
Abstract
Cyanuric acid (CA) is known as an important environmental and human toxin with low electroactivity. Therefore, it is an attractive issue to develop an electrochemical sensor for CA determination. Here, we reported a novel electrochemical sensor for the indirect electrochemical determination of CA using the complex formation between CA and melamine (MEL), for the first time. Firstly, the electrochemical conversation of MEL to electroactive polymelamine was conducted using a glassy carbon electrode modified with overoxidized Poly-(para-aminophenol) film. MEL was chosen as a probe for indirect determination, and its current intensity was recorded. CA was then added to the medium, and the decline in the current intensity was considered as the sensor response. The calibration curve within the range of 100–2500 µmol L−1 was constructed from sensor response against CA concentration using square wave voltammetry. The limit of detection was found to be 80.28 µmol L−1. Moreover, the proposed mechanism for the indirect determination of CA was investigated based on the quantum theory of atoms in molecules analysis. Finally, the developed method was able to exhibit an excellent recovery range (91.2–101.0%) for the real sample analysis.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cyanuric acid (CA) or 2,4,6-trihydroxy-1,3,5-triazine, is a structural analog of melamine (MEL) which is widely utilized in the production of chlorine stabilizers, household bleaches, disinfectants, and, automatic dishwasher compounds [1]. It should be pointed out that MEL can be hydrolyzed sequentially to CA and other analogs, such as ammeline, and ammelide [2]. Owing to its high nitrogen content, MEL is deliberately added to food ingredients to enhance the protein content of products, especially milk products [3]. Due to its widespread application, continuous exposure to CA and its analogs through food products [4, 5] and environmental pathways (e.g., sewage sludge, swimming pool water, and farm soils) [6,7,8] is unavoidable. CA at high levels has been found to harm health by causing liver toxicity [9]. Besides, CA is a well-known kidney toxicant and, when it presents in high concentration along with MEL, it forms insoluble crystals which participate in distal renal tubules, leading to kidney damage and death [10,11,12].
Breast milk is a unique biofluid that satisfies all of the nutritional and immune requirements of infants [13]. Nevertheless, this biofluid has been regarded as the main exposure source of toxic chemicals in infants, including CA [14]. Considering the adverse health effect of CA, its quantification in breast milk is indispensable.
Up to now, chromatographic techniques, including high-performance liquid chromatography (HPLC) [15], liquid chromatography coupled to mass spectrometry (LC–MS) [16], and gas chromatography coupled to mass spectrometry (GC–MS) [17] are the main techniques to determine CA, Unfortunately, these methods have some drawbacks, including time-consuming, tedious, and need of expensive instruments and hazardous solvents (in the case of LC methods). Contrastly, electrochemical sensors are able to overcome these problems to some degree. These sensors are an interesting variation of sensors because of their unique properties, including versatility, selectivity, sensitivity, and low-cost. Electrochemical sensors are low-cost and simple in comparison to chromatographic methods. Also, they can rapidly detect the target compounds without the need for organic solvent, so these are greener and faster than chromatographic techniques [18]. The electrochemical sensors are usually designed based on two strategies: direct and indirect methods. Yilmaz et al. [19] applied differential pulse polarography (DPP) to determine the CA amount in swimming pool water. Pei et al. [20, 21] modified the surface of a glassy carbon electrode (GCE) with polyaniline/CuGeO3 nanowire, and employed it for direct determination of CA using cyclic voltammetry (CV). These direct electrochemical methods suffer from some disadvantages. Since DPP uses the dropping Hg electrode as the working electrode, it suffers from the disadvantages of Hg in toxicity, storage, and disposal [22, 23]. Moreover, it should be noticed that the CV method is not recommended for electroanalytical applications [24]. Indirect strategy can be an applicable method when the aim is the determination of a target compound with low electroactivity. This strategy is based on the interaction between an analyte with low electroactivity and an electroactive substance as the electrochemical probe, and consequently, the signal variations of the probe are monitored. For instance, Sağlam et al. [25] developed an indirect electrochemical method to determine boron based on the complex formation between boric acid and mannitol. Boron and mannitol are electrochemically inactive and active compounds, respectively. So, boron can be determined indirectly using electroactive complexing agents such as mannitol. Similarly, as CA shows a low electroactivity, it can be determined using the indirect strategy. Many researchers have proved the formation of a 1:1 complex between MEL and CA through hydrogen binding interactions [26,27,28,29]. This fact motivated us toward the use of the indirect electrochemical strategy based on the complexation between CA and MEL to determine CA. Figure 1 shows the structures of CA, MEL, and CA-MEL complex. Our research group in 2021 has developed a simple electrochemical preconcentration method for the effective quantification of MEL [30]. In this work, a GCE was modified with overoxidized Poly-(para-aminophenol) film (OX-PPAP/GCE) to preconcentrate MEL through the potentiodynamic technique. The highlighted results revealed that MEL could be an appropriate probe for the indirect determination of CA.

The structure of CA, MEL, and CA-MEL complex
In the present work, a novel electrochemical method for the indirect determination of CA was developed based on the complex formation between CA and MEL. The MEL with low electroactivity was converted to electroactive polymelamine (PMEL) through polymerization onto the OX-PPAP/GCE surface as the modified electrode. However, the MEL current was measured using the modified electrode before and after the CA addition, which formed a 1:1 complex with MEL. Consequently, the current decrement was considered as the sensor response and recorded versus the CA concentration. Also, the proposed electrochemical sensor was successfully applied to CA determination in breast milk samples.
2 Experimental
2.1 Chemicals and reagents
All chemicals were analytical reagent grade and used without further purifications. Para-aminophenol (PAP) was obtained from Sigma Aldrich (Steinheim, Germany). CA, MEL, sodium hydroxide (NaOH), sodium dodecyl sulfate (SDS), acetic acid (HOAC), hydrochloric acid (HCl), sulfuric acid (H2SO4), potassium hexacyanoferrate (K3[Fe(CN)6]), and zinc acetate dihydrate (Zn(CH3CO2)2.2H2O) were purchased from Merck Company (Darmstadt, Germany). Stock standard solution of CA was prepared at a concentration of 0.01 mol L−1. Working solutions were daily made by diluting the stock solution of CA with deionized water. Carrez solution (I) was prepared by dissolving 5.3 g of K3[Fe(CN)6] in 50 mL of deionized water. To prepare Carrez solution (II), 11.0 g of Zn(CH3CO2)2.2H2O was mixed with 1.5 mL HOAC, and then its volume was adjusted to 50 mL by deionized water.
2.2 Preparation of OX-PPAP/GCE
A potentiostat/galvanostat (797 VA Computrace polarography system, Metrohm, Herisau, Switzerland) was employed to perform all electrochemical experiments by taking a lab-made cell consisting of a KCl-saturated calomel electrode (SCE), a platinum rod, and a GCE (Azar Electrode Co., Iran) as the reference, counter, and working electrodes, respectively. Before any modification, the GCE surface was well-polished and activated using wet alumina slurries (0.3 μm) on silk, followed by sonicating in ethanol solution (50%) for 10 min.
OX-PPAP/GCE was fabricated based on the manner reported in our previously published report [30]. Briefly, the PAP electropolymerization on the GCE surface was carried out in the aqueous electrolyte solution containing 50 mmol L−1 of PAP monomer, 1 mol L−1 HCl, and 7 mmol L−1 SDS. Uniform PPAP film deposited on the GCE (denoted as PPAP/GCE) was obtained at an optimized potential range from − 0.6 and + 2.0 V/SCE with 100 mV s–1 scan rate using 15 cycles. The overoxidation of as-fabricated PPAP/GCE was then conducted by dipping the modified GCE into NaOH solution (0.1 mol L−1) and applying + 1.2 V for 450 s. Afterward, the as-prepared modified electrode (denoted as OX-PPAP/GCE) was rinsed several times with deionized water.
2.3 Electrochemical determination of CA
The OX-PPAP/GCE electrode was positioned in an aqueous solution containing MEL (60 µmol L−1), NaCl (0.03 mol L−1), and H2SO4 (0.15 mol L−1). The potentiodynamic method was conducted from − 0.25 to + 1.55 V at a scan rate of 100 mV s−1 to pre-concentrate MEL on the modified GCE surface. After rinsing the electrode with the deionized water, the square wave voltammetry (SWV) technique was used to detect MEL at the optimized parameters of frequency 140 Hz, voltage step of 2 mV, and pulse amplitude 50 mV in the solution containing 0.1 mol L−1 NaNO3, and 0.05 mol L−1 H2SO4 [30]. To quantify CA using the indirect electrochemical method, the current intensity of MEL as the probe was measured before and after CA addition. After the addition of CA, MEL reacted with CA, and the current of free MEL was measured. The sensor response can be expressed as the following equation:
where I0 and I represent the current of MEL before and after CA addition, respectively. ΔIMEL (µA) represents the sensor response observed at 0.58 V.
2.4 HPLC analysis
The chromatographic analysis of CA was conducted with an HPLC system (Knauer, Berlin, Germany), equipped with a UV detector Model K-2600 (KNAUER, Smartline, Germany) with a K-1001 piston pump. An analytical octadecylsilyl column (ODS, 4.6 mm × 250 mm, 5 µm) was used to separate CA. The column temperature was 25 °C, and the injection volume was 20 µL. The CA separation was obtained using the mobile phase consisting of phosphate buffer: methanol, v/v (95:5 v/v, pH = 7.2–7.4) at a 1.0 ml min−1 flow rate. The UV monitoring wavelength was set at 213 nm [31].
2.5 CA extraction
Two breast milk samples were collected from mothers living in different areas of Hamedan, Iran. Before the real sample analysis by the developed method, a total of 2 mL of breast milk was transferred into a conical tube, and fortified with a known concentration of MEL standard, as the probe. Afterward, the samples were extracted using 2% H2SO4 and de-proteinated by adding 1 mL of both Carrez solutions. Subsequently, each sample mixture was shacked for 5 min and centrifuged at 4000 rpm for 20 min. The supernatant was analyzed using the developed method.
2.6 Computational details
Full geometry optimizations of CA, MEL, and CA-MEL complex were carried out using density functional theory (DFT). Gaussian 09W software [32] was employed for all computational procedures at the B3LYP/6-31G (d,p) level of theory. Bader’s QTAIM [33] analysis was conducted using the AIM2000 package [34] to study the interactions.
3 Results and discussion
3.1 Characterizations of the modified GCE
The characterization of PPAP/GCE and OX-PPAP/GCE electrodes using field emission scanning electron microscopy, Fourier transform infrared spectroscopy, CV, and electrochemical impedance spectroscopy techniques have been described in our previous study in detail [30]. Figure 2A displays CV curves of the electropolymerization process of PPAP in the potential range between − 0.6 to + 2.0 V/SCE at a scan rate of 100 mV s−1. The first cycle exhibits two oxidation peaks (at 0.64 and 1.70 V), and one reduction (at 0.4 V), which are ascribed to the electroformation of intermediate species [35, 36]. The reaction of formed species with PAP monomers, and consequently, the polymer growth occurs with the increasing cycle numbers [37]. The shift of the oxidation peak to more positive potentials observed with the increasing cycles is strong evidence for the continuous growth of PPAP polymer [35]. In the following, the overoxidation of as-fabricated PPAP/GCE in 0.1 mol L−1 NaOH was conducted to improve the conductivity and obtain a more porous surface. Finally, an adherent and homogeneous film, named OX-PPAP/GCE was coated on the GCE surface.

Voltammograms of A the PAP electropolymerization on the surface of bare GCE (the inset shows the cycle number 1), B the electrochemical preconcentration of 60 μmol L−1 MEL at OX-PPAP/GCE
3.2 Electrochemical determination of CA
The mechanism of preconcentration of MEL at the OX-PPAP/GCE surface was briefly discussed. The mechanism contains two steps: (i) the electro-generation of active chlorine species from chloride ions, (ii) The electro-assisted polymerization of MEL to form electro-active PMEL (the formation of preconcentrated MEL). To confirm the proposed mechanism, the CV method was employed. Figure 2B shows the CV curves obtained during the MEL preconcentration on the modified electrode within − 0.25 to + 1.55 V at the scan rate of 100 mV s−1 and in the electrolyte containing NaCl (0.03 mol L−1) and H2SO4 (0.15 mol L−1). The irreversible anodic peak that appeared at around + 1.55 V/SCE, was ascribed to the electro-generation of active chlorine species [38,39,40]. It should be stressed that the observed irreversible anodic peak at high positive potentials (up to + 1.55 V) corresponded to the electro-oxidation of chloride ions into active chlorine species, such as HOCl, OCl−, Cl2●, Cl●, and ClO● [38,39,40]. Chen et al. previously reported that these active chlorine species play a critical role in the electropolymerization of MEL [41]. The reversible peaks present at + 0.61 V/SCE corresponded to the transformation of –NH– sites into azo sites in PMEL [42].
Since electro-active PMEL exhibits a reversible electrochemical behavior, this can be proportional to the initial concentration of MEL. Hence, the SWV method was engaged to determine the MEL concentration.
As CA is a specie with low electroactivity, it can be indirectly determined using electrochemical methods through reaction with a suitable reagent. In this work, the current intensity of MEL was recorded before and after the addition of CA in the solution using the SWV method (Fig. 3). The CA determination can be made based on the formation of the MEL-CA complex because of the complex formation of CA with MEL at 1:1 mol ratio through OCA… H-NMEL, O-HCA…NMEL, and OCA…H-NMEL hydrogen bonds [43,44,45].

Square wave voltammograms of 60 µmol L−1 MEL with the addition of 2000 µmol L−1 CA recorded with OX-PPAP/GCE electrode
3.3 Optimization of experimental conditions
To achieve the best analytical performance, important parameters, including MEL concentration, reaction time, and reaction temperature were optimized, as seen in Fig. 4. Remarkably, in all experiments, SWV was performed in the range of + 0.40 V to + 0.9 V, pulse amplitude of 50 mV, frequency of 140 Hz, and in the supporting electrolyte containing 0.1 mol L−1 of NaNO3 and 0.03 mol L−1 of H2SO4. Also, ΔIMEL was considered as the sensor response.

Effect of A the concentration of MEL (condition: CA: 1000 µmol L−1, temperature: 25 °C, time: 5 min), B reaction temperature (condition: MEL: 60 µmol L−1, CA: 1000 µmol L−1, time: 5 min), and C reaction time (condition: MEL: 60 µmol L−1, CA: 1000 µmol L−1, temperature: 25 °C)
3.4 Optimization of MEL concentration
Since, in this work, the indirect determination of CA was based on its complexation with MEL, the optimum concentration of MEL needed to be determined. Clearly, IMEL increased up to 60 μmol L−1 MEL, which leads to enabling the maximum response (ΔIMEL) in the indirect determination of CA. Hence, 60 µmol L−1 was chosen as the optimum concentration of MEL as the probe. This is consistent with our previous work that the corresponding calibration curve of GCE/OX-PPAP toward Mel shows good linearity in the concentration range of 2.0–60 μmol L−1 of MEL [30].
3.5 Optimization of reaction temperature
The reaction temperature is a critical factor in the proposed strategy for the CA determination because it influences the stability of the CA-MEL complex. The temperature effect on the method response was studied over the range of 10–50 °C. As can be seen in Fig. 4B, ΔIMEL increases up to 25 °C, and then decreases at higher temperatures. This result arises from the fact that lengths, strengths, and the number of hydrogen bonds are strongly affected by temperature. Since these interactions are so weak, their number and strange decrease with increasing temperature [46, 47]. Hence, the optimal temperature was selected 25 °C.
3.6 Optimization of reaction time
The effect of reaction time on the sensor response was investigated in the range of 0–10 min. Figure 4C shows the results of the time investigation in which ΔIMEL was increased with increasing the reaction time up to 5 min, followed by its decrease with further increasing time. This trend can be attributed to enhancing the growth rate of the CA-MEL complex with increasing time [48]. As hydrogen bonding is possibly weak, it can break easily at a higher time. Considering the results depicted in Fig. 4C, the reaction time of 5 min was selected as the optimum reaction time between MEL and CA.
3.7 Analytical performance
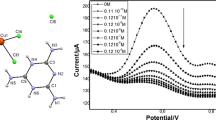
The CA determination was performed using a calibration curve with the corresponding analytical performance parameters. Under optimum conditions, the current declines of MEL exhibited a linear dependence over the CA concentration range of 100–2500 µmol L−1 (Fig. 5). SWV voltammograms related to CA determination were presented in the inset of Fig. 5. Limit of detection (LOD = 3σb m−1, where σb and m symbolized the standard deviation of blank and the calibration curve slope, respectively) was found to be 80.28 µmol L−1. Although the obtained LOD is relatively high, it is satisfactory for the determination of MEL in real milk samples. According to several studies, analytical methods with relatively high LOD values could employ in the quantification of different analytes in real samples [49,50,51,52,53]. Besides, it should be emphasized that only a few papers [19,20,21], exist on the direct electrochemical determination of CA. Moreover, to the best of our knowledge, no effort has been devoted to developing an indirect electrochemical method for CA determination. However, our results suggested that further studies presenting lower LOD values, and higher sensitivities are needed to develop electrochemical methods for the determination of CA. To evaluate the reproducibility of the proposed method, the relative standard deviation (RSD) of ΔIMEL from 10 repeated experiments was calculated, which was 4.7%. Fig. S1 depicts square wave voltammograms of CA related to the reproducibility experiment. Noticeably, the modified GCE exhibited excellent long-term stability, and the initial response was kept at about 82% after three weeks of storage (Fig. S2A). Fig. S2B shows the voltammograms related to stability.

The CA calibration curve under the optimum condition. Inset shows the voltammograms related to different concentrations of CA
3.8 Effect of common ions in breath milk on the electrochemical determination of CA
The potential interference of vitamin D, vitamin E, thiamin, sucrose, glucose, lactose, maltose, fructose, calcium carbonate, and magnesium nitrate (common compounds in the real sample of interest) was investigated in the indirect determination of CA. The method response was monitoring. Table 1 presents the obtained results. To explain the interference results, the tolerance limit of ± 5%, which is established as interference concentration producing a relative error less than ± 5% error in the analyte response, was considered. These compounds did not show any interference in the determination of CA (1000 µmol L−1) at 1000-fold concentrations. The interference results confirmed the proposed mechanism of indirect determination of CA, in which CA could form the complex with MEL.
3.9 Computational analysis
As the formation of hydrogen-bond interaction between CA and MEL was considered the basis of the proposed strategy for CA determination, the computational analysis was conducted to give a deep insight into the interaction between them. Figure 6 displays the optimized geometries of CA, MEL, and the CA-MEL complex. As can be seen in Fig. 3, there are three hydrogen bonds between CA and MEL molecules, including a hydrogen-bond between oxygen atoms of carboxylic groups in CA…hydrogen atoms of MEL (OCA…HMEL), and hydrogen of amine group in CA…nitrogen of MEL (N-HCA…NMEL).

A Optimized geometry of the CA-MEL complex, B The bond paths (pink lines), BCPs (red points), and RCPs (yellow points) for the interactions between CA and MEL in the complex. Colors assigned to each element: red: oxygen, blue: nitrogen, black: carbon, gray: hydrogen. (Color figure online)
The values of zero-point corrected binding energies were obtained − 317,538, − 280,129, and − 597,710 kcal mol−1 for CA, MEL, and their complex, respectively. These results indicate that the CA-MEL complex is more stable than CA and MEL with an energy difference of − 43.8704 kcal mol−1. This stability can be attributed to the hydrogen-bond interaction between CA and MEL.
To give more information about the formation mechanism of the CA-MEL complex, the QTAIM analysis was conducted. In QTAIM, the bond path and the local topological properties at the bond critical point (BCP), and ring critical points (RCP), including Laplacian of the electron density (∇2ρb), charge density (ρb), and local potential electron density (VC), local kinetic electron energy density (GC), and the total electron energy density (Hb) are important parameters which are mainly used to describe the strength of a bond [54]. Table 2 summarizes the calculated parameters for the CA-MEL complex. Figure 6 displays the interaction between the optimized geometries of CA and MEL, the bond paths (pink lines), BCPs (red points), and RCP (yellow points) for the CA-MEL complex.
The QTAIM results reveal the presence of three principal interactions with different bond paths, including O24… H8-N7, O20-H27…N2, and O23…H12-N10 between CA and MEL. It is interesting to point out that two RCP that appear in the topological analysis, are related to the ring formation between MEL and CA. These rings lead to more stability of the CA-MEL complex.
The GC/VC ratio is an indicator for investigating the nature of chemical bonds. A ratio higher than 1 indicates the non-covalent nature of the bond, while a ratio lower than 0.5 represents the covalent nature of the chemical bond. In general, larger values of ρb and ∇2ρb demonstrate stronger interactions [55]. Thanks to its higher ρb and ∇2ρb values, the-N2…H27-O20 bond is stronger than N7-H8…O24 and N10-H12…O23 bonds, which is in line with the results obtained from GC/VC ratio. The results presented in Table 2 suggest that hydrogen bonding interactions between CA and MEL are partially covalent interactions.
3.10 Real sample
To further study the practical applicability of the developed method, it was tested with breast milk samples as real samples. Table 3 presents the found and recovery values calculated for specific concentrations of the real spiked samples. The recoveries ranging from 91.2% to 101.0% for the analysis of CA were obtained using the developed indirect method (Table 3), confirming the reliability and efficiency of the proposed method as a novel and powerful electrochemical method.
In the validation study, CA was determined in breast milk samples using HPLC–UV/Vis method as the reference method (Table 3). Comparing the results obtained from the developed method with those achieved by HPLC–UV/Vis revealed a good agreement between the results of both methods. This confirms the applicability of the developed electrochemical sensor for CA determination in real samples.
4 Conclusion
CA is an important compound due to its potential impact on the environment and human health. As CA shows low electroactivity, the development of effective electrochemical methods for its determination has attracted much interest. Thus, we developed a simple and reliable indirect electrochemical method for the CA determination using OX-PPAP/GCE as the modified electrode and MEL as the probe. The CA-MEL complexation was used as the basic principle for the indirect determination of CA, which has low electroactivity, using the SWV method. The proposed method measured the current of uncomplexed MEL, and the current difference of MEL in the absence and presence of CA attributed to CA concentration added to the medium. The CA determination was successfully performed by recording current declines of MEL as a linear function of increasing CA concentrations. The proposed method was able to quantify CA in the real samples with acceptable recoveries. The commonly studied compounds did not interfere with the CA determination. Briefly, the proposed indirect electrochemical method is simple, low-cost, and applicable to CA determination.
References
Dorne JL, Doerge DR, Vandenbroeck M, Fink-Gremmels J, Mennes W, Knutsen HK, Vernazza F, Castle L, Edler L, Benford D (2013) Recent advances in the risk assessment of melamine and cyanuric acid in animal feed. Toxicol Appl Pharmacol 270:218–229
Gong H, Tang S, Zhang T (2016) Catalytic hydrolysis of waste residue from the melamine process and the kinetics of melamine hydrolysis in NaOH solution. React Kinet Mech Catal 118:377–391
Hu X, Shi J, Shi Y, Zou X, Arslan M, Zhang W, Huang X, Li Z, Xu Y (2019) Use of a smartphone for visual detection of melamine in milk based on Au@ Carbon quantum dots nanocomposites. Food Chem 272:58–65
Zhu H, Kannan K (2019) Melamine and cyanuric acid in foodstuffs from the United States and their implications for human exposure. Environ Int 130:104950
Hong M, Sai F, Yong-Ning W, Zhang L, Ping-Ping Z, Hui-Jing C, Yun-Feng Z, Jing-Guang L (2009) Simultaneous determination of melamine, ammelide, ammeline, and cyanuric acid in milk and milk products by gas chromatography-tandem mass spectrometry. Biomed Environ Sci 22:87–94
Zhu H, Halden RU, Kannan K (2019) A nationwide survey of the occurrence of melamine and its derivatives in archived sewage sludge from the United States. Environ Pollut 245:994–999
Zhu H, Wang Y, Sun H, Kannan K (2019) Fertilizers as a source of melamine and cyanuric acid in soils: a nationwide survey in China. Environ Sci Technol Lett 6:55–61
Liu X, Silverman AD, Alam KK, Iverson E, Lucks JB, Jewett MC, Raman S (2019) Design of a transcriptional biosensor for the portable, on-demand detection of cyanuric acid. ACS Synth Biol 9:84–94
Karns JS (1999) Gene sequence and properties of an s-triazine ring-cleavage enzyme from Pseudomonas sp. strain NRRLB-12227. Appl Environ Microbiol 65:3512–3517
Brown CA, Jeong KS, Poppenga RH, Puschner B, Miller DM, Ellis AE, Kang KI, Sum S, Cistola AM, Brown SA (2007) Outbreaks of renal failure associated with melamine and cyanuric acid in dogs and cats in 2004 and 2007. J Vet Diagn 19:525–531
Liu CC, Hsieh TJ, Wu CF, Tsai YC, Huang SP, Lee YC, Huang TY, Shen JT, Chou YH, Huang CN (2017) Urinary melamine excretion and increased markers of renal tubular injury in patients with calcium urolithiasis: a cross-sectional study. Environ Pollut 231:1284–1290
WH Organization (2009) Toxicological and health aspects of melamine and cyanuric acid: report of a WHO expert meeting in collaboration with FAO. Health Canada, Ottawa, pp 1–4
de Oliveira TV, de Carvalho PP, Petronilho S, Santelli RE, Braz BF, Freire AS, Saunders C, da Rocha HF, Sanz-Medel A, Fernández-Sánchez ML (2020) Total metal content and chemical speciation analysis of iron, copper, zinc and iodine in human breast milk using high-performance liquid chromatography separation and inductively coupled plasma mass spectrometry detection. Food Chem 326:126978
Zhu H, Kannan K (2019) Occurrence of melamine and its derivatives in breast milk from the United States and its implications for exposure in infants. Environ Sci Technol 53:7859–7865
Chao YY, Lee CT, Wei YT, Kou HS, Huang YL (2011) Using an on-line microdialysis/HPLC system for the simultaneous determination of melamine and cyanuric acid in non-dairy creamer. Anal Chim Acta 702:56–61
Braekevelt E, Lau BY, Feng S, Ménard C, Tittlemier S (2011) Determination of melamine, ammeline, ammelide and cyanuric acid in infant formula purchased in Canada by liquid chromatography-tandem mass spectrometry. Food Addit Contam 28:698–704
Tzing SH, Ding WH (2010) Determination of melamine and cyanuric acid in powdered milk using injection-port derivatization and gas chromatography–tandem mass spectrometry with furan chemical ionization. J Chromatogr A 1217:6267–6273
Mehrabi A, Rahimnejad M, Mohammadi M, Pourali M (2021) Electrochemical detection of Flutamide as an anticancer drug with gold nanoparticles modified glassy carbon electrode in the presence of prostate cancer cells. J Appl Electrochem 51(4):597–606
Yilmaz ÜT, Yazar Z (2010) Determination of cyanuric acid in swimming pool water and milk by differential pulse polarography. Clean: Soil, Air, Water 38:816–821
Pei L, Xie Y, Pei Y, Cai Z, Fan C (2013) Determination of cyanuric acid by electrochemical cyclic voltammetry method using CuGeO3 nanowires as modified electrode materials. IJNME. https://doi.org/10.1115/1.4026024
Pei L, Liu H, Lin N, Xie Y, Cai Z (2015) Electrochemical analysis of cyanuric acid using polyaniline/CuGeO3 nanowires as electrode modified materials. Curr Pharm Anal 11:16–24
Han H, Pan D, Pan F, Hu X, Zhu R (2021) A functional micro-needle sensor for voltammetric determination of iron in coastal waters. Sens Actu B Chem 327:128883
Piech R (2011) Study on simultaneous measurements of trace gallium (III) and germanium (IV) by adsorptive stripping voltammetry using mercury film electrode. J Appl Electrochem 41(2):207–214
Bard AJ, Faulkner LR, White HS (2022) Electrochemical methods: fundamentals and applications. Wiley
Sağlam Ş, Üzer A, Erçağ E, Apak R (2021) Electrochemical determination of boron using the signal suppression of mannitol on a gold nanoparticle coated p-Aminothiophenol polymer electrode. Microchem J 166:106252
Zerkowski JA, MacDonald JC, Seto CT, Wierda DA, Whitesides GM (1994) Design of organic structures in the solid state: molecular tapes based on the network of hydrogen bonds present in the cyanuric acid. cntdot. melamine complex. J Am Chem Soc 116:2382–2391
Ji H-F, Xu X (2010) Hexagonal organic nanopillar array from the melamine—cyanuric acid complex. Langmuir 26:4620–4622
Wang C, Qin X, Huang B, He F, Zeng C (2010) Hemolysis of human erythrocytes induced by melamine–cyanurate complex. Biochem Biophys Res Commun 402:773–777
Mehra N, Jeske M, Yang X, Gu J, Kashfipour MA, Li Y, Baughman JA, Zhu J (2019) Hydrogen-bond driven self-assembly of two-dimensional supramolecular melamine-cyanuric acid crystals and its self-alignment in polymer composites for enhanced thermal conduction. ACS Appl Polym Mater 1:1291–1300
Esmaeily Z, Madrakian T, Afkhami A, Ghoorchian A, Ghasemzadeh-Mohammadi V (2021) Electropolymerization as an electrochemical preconcentration approach for the determination of melamine in milk samples. Electrochim Acta 390:138897
Cantú R, Evans O, Kawahara FK, Shoemaker JA, Dufour AP (2000) An HPLC method with UV detection, pH control, and reductive ascorbic acid for cyanuric acid analysis in water. Anal Chem 72(23):5820–5828
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Petersson G, Nakatsuji H (2016) Gaussian 16 revision a. 03. 2016. Gaussian inc, Wallingford
Bader R, Biegler-König F, Schönbohm J (2002) AIM2000 program package, Ver. 2.0. McMaster University, Hamilton
Bader R (2000) AIM2000 program, v. 2.0. McMaster University, Hamilton
Koçak ÇC, Dursun Z (2013) Simultaneous determination of ascorbic acid, epinephrine and uric acid at over-oxidized poly (p-aminophenol) film modified electrode. J Electroanal Chem 694:94–103
Ojani R, Raoof J-B, Fathi S (2009) Poly (o-aminophenol) film prepared in the presence of sodium dodecyl sulfate: application for nickel ion dispersion and the electrocatalytic oxidation of methanol and ethylene glycol. Electrochim Acta 54:2190–2196
Chen C, Sun C, Gao Y (2008) Electrosynthesis of poly (aniline-co-p-aminophenol) having electrochemical properties in a wide pH range. Electrochim Acta 53:3021–3028
Chen S, Huang W, Zheng J, Li Z (2011) Study on the electrodissolution and roughening of a palladium electrode in chloride containing solutions. J Electroanal Chem 660(1):80–84
Panizza M, Cerisola G (2003) Electrochemical oxidation of 2-naphthol with in situ electrogenerated active chlorine. Electrochim Acta 48(11):1515–1519
Szpyrkowicz L, Radaelli M, Daniele S (2005) Electrocatalysis of chlorine evolution on different materials and its influence on the performance of an electrochemical reactor for indirect oxidation of pollutants. Catal Today 100(3–4):425–429
Chen S, Liu S, Wen A, Zhang J, Nie H, Chen J, Zeng R, Long Y, Jin Y, Mai R (2018) New insight into electropolymerization of melamine. I: chloride promoted growth of polymelamine in different pH medium. Electrochim Acta 271:312–318
Kesavan S, Revin SB, John SA (2014) Potentiodynamic formation of gold nanoparticles film on glassy carbon electrode using aminophenyl diazonium cations grafted gold nanoparticles: determination of histamine H2 receptor antagonist. Electrochim Acta 119:214–224
Praus P, Smýkalová A, Foniok K, Novák V, Hrbáč J (2021) Doping of graphitic carbon nitride with oxygen by means of cyanuric acid: properties and photocatalytic applications. J Environ Chem Eng 9:105498
Conn MM, Rebek J (1997) Self-assembling capsules. Chem Rev 97:1647–1668
Ma M, Bong D (2011) Determinants of cyanuric acid and melamine assembly in water. Langmuir 27:8841–8853
Ohtaki H (2003) Effects of temperature and pressure on hydrogen bonds in water and in formamide. J Mol Liq 103:3–13
Mizan TI, Savage PE, Ziff RM (1996) Temperature dependence of hydrogen bonding in supercritical water. J Phys Chem A 100:403–408
Li R, Cui X, Bi J, Ji X, Li X, Wang N, Huang Y, Huang X, Hao H (2021) Urea-induced supramolecular self-assembly strategy to synthesize wrinkled porous carbon nitride nanosheets for highly-efficient visible-light photocatalytic degradation. RSC Adv 11:23459–23470
Ghoorchian A, Afkhami A, Madrakian T, Rameshan R, Rameshan C, Hajian A (2020) Absorbance-based spectroelectrochemical sensor for determination of ampyra based on electrochemical preconcentration. Sens Actu B Chem 324:128723
Ngamchuea K, Moonla C, Watwiangkham A, Wannapaiboon S, Suthirakun S (2022) Electrochemical and structural investigation of copper phthalocyanine: application in the analysis of kidney disease biomarker. Electrochim Acta 428:140951
Ding L, Yan J, Zhao Z, Li D (2019) Synthesis of NiGa2O4 nanosheets for non-enzymatic glucose electrochemical sensor. Sens Actu B Chem 296:126705. https://doi.org/10.1016/j.snb.2019.126705
Lin S, Liu S, Dai G, Zhang X, Xia F, Dai Y (2021) A click-induced fluorescence-quenching sensor based on gold nanoparticles for detection of copper(II) ion and ascorbic acid. Dyes Pigments 195:109726. https://doi.org/10.1016/j.dyepig.2021.109726
Lin Y, Kannan P, Zeng Y, Qiu B, Guo L, Lin Z (2019) Enzyme-free multicolor biosensor based on Cu2+-modified carbon nitride nanosheets and gold nanobipyramids for sensitive detection of neuron specific enolase. Sens Actu B Chem 283:138–145. https://doi.org/10.1016/j.snb.2018.12.007
Zalazar MF, Paredes EN, Romero Ojeda GD, Cabral ND, Peruchena NM (2018) Study of confinement and catalysis effects of the reaction of methylation of benzene by methanol in H-Beta and H-ZSM-5 zeolites by topological analysis of electron density. J Phys Chem C 122:3350–3362
Nasiri M, Shakourian-Fard M, Fattahi A (2012) Influence of the hydrogen bonding on the basicity of selected macrocyclic amines. J Phys Org Chem 25:803–810
Acknowledgements
This work has been supported by grants from the Bu-Ali Sina University Research Council. This research is also jointly supported by the Hamadan University of Medical Sciences, and the Centre of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) which are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no financial or proprietary interests in any material discussed in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hashemi, S., Afkhami, A., Kamalabadi, M. et al. Electrochemical determination of cyanuric acid using the signal suppression of melamine on an overoxidized Poly-(para-aminophenol) coated glassy carbon electrode. J Appl Electrochem 53, 2483–2493 (2023). https://doi.org/10.1007/s10800-023-01931-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-023-01931-x