schematic diagram of the crystal structure of V2O5/C showing different oxygen sites (O1, O2, O3, and O4) bonded with vanadium; c Raman spectra of V2O5 and V2O5/C samples; d TGA curve of V2O5/C sample
Figure 3a shows the X-ray diffraction (XRD) patterns of the V2O5 and V2O5/C samples. All diffraction peaks correspond well to those of the layered structure V2O5 with lattice constants a = 11.516 nm and c = 4.3727 nm, which accord with the literature values (PDF#41-1426), and no obvious peaks of additional contaminants are observed. The results indicate that both samples are crystalline-phase V2O5. However, the diffraction peaks of the (110) and (310) planes of V2O5/C gradually weaken with increasing C6H12O6 content. This implies that the addition of C6H12O6 surfactant would significantly hinder the preferred development of (110) planes, indicating a decreasing degree of crystallinity. However, it is evident from the graph that the peaks in the V2O5/C have shifted to the left. The leftward shift of XRD peaks indicates a movement toward smaller angles. According to the Bragg’s equation, a decrease in angle corresponds to an increase in the interplanar spacing (d-spacing) and an increase in the lattice parameters. This suggests that in the case of the carbon addition, some forms of interaction between C and V2O5 may cause a change in the crystal structure, resulting in an enlargement of its lattice parameters. No carbon peak was observed in the XRD of the V2O5/C sample, indicating that carbon may exist in an amorphous form.

a XPS spectra; b, c V 2p and O1s XPS spectra of V2O5 and V2O5/C
Figure 3c depicts the Raman spectra of the various V2O5 samples. The α-V2O5 is responsible for the characteristic peaks found at 144, 198, 286, 306, 407, 473, 524, 693, and 995 cm−1 [31]. The peaks at 995 and 693 cm−1 correspond to the stretching modes of the V=O1 bonds and V–O3 bonds, respectively, while the peak at 524 cm−1 corresponds to the stretching vibration of the V–O4 bond (Fig. 3b). The peaks at 473 and 306 cm−1 can be attributed to the bending vibrations of V–O2 and V–O4 bonds, respectively. In contrast, the vibrations at 407 and 286 cm−1 are attributable to the bending of the V=O1 bonds. The [VO5]–[VO5] vibrations correspond to the two Raman bands at 194 and 141 cm−1. After adding C6H12O6, most of the vibrations of V–O2 (473 cm−1) and V–O4 (524 cm−1) remain intact. The Raman peak at 693 cm−1 decreases by 32%, indicating that the oxygen vacancy site is primarily at the V–O3 bridge sites [32, 33]. The vanadium oxide matrix containing C6H12O6 as carbon sources exhibits two major Raman scattering peaks at around 1350 and 1580 cm−1. The 1350 cm−1 Raman scattering peak (D-band) is caused by the imperfection and disorder of carbon [13], while the 1580 cm−1 Raman scattering peak (G-band) is due to the SP2 atomic motion of stretching in the carbon ring or long chain, indicating the degree of graphitization of carbon-based materials [34, 35]. This illustrates the existence of carbon. Figure 3d shows TGA curve of V2O5/C in the temperature range of 30–600 °C. As it can be seen, the curve reveals that the weight loss below 150 °C resulted from the water in the V2O5/C. And the weight loss in the 200 to 400 °C temperature range shows that the content of carbon in the composite is 0.64 wt %.
The chemical composition of the samples was identified using X-ray photoelectron spectroscopy (XPS), as shown in Fig. 4a. It is important to note that the vanadium valance seen in Fig. 4b was estimated based on the XPS analysis. The V 2p3/2 peaks of V2O5 were observed at 516.3 eV (V4+) and 517.5 eV (V5+) (V4+/V5+ = 0.05:1). In the XPS spectrum of V2O5/C, the peaks for V5+ become faint, while the peaks for V4+ increase (V4+/V5+ = 0.1:1). This increase in the V4+ ratio may be attributed to the introduction of oxygen vacancies in V2O5/C, which is consistent with the Raman analysis.

a The third cycle of the CV curves of the V2O5/C electrode and V2O5 electrode at a scanning rate of 0.1 mV s−1 in the potential range between 0.2 and 1.6 V (vs. Zn2+/Zn); b First three cycles of the CV curves of the V2O5/C electrode at a scanning rate of 0.1 mV s−1 in the potential range between 0.2 and 1.6 V (vs. Zn2+/Zn); c CV curves of the V2O5/C composite electrodes at different scan rates; d Log i vs. log v plot of peak currents in the CV curves
To investigate further, the O1s spectra of the V2O5/C and V2O5 samples are shown in Fig. 4c, where two distinct peaks can be observed. The OI peak, which corresponds to the lattice oxygen of V2O5/C (V2O5), has a binding energy of 527.3 eV, while the OII peak at 528.4 eV is attributed to the oxygen vacancies in the metal oxide matrix [36, 37]. The V2O5/C sample has 7.5% more oxygen vacancies than V2O5, which could enhance its electrochemical characteristics .Oxygen vacancies in cathode materials can act as active sites for the oxygen reduction reaction, which is a crucial step in electrochemical processes. Additionally, these vacancies can increase the ionic and electronic conductivity of cathode materials, thereby leading to better electrochemical performance. By providing additional pathways for ion and electron transport, oxygen vacancies enhance the diffusion of charge carriers within the material. Moreover, oxygen vacancies have the ability to modify the redox properties of cathode materials, expanding their electrochemical stability window and improving their capacity retention. Consequently, the presence of oxygen vacancies can create more redox sites within the material, allowing for reversible redox reactions and avoiding irreversible side reactions that may degrade the cathode performance [36].

a GCD charge-discharge curves of V2O5 electrode at different cycles; b GCD charge-discharge curves of V2O5/C electrode at different cycles
Figure 5a shows the CV curves of V2O5/C and V2O5 at a scan rate of 0.1 mV s−1 and a voltage range of 0.2−1.6 V. The quasi-rectangular curve of the V2O5/C and V2O5 electrodes exhibits multiple pairs of redox peaks resulting from the redox reactions of vanadium in various valence states. Compared to V2O5, V2O5/C shows a higher current density, which can be attributed to its larger surface area. This suggests that the former has a greater electrochemical reactivity and capacity than the latter [38].

a Nyquist plots of the pristine V2O5 and V2O5/C electrodes with an AC amplitude of 10 mV (inset showing the corresponding equivalent circuit); b The relationship between the real part of the impedance and low frequencies; c Rate performance of V2O5/C and V2O5 cathodes; d Cycling performance and Coulombic efficiency of V2O5/C and V2O5 at a current density of 4 A g−1
Figure 5b illustrates the first three cycles of CV plots for the V2O5/C electrode within a voltage window of 0.2–1.6 V at a scan speed of 0.1 mV s−1. In the forward scan, a slight shoulder at 0.93 V is followed by two strong peaks at 0.87 V and 0.59 V, which demonstrates the electrochemical intercalation of Zn2+ into the layered structure. In the reverse scan, three peaks at 0.76, 1.05, and 1.14 V correspond to the deintercalation of Zn2+ ions from the multilayer framework. The peaks in the forward scan may be due to continual decreases from V5+ to lower oxidation states, whereas the reverse peaks may occur in the reverse scan. The first cycle in the CV profile shows slightly different peak locations than the other cycles, which can be attributed to the slow activation of the new electrode [17]. Additionally, following the first cycle, the subsequent CV curves exhibit strong repeatability and similarity, indicating the excellent electrochemical reversibility of the electrode.
As shown in Fig. 5c, the electrochemical kinetics of V2O5/C were studied by collecting cyclic voltammetry (CV) curves at different scan rates. As the scan rate increased from 0.2 mV s−1 to 1.0 mV s−1, the shape of the CV curves remained highly similar. The curves exhibited similar morphologies at different scan rates, with broadening of the peaks in the CV curves, indicating pseudocapacitive behavior. This is beneficial for facilitating rapid electrochemical reactions. The electrochemical kinetics process [36] can be calculated using Eq. (1):
The i and v represent the peak current and scan rate, respectively, while a and b are variable parameters. The value of b can be calculated by obtaining the slope of the logarithmic plot of log(v) versus log(i) (Fig. 5d).
For the given system, the value of b reflects the charge storage kinetics of the electrode. A lower limit of 0.5 indicates a diffusion-controlled process, while the b value of 1.0 indicates a capacitance-controlled process, including the double-layer effect and pseudocapacitance occurring near the surface of the electrode material. When the b is bigger than 0.5 and less than 1, it represents a combination of capacitance and diffusion-controlled processes [39]. In the CV curves of V2O5/C at different scan rates, the values of peak 1, peak 2, peak 3, and peak 4 for V2O5/C are 0.980, 0.822, 0.842, and 0.968, respectively. This indicates that the electrochemical kinetics of the V2O5/C electrode result from a combination of diffusion-controlled and capacitance-controlled processes.
Figure 6a and b show the galvanostatic charge-discharge (GCD) curves of V2O5 and V2O5/C electrodes, respectively, at a current density of 4 A g−1. It can be observed that the discharge specific capacity of the V2O5 electrode gradually increases from 124 to 178 mAh g−1 during the initial fifty cycles. In contrast, the discharge specific capacity of the V2O5/C electrode increases from 119 to 221 mAh g−1 during the initial fifty cycles. This process can be attributed to the activation of the active material in the cathode.
To better understand the dynamics of interfacial transport in the V2O5/C nanosheets, electrochemical impedance spectra (EIS) were conducted. As depicted in Fig. 7a, Nyquist plots were generated for both pristine V2O5 and V2O5/C sheets, revealing a depressed semicircle in the high-to-medium frequency range and an angled line in the low-frequency range. The Nyquist charts presented in the inset of Fig. 7a were analyzed using an analogous circuit. By using Eq. (2), it was possible to calculate the diffusion coefficient of Zn2+:
where A represents the surface area, n is the valence of ions, C is the concentration of zinc ions in the electrolyte, F represents the Faraday constant, R is the gas constant, T is the experimental temperature, and δ is the slope determined from the fitting lines between Zim and ω−1/2.
Table 1 displays the resistance of these electrodes. V2O5/C exhibits smaller values of Rs and Rct than pristine V2O5 due to the oxygen vacancies in the V2O5/C electrode, which increase the carrier concentration and enhance its electrical conductivity [40]. The Zn-ion diffusion coefficients can be calculated using the relationship between low frequencies and the real component of impedance (Fig. 7b). The Zn2+ diffusion coefficient of V2O5/C (3.85 × 10−13 cm2 s−1) is higher than that of pristine V2O5 (1.19 × 10−13 cm2 s−1). This increased Zn2+ diffusion after hydrogenation is attributed to enhanced conductivity and smoother Zn2+ tunnels resulting from oxygen vacancies [41, 42].
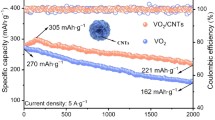
Figure 7c and d illustrate the comparisons of rate capability and cycle stability. Figure 7c shows the rate performances of V2O5/C and V2O5. The V2O5/C demonstrated higher rate capability, with average specific discharge capacities of 287, 270, 248, and 221 mAh g−1 at current densities of 1, 2, 4, and 8 A g−1, respectively. Figure 7d shows that the efficiency of the V2O5/C electrode and the V2O5 electrode increased by more than 100% during the first few dozen cycles due to the activation of the drainage zinc-ion batteries at the beginning. The activation procedure can be broken down into two steps. First, as the charge and discharge operation proceeds, the electrolyte enters the interior space of V2O5 [43]. Second, the insertion/extraction of Zn2+ and the phase transition of V2O5 produce additional new active sites [43, 44]. The V2O5/C electrode also exhibits better cycling capability than the V2O5 electrode. The specific capacity of V2O5/C is maintained around 220 mAh g−1 at 4 A g−1, gradually decreasing to 188 mAh g−1 (85.5%) after 500 cycles with nearly 100% Coulomb efficiency. In contrast, the specific capacity of V2O5 is maintained at around 180 mAh g−1 at 4 A g−1, exhibiting a capacity of 139 mAh g−1 (77.2%) at 4 A g−1 after 500 cycles. Table 2 presents a comparison of the cycling performance between previous studies and this work. The enhancement of V2O5/C composite rate capacity and cycle stability is attributed to the larger specified surface area and better electrical conductivity due to the addition of carbon. The oxygen defect concentration also improves the electrochemical characteristics of the V2O5/C electrode.
4 Conclusion
In summary, V2O5/C sheets were effectively synthesized as cathode materials for aqueous ZIBs using a simple solution combustion synthesis method. EIS, XPS, and CV techniques were used to demonstrate that V2O5/C with mixed vanadium valences exhibits more electrochemical activity, lower polarization, quicker ion diffusion capacity, and greater electrical conductivity than V2O5. The addition of carbon as a source potentially improves electrical and ionic conductivity, while the concentration of oxygen flaws might enhance the V2O5/C electrode’s electrochemical characteristics. V2O5/C maintains a higher specific capacity of 188 mAh g−1 (85.5%) at 4 A g−1 after 500 cycles with nearly 100% Coulomb efficiency compared to V2O5, which has a specific capacity of 139 mAh g−1 (77.2%). This approach is applicable to ZIBs’ electrodes and has the potential to be used in other energy storage applications due to its simplicity and scalability.
References
Yan XT, Feng XC, Hao BY, Liu JJ, Yu YR, Qi JJ, Wang HH, Wang ZY, Hu YQ, Fan XB, Li CL, Liu JP (2022) Enhancing the kinetics of vanadium oxides via conducting polymer and metal ions co-intercalation for high-performance aqueous zinc-ions batteries. J Colloid Interface Sci 628:204–213. https://doi.org/10.1016/j.jcis.2022.08.064
Bu F, Li C, Wang QZ, Liu XY (2022) Ultraviolet-assisted printing of flexible all-solid-state zinc batteries with enhanced interfacial bond. Chem Eng J 449:137710. https://doi.org/10.1016/j.cej.2022.137710
Zhou T, Zhu LM, Xie LL, Han Q, Yang XL, Chen L, Wang GK, Cao XY (2022) Cathode materials for aqueous zinc-ion batteries: a mini review. J Colloid Interface Sci 605:828–850. https://doi.org/10.1016/j.jcis.2021.07.138
Zhang N, Jia M, Dong Y, Wang YY, Xu JZ, Liu YC, Jiao LF, Cheng FY (2019) Hydrated layered vanadium oxide as a highly reversible cathode for rechargeable aqueous zinc batteries. Adv Funct Mater 29:1807331. https://doi.org/10.1002/adfm.201807331
Min X, Guo MM, Li K, Gu JN, Hu XF, Jia JP, Su TH (2022) Performance of toluene oxidation on different morphologies of α-MnO2 prepared using manganese-based compound high-selectively recovered from spent lithium-ion batteries. Environ Res 215:114299. https://doi.org/10.1016/j.envres.2022.114299
Yin CJ, Pan CL, Liao XB, Pan YS, Yuan L (2021) Coordinately unsaturated manganese-based metal-organic frameworks as a high-performance cathode for aqueous zinc-ion batteries. ACS Appl Mater Interfaces 13:35837–35847. https://doi.org/10.1021/acsami.1c10063
Chen XJ, Kong QQ, Wu XQ, An XG, Zhang J, Wang QY, Yao WT (2023) V2O3@C optimized by carbon regulation strategy for ultra long-life aqueous zinc-ion batteries. Chem Eng J 451:138765. https://doi.org/10.1016/j.cej.2022.138765
Zampardi G, La Manti F (2020) Prussian blue analogues as aqueous Zn-ion batteries electrodes: current challenges and future perspectives. Curr Opin Electrochem 21:84–92. https://doi.org/10.1016/j.coelec.2020.01.014
Wang SG, Xie Y, Hou YL, Zeng R, Qin T, Guan HB, Zhao DL (2022) Graphene aerogel encapsulated Co3O4 open-ended microcage anode with enhanced performance for lithium-ion batteries. Appl Surf Sci 605:154759. https://doi.org/10.1016/j.apsusc.2022.154759
Bin D, Wen YP, Wang YG, Xia YY (2018) The development in aqueous lithium-ion batteries. J Energy Chem 27:1521–1535. https://doi.org/10.1016/j.jechem.2018.06.004
Yan MY, He P, Chen Y, Wang SY, Wei QL, Zhao KN, Xu X, An QY, Shuang Y, Shao YY, Mueller KT, Mai LQ, Liu J, Yang JH (2018) Water-lubricated intercalation in V2O5·nH2O for high-capacity and high-rate aqueous rechargeable zinc batteries. Adv Mater 30:1703725. https://doi.org/10.1002/adma.201703725
Naresh N, Eom S, Jeong SH, Lee SJ, Park SH, Park J, Ahn JH, Kim JH, Jun YH (2022) Dendrite-free zn anodes enabled by a hierarchical zincophilic TiO2 layer for rechargeable aqueous zinc-ion batteries. Appl Surf Sci 606:154932. https://doi.org/10.1016/j.apsusc.2022.154932
Nakhanivej P, Park SK, Shin KH, Yun S, Park HS (2019) Hierarchically structured vanadium pentoxide/reduced graphene oxide composite microballs for lithium ion battery cathodes. J Power Sources 436:226854. https://doi.org/10.1016/j.jpowsour.2019.226854
Chen B, Wei J, Qiao XY, Liang D, Ji YX (2022) V2O5 plates anchored in CNTS composite materials with a network structure as polysulfide capture for lithium-sulfur battery. Ceram Int 48:32348–32356. https://doi.org/10.1016/j.ceramint.2022.07.177
Halada S, Zlatnik J, Mazur P, Charvat J, Slouka Z (2022) Fast screening of carbon-based nanostructured materials as potential electrode materials for vanadium redox flow battery. Electrochim Acta 430:141043. https://doi.org/10.1016/j.electacta.2022.141043
Zhang X, Tang YC, He PG, Zhang Z, Chen TF (2021) Edge-rich vertical graphene nanosheets templating V2O5 for highly durable zinc ion battery. Carbon 172:207–213. https://doi.org/10.1016/j.carbon.2020.10.034
Chen HZ, Qin HG, Chen LL, Wu J, Yang ZH (2020) V2O5@CNTs as cathode of aqueous zinc ion battery with high rate and high stability. J Alloys Compd 842:155912. https://doi.org/10.1016/j.jallcom.2020.155912
Wang XW, Wang LQ, Zhang B, Feng JM, Zhang JF, Ou X, Hou F, Liang J (2021) A flexible carbon nanotube@V2O5 film as a high-capacity and durable cathode for zinc ion batteries. J Energy Chem 59:126–133. https://doi.org/10.1016/j.jechem.2020.10.007
Xu N, Yan CY, He W, Xu L, Jiang ZH, Zheng AY, Wu HY, Chen M, Diao GW (2022) Flexible electrode material of V2O5 carbon fiber cloth for enhanced zinc ion storage performance in flexible zinc-ion battery. J Power Sources 15:533. https://doi.org/10.1016/j.jpowsour.2022.231358
Corpuz LMD, Corpuz RD, Somwangthanaroj A, Nguyen MT, Yonezawa T, Ma J, Kheawhom S (2019) Binder-free centimeter-long V2O5 nanobers on carbon cloth as cathode material for zinc-ion batteries. Energies 13:1. https://doi.org/10.3390/en13010031
Liu HY, Liang XP, Jiang T, Zhang YY, Liu SW, Wang XZ, Fan XW, Huai XG, Fu YD, Geng ZB, Zhang DQ (2022) High-performance self-doped V4+-V2O5 ion storage films grown in situ using a novel hydrothermal-assisted sol-gel composite method. Electrochim Acta 404:139784. https://doi.org/10.1016/j.electacta.2021.139784
Azadian F, Rastogi AC (2022) Electrochemical and energy storage properties of layer-by-layer assembled vanadium oxide electrode-based solid-state supercapacitor in n-SnO2:F/n-V2O5 heterostructure device form using ionic liquid gel electrolyte. J Solid State Electrochem 27:1–21. https://doi.org/10.1007/s10008-022-05309-5
Rui XH, Lu ZY, Yu H, Yang D, Hng HH, Lim TM, Yan QY (2013) Ultrathin V2O5 nanosheet cathodes: realizing ultrafast reversible lithium storage. Nanoscale 5:556–560. https://doi.org/10.1039/c2nr33422d
Umeshbabu E, Rao GR (2016) Vanadium pentoxide nanochains for high-performance electrochemical supercapacitors. J Colloid Interface Sci 472:210–219. https://doi.org/10.1016/j.jcis.2016.03.050
An QY, Wei QL, Mai LQ, Fei JY, Xu X, Zhao YL, Yan MY, Zhanga PF, Huang SZ (2013) Supercritically exfoliated ultrathin vanadium pentoxide nanosheets with high rate capability for lithium batteries. Phys Chem Chem Phys 15:16828–16833. https://doi.org/10.1039/c3cp52624k
Wu HY, Qin ML, Cao ZQ, Li XL, Jia BR, Chen PQ, Huang M, Qu XH (2018) Direct synthesis of vanadium oxide nanopowders by the combustion approach. Chem Phys Lett 706:7–13. https://doi.org/10.1016/j.cplett.2018.05.064
Kumar VK, Ghosh S, Ghosh S, Behera PS, Biswas S, Martha SK (2022) Enhanced electrochemical performance of O3-type NaNi0.5Mn0.3Co0.2O2 cathodes for sodium-ion batteries via Al-doping. J Alloys Compd 924:166444. https://doi.org/10.1016/j.jallcom.2022.166444
Ashok SC, Vazhayil A, Thomas J, Thomas N (2022) Enhanced electrochemical performance of facilely synthesized cobalt doped cubic NiO nanoflakes for supercapacitor application. J Energy Storage 55:105498. https://doi.org/10.1016/j.est.2022.105498
Afrooze A, Shaik D (2022) Porous Co3O4 nanospheres synthesized via solution combustion method for supercapacitors. Chem Pap 77:1–11. https://doi.org/10.1007/s11696-022-02535-z
Liu PC, Xu Y, Zhu KJ, Bian K, Wang J, Sun X, Gao YF, Luo HJ, Lu L, Liu JS (2017) Ultrathin VO2 nanosheets self-assembled into 3D micro/nano-structured hierarchical porous sponge-like micro-bundles for long-life and high-rate Li-ion batteries. J Mater Chem A 5:8307–8316. https://doi.org/10.1039/c7ta00270j
Baddou’ Hadjean R, Smirnov MB, Smirnov KS, Kazimirov VY, Gallardo-Amores JM, Amador U, Arroyo-de Dompablo ME, Pereir JP, Ramo (2012) Lattice dynamics of β-V2O5: Raman spectroscopic insight into the atomistic structure of a high-pressure vanadium pentoxide polymorph. Inorg Chem 51(5):3194–3201. https://doi.org/10.1021/ic202651b
Peng X, Zhang XM, Wang L, Hu LS, Cheng SHS, Huang C, Gao BA, Ma F, Huo KF, Chu PK (2016) Hydrogenated V2O5 nanosheets for superior lithium storage properties. Adv Funct Mater 26:784–791. https://doi.org/10.1002/adfm.201503859
Liu X, Li ZW, Fei HL, Wei MD (2015) Composite of K-doped (NH4)2V3O8/graphene as an anode material for sodium-ion batteries. Dalton Trans 44:18864–18869. https://doi.org/10.1039/c5dt03239c
Hussain S, Chen H, Zhang Z, Zheng H (2020) Vibrational spectra and chemical imaging of cyclo[18]carbon by tip enhanced Raman spectroscopy. Chem Commun 56:2336–2339. https://doi.org/10.1039/c9cc09130k
Schuepfer DB, Badaczewski F, Guerra-Castro JM, Hofmann DM, Heiliger C, Smarsly B, Klar PJ (2020) Assessing the structural properties of graphitic and non-graphitic carbons by Raman spectroscopy. Carbon 161:359–372. https://doi.org/10.1016/j.carbon.2019.12.094
Lu XH, Zeng YX, Yu MH, Zhai T, Liang CL, Xie SL, Balogun MS, Tong YX (2014) Oxygen-deficient hematite nanorods as high-performance and novel negative electrodes for flexible asymmetric supercapacitors. Adv Mater 26:3148–3155. https://doi.org/10.1002/adma.201305851
Fujii T, Groot Fd, Sawatzky GA, Voogt FC, Hibma T, Okada K (1999) In situ XPS analysis of various iron oxide films grown by NO2-assisted molecular-beam epitaxy. Phys Rev B 59:3195–3202. https://doi.org/10.1103/PhysRevB.59.3195
Chao DL, Xia XH, Liu JL, Fan ZX, Ng CF, Lin JY, Zhang H, Shen ZX, Fan HJ (2014) A V2O5/conductive-polymer core/shell nanobelt array on three-dimensional graphite foam: a high-rate, ultrastable, and freestanding cathode for lithium-ion batteries. Adv Mater 26:5794–5800. https://doi.org/10.1002/adma.201400719
Yang YQ, Tang Y, Fang GZ, Shan LT, Guo JS, Zhang WY, Wang C, Wang L, Zhou J, Lianget SQ (2019) Li+ intercalated V2O5·nH2O with enlarged layer spacing and fast ion diffusion as an aqueous zinc-ion battery cathode. Energy Environ 11:3157–3162. https://doi.org/10.1039/c8ee01651h
Zhai T, Xie SL, Yu MH, Fang PP, Liang CL, Lu XH, Tong YX (2014) Oxygen vacancies enhancing capacitive properties of MnO2 nanorods for wearable asymmetric supercapacitors. Nano Energy 8:255–263. https://doi.org/10.1016/j.nanoen.2014.06.013
Shin JY, Joo JH, Samuelis D, Maier J (2012) Oxygen-deficient TiO2-delta nanoparticles via hydrogen reduction for high rate capability lithium batteries. Chem Mater 24:543–551. https://doi.org/10.1021/cm2031009
Xia T, Zhang W, Murowchick JB, Liu G, Chen XB (2013) A facile method to improve the photocatalytic and lithium-ion rechargeable battery performance of TiO2 nanocrystals. Adv Energy Mater 3:1516–1523. https://doi.org/10.1002/aenm.201300294
Chen LL, Yang ZH, Wu J, Chen HZ, Men JL (2020) Energy storage performance and mechanism of the novel copper pyrovanadate Cu3V2O7(OH)2·2H2O cathode for aqueous zinc ion batteries. Electrochim Acta 330:135347. https://doi.org/10.1016/j.electacta.2019.135347
Qin HG, Chen LL, Wang LM, Chen X, Yang ZH (2019) V2O5 hollow spheres as high rate and long life cathode for aqueous rechargeable zinc ion batteries. Electrochim Acta 306:307–316. https://doi.org/10.1016/j.electacta.2019.03
Acknowledgements
This work is financially supported by the Natural Science Foundation of Fujian Province (2022J011269) and Industry-Academe-Research Project of Xiamen (2022XCY0402) and Open Fund of Fujian Provincial Key Laboratory of Functional Materials and Applications (Grant No. fm2022005).
Author information
Authors and Affiliations
Contributions
JC (First Author): Conceptualization, Methodology, Software, Formal Analysis, Writing - Original Draft; YY: Data Curation; SG: Investigation; WZ: Resources, Supervision; HZ: Software, Validation; ZC: Writing - Review and Editing; JM (Corresponding Author): Conceptualization, Funding Acquisition, Resources, Supervision, Writing - Review and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cui, J., Ma, J., Yuan, Y. et al. Direct synthesis of vanadium pentoxide powder with carbon recombination as aqueous zinc-ion battery cathode. J Appl Electrochem 54, 289–299 (2024). https://doi.org/10.1007/s10800-023-01971-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-023-01971-3