
Abstract
Charge carriers have been studied for use in applications such as fuel cells, redox flow batteries, and electrochemical CO2 reactors for conversion to value-added products. Here, transient-based equilibrium models are developed for two well-known charge carriers: methyl viologen (MV) and ethyl viologen (EV). The models are simulated using Butler-Volmer kinetics until steady-state is reached. EV is favored over MV due to lower dimerization, and enabling over 2 × production of reduced EV+ over MV+. MV and EV products do not appear to significantly change, except only under sufficiently acidic conditions (pH < 4). Charge and energy input requirement are used to assess total system efficiency and potential for system scale-up via chronoamperometry. The charge and energy analysis performed with EV as the charge carrier reveals that optimal charging voltage is around − 0.8 to − 0.85 V vs. Ag/AgCl, which is above the minimum reduction voltage (around − 0.6 to − 0.7 V vs. Ag/AgCl) and suggest more favorable conditions for performing such charge carrier reductions.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Electron charge carriers, or charge mediators, have served as redox indicators in various biological and biochemical processes [1,2,3,4]. Some of the direct uses of mediators include microbial fuel cells [5,6,7,8,9] and photoelectrochemical cells [10, 11]. Research on additional applications for mediators, such as in redox flow batteries and supercapacitors [12,13,14,15,16,17] will be invaluable in the future for renewable energy storage. Charge mediators have also been studied for their usefulness in assisting the selective electrochemical reduction of carbon dioxide (CO2) into value-added products [18, 19]. Most charge mediators readily dissolve into ions in water. When dissolved into a solution, the charge carrier cation can be reduced into a variety of different species [20]. The speciation of charge mediator formed depends primarily on the surrounding electrochemical potential, concentration, and pH conditions [1], which determine transfer proton/electron capacity.
Among the most commonly known electron mediators are viologens, which were first documented in 1933 [1] and subsequently used for many bio-catalytic applications [2]. Methyl viologen (MV, tradename paraquat) is perhaps the most extensively studied as a biocatalyst. Reduction voltages for the first electron transfer of viologens typically range from − 600 to − 700 mV vs. Ag/AgCl reference electrode [2, 21]. Additional reduction of viologen, which occurs at over − 950 mV vs. Ag/AgCl (− 986 mV vs. SCE) [22, 23] and has also been reported to occur at voltages even lower than − 1.1 V vs. SCE. These voltages may be less stable and hard to re-oxidize back to previous states [24]. While methyl viologen has been one of the most extensively studied charge carriers, it faces serious environmental concerns with toxicity [25] and the potential to cause lung cancer [26]. One alternative is ethyl viologen (EV, tradename diquat), which has lower toxicity [27] and is not a suspected lung carcinogen [28]. EV also displays greater conductivity and maintains nearly identical redox potentials [1, 2]. Degradation through dimerization of viologens and quinones has also been reported to be an issue as it competes with desired reaction products and can be difficult to reverse [13, 29,30,31]. Consequently, there is a need to reliably predict the state of the charge carrier, requiring more robust theoretical models which can aid in their design [29, 32, 33].
It has been stated previously that it is necessary for viologens to be optimized before commercialization. Recently, rigorous studies have been conducted on the characterization, synthesis, and long-term cycling performance of such systems [34,35,36,37,38]. While electrochemical studies reliably estimate kinetic parameters such as diffusion constant and reaction rates, the assessment of the variation in particular species long-term has not been as closely studied. To the best of the authors’ knowledge, only a few recent studies have directly examined the independent speciation of products charge mediator products [29, 32, 39]. Expanding upon existing work can enable more focused studies on experiments and relevant parametric ranges to corroborate computational models. Additionally, an electrochemical model is a fundamental tool as all parameters can be directly inserted or fit to experimental data, and modified depending on the type of mediator and operating conditions of interest [40]. This presents an opportunity to develop and expand on electrochemical models, which have not been examined as much in recent studies despite other significant advances in the field. One of the main motivations for developing the model is to quickly assess product composition over a varying range of voltage and pH values, eliminating the need for a larger number of experiments.
The understanding of long-term behavior, for applications such as batteries and capacitors, is necessary in order to optimize carrier speciation and also assess the onset of dimerization. Here, A range of electrochemical properties of MV and EV will be compared to assess optimal operating conditions for each charge carrier, including experimental design suggestions. A rate-based transient model is constructed which considers mass/charge transfer rate and electrochemical properties to demonstrate dynamic convergence to predicted thermodynamic equilibrium values at a given reaction time scale. This study primarily considers applications for MV and EV as charge carriers for CO2 capture, but as previously stated, the model can be extended to other uses such as in batteries and electrochromatics. The model is also compared with experimental chronoamperometry results to assess the accuracy of the fitted parameters; the parameters fit to experimental data are either taken from existing literature or examined through a series of cyclic voltammetry tests and varying scan rates. A product composition analysis confirms that reactions proceed quickly, and dimerization competition is present in the intermediate voltage ranges and at higher concentrations. Lastly, the total charge and energy consumption of EV are reported at various applied voltages, both of which reveal that optimal EV reduction does not necessarily occur at voltages which have previously been predicted in experiments. The model is verified for at least 1 hour of run time for voltages ranging from − 0.5 V to − 1.1 V vs. Ag/AgCl, with optimal performance for EV occurring around − 0.85 V. The primary aim of this work is to demonstrate the feasibility of the models, which simultaneously incorporate fundamental electrochemical theory and enable the refinement of parameters through experimental results. In the future, by examining different charge carriers, advantages and disadvantages of each can be more immediately predicted for applications such as electrochromic devices, batteries, and CO2 conversion fuel cells [41, 42].
2 Modeling
The electrochemical cell is modeled as a single (0-D) unit, as the primary objective is to determine how current, cell voltage, and different reaction products change with time to best connect with experimental data. To eliminate gradients, the core of the model neglects diffusion simply, allowing concentrations in the region modeled near the electrode to “disperse” to the bulk solution (well-mixed) via a moving average calculation. This is accomplished by employing a mixing fraction, denoted by f, which is further explained in the SI. The modeled area near the electrode is a perfectly mixed volume where the species diffuses much faster than they can react. Spatial variations are thus incorporated through the non-physical moving average scheme. This results in two perfectly mixed volumes: an outer bulk and inner reaction volume which communicates through the non-physical method—moving average with a mixing fraction to control the speed at which the inner volume mixes into the outer volume. Volume-averaging schemes [43, 44] and related reduction in system dimension [45,46,47] have been discussed and employed in previous electrochemical literature and the technique employed here further simplifies the model from 1-D to 0-D with appropriate experimental correlations. Thus, mass transport through a gradient is not included in the model due to the averaging scheme used, but mass transport between the cathode and anode is included. Accordingly, the system can still exhibit a limiting current due to mass transfer considerations, which are accounted for in the model and later shown to be verified with experimental results.
The thermodynamic modeling in this work is based on Nernstian equilibrium [31, 32]. Further details on the transient model are detailed in the Supporting Information; here, only an overview of the key reactions involved are discussed. The transient model developed is similar to a previous study with equilibrium conditions for CO2 reduction [48], but here the spatial variations in the product compositions are neglected.
The methyl viologen (MV) samples considered in this work consist of a dichloride hydrate anion (Fig. SI1A). Only the reaction pathways for MV are presented here; ethyl viologen (EV) is assumed to have similar reaction pathways [1, 2, 13]. Here, the EV cation is paired with a dibromide anion (Fig. SI1B). The known electrochemical reductions of the dispersed MV2+ solution to MV+ and subsequently to MV0 are as follows:
Three additional reactions occur in solution, including the dimerization reaction, salt dissociation, and protonation of the reduced MV+ radical ion:
While other products have been explored/discussed [22], they are not considered to contribute significantly to the product speciation. The majority of other reaction products are believed to occur from the dimer MV22+ and reduced ion MV0. The full set of reaction pathways for the charge carriers can be illustrated in a “scheme of squares” diagram (Fig. 1). Here the salt dissociation is omitted as only separated ion reactions are considered. Both MV and EV generally undergo two pH-independent redox reactions with the first reduction product undergoing a possible dimerization reaction (or protonation in more acidic environments). However, the sensitivity of pH to the quantity of the products shown in Fig. 1 will later be assessed due to the proton contribution in equilibrium constants.

Scheme of squares depicting reaction products for MV/EV. All processes are independent of pH. The salt dissociation of MV is not included here due to its relatively minimal contribution
The equations provided in greater detail in the Supporting Information were solved in MATLAB using the Eulerian forward discretization scheme used for ODEs. As the set of equations could not be solved analytically, it was not possible to rigorously determine the error in this time step. The time step chosen was sufficiently small such that it would not cause a system instability. This increased the accuracy of the computation while averaged values were considered in order to neglect concentration/potential gradients.
3 Experimental
Cyclic voltammetry (CV) tests were run in a simple reactor with cathode (working), anode (counter), and reference electrodes in a single chamber of volume 30 mL. Electrochemical tests were conducted on a Gamry REF-600 Potentiostat using current interrupt (CI) to compensate for ohmic resistances. The CV tests were swept over a voltage range from − 1.5 to 1.5 V vs. Ag/AgCl, to initiate the system and more clearly illustrate the locations of the redox peaks. Then, the solution was swept over a range of − 1.0 to 0 V vs. Ag/AgCl at various scan rates to analyze specific locations of redox peaks. For the CV tests only, electrolytes were used corresponding to prior literature studies to ensure similar redox values could be replicated [32, 49]. For MV, that electrolyte was 0.1 M KCl (Cole-Parmer Ionic Strength Adjustors). Both charge carrier/electrolyte combinations were assessed at scan rates of 5, 10, 20, and 50 mV/s to determine sensitivity to redox peak locations as well as current magnitudes. A mass of approximately 180 mg carbon felt (AvCarb G100 Soft Graphite Battery Felt, Fuel Cell Store) was used as the working electrode. The counter electrode consisted of an approximately 520 mg platinum wire (99.9%, 1 mm diameter, Sigma-Aldrich) submerged in the cathode solution, and Ag/AgCl was the reference electrode (Basi MF-2052 Ag/AgCl Reference Electrode, 3 M NaCl).
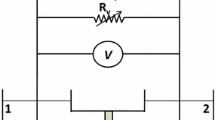
In all other experiments involving direct applied voltage electrolysis, an acrylic batch cell (Fig. 2) was used. The cathode chamber consisted of a carbon felt working electrode, 200 mM potassium phosphate buffer solution, and a 10 mM charge carrier. The anode chamber functioned primarily to supply protons and consisted of a Pt wire (same wire as mentioned above) counter electrode in a 1 mM sulfuric acid (ACROS Organics) solution. The cathode chamber was stirred consistently to maintain uniform composition inside the batch volume for the duration of the experiment. The volumes used here were 10 mL for each compartment. Ag/AgCl was used as the reference electrode for the applied cathode voltage, and the two sides were separated by a Nafion 115 (Fuel Cell Store) proton exchange membrane (PEM) that had been soaked in DI water for at least 24 h prior to the start of the experiment. Prior to applying any external voltage/current, the cell was purged with either CO2 or N2 gas to minimize oxygen interference, allowing an additional 30 min for the Nafion membrane to equilibrate in the cell. For the N2, it was expected that pH would remain balanced at pH 7 since it is an inert gas, but the CO2 purge yields a lower pH (Fig. SI3) due to some formation of carbonic acid and other carbonates.

Schematic of batch cell setup
The charge carriers used for the experiments were methyl viologen dichloride hydrate (MV, 98%, Sigma-Aldrich) and ethyl viologen dibromide (EV, 99%, Sigma-Aldrich). The buffer consisted of a 50%/50% mixture of potassium phosphate monobasic (Fisher Chemicals, 99.9%) and potassium phosphate dibasic (Anhydrous, MP Biomedicals). The experiments were purged with CO2 (99% Purity Grade) to decrease the system concentration of dissolved oxygen (DO), which can lead to unwanted secondary reactions, including charge loss through oxidation.
In both the model and experiments, charge and energy values were calculated as integrated current and total voltage values in the cell over the operational time:
where the current density is multiplied by area Acell to convert charge and energy into units of Coulombs and Joules, respectively.
4 Results and discussion
4.1 Transient-based modeling
The mixing parameter delay due to the presence of the mixing fraction f is the primary reason that the total cell current decays to a non-zero value to reflect the current leakage. The delay caused by the mixing fraction slows down the equilibrating time, which requires additional current input to offset the delay. Oxygen-mediated chemical reactions may also aid the re-oxidation of the charge carrier within the bulk solution [50]. Thus, as this mixing fraction decreases, the total cell current may be higher at first due to the entire charge carrier reacting, but will quickly decay to a lower value (Fig. SI4A). This process may take a longer time before equilibrium is seen depending on the selected operating conditions, but nonetheless is an obstacle in decreasing the required input charge.
Experimentally, many factors can go into how averaging parameters such as mixing fraction f are determined, some of which are not replicated computationally. One such factor that should be considered is the geometry of the setup, as the orientation of the reactor chamber and electrode will affect rate of transport of charge carrier within the reactor. The size, mixing rate, and spatial effects inside the reactor can also affect the ability of the charge carrier to react [51,52,53]. Such spatial effects are beyond the scope of the model, but the volume of the reactor is considered. Clearly, with more charge carrier mass at higher volumes, more charge input is needed to maintain concentrations at electrochemical equilibrium (Fig. SI4B). The computational model and Eq. SI24 for bulk volume are shown to decrease reduction currents at lower volumes, an important consideration when looking ahead at system scale-up.
The exact choice of reaction rates primarily depended on attaining a reliable fit to existing experimental data. The values selected appeared in the range of published kinetic reaction rates [21, 29, 49, 54], along with an estimation based on experimental CV data, redox peak separation, and diffusion constants. All kinetic values shown in Table SI1 were obtained in the range of 10–4–10–2 m s−1, so selected values within that range proved reasonable for the time that it should take for the charge carrier reactions to proceed. Examining concentration-based changes computationally, the dimerization of MV occurs much more rapidly and at several orders of magnitude above that of EV (Fig. 3A, B). At 10 mM, dimerization favors the reduced MV by about 0.5 mM, but since dimer composes twice the amount of MV, this is effectively over two-thirds the total MV in solution that is dimerized. Furthermore, the dimerization occurs at nearly the same rate as MV+, and the running time of the experiment cannot be altered to favor MV+ over dimerization. The model also shows a trace amount of secondary electrochemical reduction to MV0, despite a voltage insufficient to trigger reduction to that particular species. This discrepancy is due to the iterative solution technique of the thermodynamic equilibrium model, where the MV0 product begins to appear in trace amounts (1 × 10–6 mM) at only − 0.6 V for MV and − 0.7 V for EV. Thus, the redox voltages in the model are primarily equilibrium voltages and not true cutoff threshold values below which a charge carrier product would be unable to form [1].

Steady-state convergence of selected charge carrier products over a time of 1.5 h at an applied voltage of − 0.75 V vs. Ag/AgCl and an operating pH of 6.3. A 10 mM MV, B 10 mM EV, C 0.1 mM MV, D 0.1 mM EV
When comparing the viologens, values of KD notably decrease the extent to which dimerization occurs. In EV, this suggests that a higher dimerization constant suppresses dimerization as the values vary inversely with the amount of dimer formed (Fig. 3C, D). As a result, EV+ is now more heavily favored, with only about 9% of the initial dissociated EV2+ unreacted. The undissociated salts here are considered negligible; while EVBr2 continues to increase, its significance as a steady-state value is not reflected in the full equilibrium solution for either viologen. For both viologens, the convergence to the final products predicted by equilibrium conditions is only 15 min on average.
4.2 Transient convergence to steady-state
When compared with steady-state equilibrium models at different voltages, the final product values all fully converge after reaching steady-state at sufficient operation time, as would be expected from comparing the forward and reverse reaction rates. The MV and EV values are similar to existing literature [1, 2, 32], with peak MV+ production occurring at nearly the midpoint between the first and second electrochemical reductions (Fig. 4A, B). Notably, dimerization is only sufficiently present in MV where in EV it is decreased significantly due to the differing dimerization constant value. For clarity, the steady-state model is also shown at a range of different concentrations (Fig. SI5), confirming that the dimerization only impacts MV more significantly in the intermediate voltage range when concentration is higher. For completion, the maximum viologen concentration is extended to 100 mM. For MV as well as EV (Fig. SI6), some of the initial unreacted viologen salt will remain if the applied voltage is insufficient.

Final product composition of charge carriers as a function of applied cathodic voltage, C0 = 10 mM, pH 6.3. A MV, B EV. Symbols represent values from transient simulations after 1.5 h, and lines are from steady-state equilibrium model. Values below 1 × 10–6 mM are neglected as below tolerance errors
When considering pH variations, only the low acidic-neutral values were considered and formation did not change significantly when pH was raised to 7. Carbonate-based buffer with potassium salt can enable a more basic pH range of operations. However, the higher pH values may not be permitted in certain biological applications or in acid-based batteries. Conversely, at other pH ranges, product formation does not change appreciably (Fig. SI7). The protonation of viologen product, while always factored into equilibrium operation, is only present in acidic (pH < 4) environments.
4.3 Cyclic voltammetry
MV and EV plots reveal peaks at values approximate to those shown in previous studies (Fig. 5) [30, 55]. This confirms the validity of the redox data consistent with literature and also provides guidance on the optimal operating voltages. The MV charge carrier displayed clearer results, but the EV showed greater current magnitudes. In both cases, the redox peaks are seen more clearly when scan rate is decreased to 5 mV s−1, indicating that a small current should be reached at a stable convergence time when all of the product is reduced.

Charge carrier CV results run in a single-cell configuration at various scan rates. A 10 mM MV with 0.1 M KCl buffer, B 10 mM EV with 0.1 M KCl buffer
Randles–Sevcik analyses have been conducted in several recently published works to reliably estimate parameters such as diffusion coefficients [36, 56,57,58]. A Randles–Sevcik analysis was conducted on each of the CV tests to determine the extent of diffusion coefficients for both the MV and EV redox reactions. Here, only the first reduction and oxidation stages (MV2+/EV2+ to MV+/EV+) were considered as it was most desired to compare the relevant reactions used for most future chronoamperometry studies. The scan rate was plotted against the current for each redox peak and fit to the Randles–Sevcik equation:
where ip is the peak current in amps, A is the electrode area in cm2, C is the solution concentration in mol mL−1, F is the Faraday constant, D is the viologen diffusion coefficient in cm2 s−1, ν is the scan rate in V s−1, R is the gas constant in J mol−1 K−1, and T is the temperature in K, here assumed as 293 K room temperature.
To improve the accuracy of the method, the peak currents were measured relative not to 0 V, but to the averaged value of the current during the entire CV scan. In all of the tests, the averaged CV current was negative (Table SI2), with the EV currents notably more negative (− 24 to − 32 mA) than the MV currents (− 6 to − 8 mA). Despite considering this adjusted baseline current, the EV peaks at each scan rate were always greater than the peaks for the MV currents. This coincides with the greater electrical conductivity of EV (see SI), suggested and shown in the chronoamperometry tests.
For the CV tests, the chosen solution concentration was 200 mM, because although the viologen concentration was lower at 10 mM, using a higher concentration enabled a more proper alignment with diffusion values. The completed Randles–Sevcik analysis (Fig. 6) yielded most diffusion coefficients on the order of 10–9 cm2 s−1 as predicted from previous studies [59, 60]. Despite the baseline adjustment, all of the EV diffusion coefficients obtained were greater than those for MV. The maximum diffusion coefficient observed was for the oxidation of the EV+, which may further reflect the change in the average current when this particular species is re-oxidized after charging. In the model, diffusion coefficients were not immediately considered due to the lack of strong concentration gradients, but could still be implemented to account for limiting currents.

Calculation of diffusion coefficients for MV and EV redox peaks by plotting scan rate vs. peak current and fitting using Randles–Sevcik analysis
4.4 Chronamperometry and open-circuit voltage
The chronoamperometry tests at − 0.75 V vs. Ag/AgCl for MV and EV were compared against a blank sample, which otherwise employed the same chemistry and operating conditions as the viologen experiments. CO2 and N2 were used as purge gases while the cell was charged for 1.5 h and then discharged for 3 h. The initially higher current seen in Fig. 7A suggests favorability toward product formation, and the subsequent current decrease helps minimize input energy. When purged with CO2 the steady-state current magnitude in all cases remains below 5 mA, reaching these values within the first 10 min of the experiment. In the blank sample, however, the current is initially smaller and gradually increases, probably because it is only possible for minor side reactions to occur in this case. The gradual current uptick in Fig. 7A may explain why in Fig. 7B the blank sample retained the higher initial voltage when left in an open circuit (OCV) configuration. Conversely, the viologens, while charged to a much greater extent than in the blank sample, could not sustain this charge for more than 10 min at OCV, indicating the challenge of retaining reduced viologen products in an open-air environment and reinforcing the need to keep the system charged.

Charging and OCV data for 10 mM charge carrier samples in 200 mM phosphate buffer solution with carbon felt electrode. Anode consisted of platinum wire in a 1 mM sulfuric acid solution. A Charging data purged with CO2, B OCV data purged with CO2, C charging data purged with N2, D discharging data purged with N2
When purged with N2, Fig. 7C, D showed similar results, but initial current for EV was more severely diminished, leading to a more consistent decay throughout. At steady-state, the current magnitudes for MV and EV were − 2.0 and − 0.7 mA, respectively, a decrease in magnitude over the CO2 purging by over 2 × in both cases. Generally, the CO2 exhibits lower overall currents, decreasing required charge input, but with this comes a lower retention of the potential during the OCV. Thus, it may be preferable to purge N2 instead since the system will remain inert, with no hydrocarbons or other products of interest produced. Both gas purges with N2 and CO2 do show a general increase in the current from MV to EV.
In any case, the data between CO2 and N2 in the viologen studies is mostly consistent, with EV requiring a higher input charge but also subsequently releasing it quicker in both cases. In both Fig. 7B and D, a more rapid release is displayed, indicating that EV reduction is not as stable for the reduced species EV+. This is to be expected since EV requires a higher voltage to reduce, thus illustrating the greater current reflected in Fig. 7A–C.
4.5 Connecting theory with experiment
The experiments to measure electrochemical performance for a given charge carrier reduction can be fit to the model, accounting for a molecular oxygen contribution that limits mass transfer. When considering all of the model parameters in Table SI1, testing with a charge of − 0.75 V vs. Ag/AgCl yields a strong fit to both the MV and EV experiments (Fig. SI8). Here, EV is selected for the charge/energy analysis as well as the UV–Vis data shown in the Supporting Information (Figs. SI9, 10), due to the decreased competition with dimerization. The contribution of molecular oxygen is important because it explains the continued rise in the overall system current even after the redox peak value necessary to drive the single and double electron reactions are reached. This rise in current is not seen from the kinetic equations alone, which would otherwise lead to unrealistically high viologen conversion values for a minimal current.
Charge/energy data during the charge time is analyzed and resulting trends in the data as a function of applied cathodic voltage is discussed. A key result demonstrated in Fig. 8A is that the voltage range is divided into three regions: (1) before the initial EV redox peak of − 0.685 V, in which the model and experiment data closely align since insufficient current does not allow O2 to contribute significantly; (2) above the second peak region, at which mass transfer limitations come into play for the contribution of O2; (3) in between the two peaks which is the critical region in which desired EV+ is formed. This region also appears to be the most challenging to currently capture the behavior of the total current for. The fact that the model is overpredicting the experimental charge for most values suggests that an even lower O2 contribution may be assumed. Nevertheless, a contribution of below 0.5 mM total is considered a minimally invasive amount when compared with the value of CO2 or N2 when saturated into an aqueous solution at standard operating temperatures and pressures.

Comparison of modeling charge and energy input data with experiments for 10 mM EV charge carrier at different applied voltages. A Charge, B Energy. Experiments run and simulated for 1 h using CO2 purging
According to the model, a maximum charge value should occur at around − 0.7 V as the activation toward EV+ product occurs (consistent with the CV data), but this is not present in the experimental data. Additionally, a charge minimum is predicted by the model around − 0.8 V which would suggest a peak performance in efficiency (still within the region between the redox peak), which suggests that our initially tested voltage of − 0.75 V may not be the highest for optimized production of EV+. The energy results replicate the charge data, demonstrating that the model can effectively predict this transient data well at higher voltages (Fig. 8B).
While thermodynamic equilibrium data under applied voltage conditions were successfully replicated from the model, the charge/energy analysis also proved useful to replicate charge carrier efficiency. Except for the aforementioned interior region, the model was a good fit to the trend seen experimentally, and the fitting parameters found are comparable to those already found in existing literature. Improvements that could be made to the model include considerations about the products from side reactions such as oxygen evolution [2, 50] and oxygen reduction at the cathode due to outside air, which can increase input charge in reactor systems open to the atmosphere. In addition to more sophisticated modeling revisions, developing a more airtight reactor to mitigate molecular oxygen is crucial to ensuring charge carrier stability and improving efficiency. Finally, the model can be improved upon to include additional reactions to account for inefficiencies once their contributions are more clearly understood. To develop a deeper understanding of additional reaction products occurring, experiments should not only consider UV–Vis, but also other techniques, such as Raman spectroscopy or nuclear magnetic resonance (NMR), to analyze full product composition.
5 Conclusion
A transient-based equilibrium model is developed to predict changes in charge carrier product composition to (1) connect with existing experimental data, and (2) ensure a reasonable time scale at which convergence to thermodynamic equilibrium conditions is met. With all three charge carriers, appropriate selection of the model parameters enabled strong correlations to experimental data for current density and published work on concentration changes, through further experimental validation is needed to assess the exact formation of more complicated products. Under tested experimental operating conditions of − 0.75 V vs. Ag/AgCl and pH 6.3, MV and EV reached steady-state convergence within approximately 15 min. From both experimental and modeling data, an optimal voltage range is found in which charge and energy input are minimized due to activating the redox potential of the viologen (EV used for this analysis), but before the contribution of molecular side reactions becomes significant. This optimal performance is reached around − 0.8 to − 0.85 V vs. Ag/AgCl, which is about 15% additional voltage than the − 0.75 V which has typically been used in past studies involving viologens. The analysis developed in this study examines a viologen charge carrier shuttling can be tuned while minimizing their experimental use to the environmental toxicity. Designing charge carrier reduction experiments around such conditions predicted theoretically, rather than only at − 0.75 V vs. Ag/AgCl, may enable more rapid and reliable charge carrier production to assist in applications such as fuel cells, batteries, and CO2 conversion to value-added products.
References
Michaelis L, Hill ES (1933) The viologen indicators. J Gen Physiol 16(6):859
Bird C, Kuhn A (1981) Electrochemistry of the viologens. Chem Soc Rev 10(1):49–82
Bruinink J, Kregting C, Ponjee J (1977) Modified viologens with improved electrochemical properties for display applications. J Electrochem Soc 124(12):1854
Guo W, Duan Z, Mabayoje O, Chemelewski WD, Xiao P, Henkelman G et al (2016) Improved charge carrier transport of hydrogen-treated copper tungstate: photoelectrochemical and computational study. J Electrochem Soc 163(10):H970
Delaney GM, Bennetto HP, Mason JR, Roller SD, Stirling JL, Thurston CF (1984) Electron-transfer coupling in microbial fuel cells. 2. Performance of fuel cells containing selected microorganism—mediator—substrate combinations. J Chem Technol Biotechnol 34(1):13–27
Sund CJ, McMasters S, Crittenden SR, Harrell LE, Sumner JJ (2007) Effect of electron mediators on current generation and fermentation in a microbial fuel cell. Appl Microbiol Biotechnol 76(3):561–568
Wang Y-F, Tsujimura S, Cheng S-S, Kano K (2007) Self-excreted mediator from Escherichia coli K-12 for electron transfer to carbon electrodes. Appl Microbiol Biotechnol 76(6):1439–1446
Picioreanu C, van Loosdrecht MC, Curtis TP, Scott K (2010) Model based evaluation of the effect of pH and electrode geometry on microbial fuel cell performance. Bioelectrochemistry 78(1):8–24
Martinez CM, Alvarez LH (2018) Application of redox mediators in bioelectrochemical systems. Biotechnol Adv 36(5):1412–1423
Okano M, Iida T, Shinohara H, Kobayashi H, Mitamura T (1984) Water photolysis by a photoeleetrochemical cell using an immobilized chloroplasts-methyl viologen system. Agric Biol Chem 48(8):1977–1983
Hagfeldt A, Cappel UB, Boschloo G, Sun L, Kloo L, Pettersson H et al (2012) Dye-sensitized photoelectrochemical cells. Practical Handbook of Photovoltaics. Elsevier, Cambridge, pp 479–542
Huskinson B, Marshak MP, Suh C, Er S, Gerhardt MR, Galvin CJ et al (2014) A metal-free organic–inorganic aqueous flow battery. Nature 505(7482):195–198
Sathyamoorthi S, Kanagaraj M, Kathiresan M, Suryanarayanan V, Velayutham D (2016) Ethyl viologen dibromide as a novel dual redox shuttle for supercapacitors. J Mater Chem A 4(12):4562–4569
Gerhardt MR, Tong L, Gómez-Bombarelli R, Chen Q, Marshak MP, Galvin CJ et al (2017) Anthraquinone derivatives in aqueous flow batteries. Adv Energy Mater 7(8):1601488
Goulet M-A, Tong L, Pollack DA, Tabor DP, Odom SA, Aspuru-Guzik A, Kwan EE, Gordon RG, Aziz MJ (2019) Extending the lifetime of organic flow batteries via redox state management. J Am Chem Soc 141(20):8014–8019
Chen Q, Gerhardt MR, Hartle L, Aziz MJ (2015) A quinone-bromide flow battery with 1 W/cm2 power density. J Electrochem Soc 163(1):A5010
Lorger S, Usiskin R, Maier J (2019) Transport and charge carrier chemistry in lithium oxide. J Electrochem Soc 166(10):A2215
Amao Y (2018) Formate dehydrogenase for CO2 utilization and its application. J CO2 Util 26:623–641
Ikeyama S, Amao Y (2016) Novel artificial coenzyme based on the viologen derivative for CO2 reduction biocatalyst formate dehydrogenase. Chem Lett 45(11):1259–1261
Monk PM, Hodgkinson NM (1998) Charge-transfer complexes of the viologens: effects of complexation and the rate of electron transfer to methyl viologen. Electrochim Acta 43(3–4):245–255
Akbar S, Naqvi I, Mohammad M (2014) Electron transfer reactions associated with ethyl viologen-β-cyclodextrin complexation: equilibrium and kinetic aspects. J Iran Chem Soc 11(3):615–621
Heyrovský M (1987) The electroreduction of methyl viologen. J Chem Soc Chem Commun 24:1856–1857
Ding J, Zheng C, Wang L, Lu C, Zhang B, Chen Y et al (2019) Viologen-inspired functional materials: synthetic strategies and applications. J Mater Chem A 7(41):23337–23360
Landrum HL, Salmon RT, Hawkridge FM (1977) A surface-modified gold minigrid electrode which heterogeneously reduces spinach ferredoxin. J Am Chem Soc 99(9):3154–3158
Olufunsho A, da Silva JAT, Akintonwa A (2011) Toxicity testing of paraquat and 2, 4-dimethylamine on Lumbricus terristris, Palaemonetes africanus and Tilapia guineensis. Terr Aquat Env Toxicol 5(1):59–60
Dusinská M, Kovaciková Z, Vallová B, Collins A (1998) Responses of alveolar macrophages and epithelial type II cells to oxidative DNA damage caused by paraquat. Carcinogenesis 19(5):809–812
Krieger R (2001) Handbook of pesticide toxicology: principles and agents. Academic press, Cambridge
Pomeroy A (2012) Biochemical mechanisms of paraquat toxicity. Elsevier, Amsterdam
Naz L, Mohammad M (2017) Kinetics of dimerization of the reduced products of methyl viologen dication. J Chem Soc Pak 39(4):506–515
Carney TJ, Collins S, Moore JS, Brushett FR (2017) Concentration-dependent dimerization of anthraquinone disulfonic acid and its impact on charge storage capabilities. Meet Abstr.
Wiberg C, Carney TJ, Brushett F, Ahlberg E, Wang E (2019) Dimerization of 9, 10-anthraquinone-2, 7-disulfonic acid (AQDS). Electrochim Acta 317:478–485
Orgill JJ, Chen C, Schirmer CR, Anderson JL, Lewis RS (2015) Prediction of methyl viologen redox states for biological applications. Biochem Eng J 94:15–21
Kathiresan M, Ambrose B, Angulakshmi N, Mathew DE, Sujatha D, Stephan AM (2021) Viologens: a versatile organic molecule for energy storage applications. J Mater Chem A 9(48):27215–27233
Madasamy K, Velayutham D, Suryanarayanan V, Kathiresan M, Ho K-C (2019) Viologen-based electrochromic materials and devices. J Mater Chem C 7(16):4622–4637
Martínez-González E, Laguna HG, Sánchez-Castellanos M, Rozenel SS, Ugalde-Saldivar VM, Amador-Bedolla C (2020) Kinetic properties of aqueous organic redox flow battery anolytes using the Marcus-Hush theory. ACS Appl Energy Mater 3(9):8833–8841
Martinez-Gonzalez E, Flores-Leonar MM, Amador-Bedolla C, Ugalde-Saldivar VM (2021) Concentration effects on the first reduction process of methyl viologens and diquat redox flow battery electrolytes. ACS Appl Energy Mater 4(7):6624–6634
Wu W, Luo J, Wang F, Yuan B, Liu TL (2021) A self-trapping, bipolar viologen bromide electrolyte for redox flow batteries. ACS Energy Lett 6(8):2891–2897
Chen M, Liu L, Zhang P, Chen H (2021) A low-cost and high-loading viologen-based organic electrode for rechargeable lithium batteries. RSC Adv 11(39):24429–24435
Chidichimo G, Imbardelli D, De Simone BC, Barone P, Barberio M, Bonanno A et al (2010) Spectroscopic and kinetic investigation of ethyl viologen reduction in novel electrochromic plastic films. J Phys Chem C 114(39):16700–16705
Kwabi DG, Ji Y, Aziz MJ (2020) Electrolyte lifetime in aqueous organic redox flow batteries: a critical review. Chem Rev 120(14):6467–6489
Watt GD (2014) Kinetic evaluation of the viologen-catalyzed carbohydrate oxidation reaction for fuel cell application. Renew Energy 63:370–375
Bahari M, Malmberg MA, Brown DM, Nazari SH, Lewis RS, Watt GD et al (2020) Oxidation efficiency of glucose using viologen mediators for glucose fuel cell applications with non-precious anodes. Appl Energy 261:114382
Roos M, Batawi E, Harnisch U, Hocker T (2003) Efficient simulation of fuel cell stacks with the volume averaging method. J Power Sources 118(1–2):86–95
Gabitto J, Tsouris C (2015) Volume averaging study of the capacitive deionization process in homogeneous porous media. Transp Porous Media 109(1):61–80
Yamada H, Bandaru PR (2016) Electrochemical kinetics and dimensional considerations, at the nanoscale. AIP Adv 6(6):065325
Stetzel KD, Aldrich LL, Trimboli MS, Plett GL (2015) Electrochemical state and internal variables estimation using a reduced-order physics-based model of a lithium-ion cell and an extended Kalman filter. J Power Sources 278:490–505
Sharma AK, Ling C, Birgersson E, Vynnycky M, Han M (2015) Verified reduction of dimensionality for an all-vanadium redox flow battery model. J Power Sources 279:345–350
Morrison AR, van Beusekom V, Ramdin M, van den Broeke LJ, Vlugt TJ, de Jong W (2019) Modeling the electrochemical conversion of carbon dioxide to formic acid or formate at elevated pressures. J Electrochem Soc 166(4):E77–E86
Bhatti NK, Subhani MS, Khan AY, Qureshi R, Rahman A (2006) Heterogeneous electron transfer rate constants of viologen at a platinum disk electrode. Turk J Chem 29(6):659–668
Chen L, Lin C, Compton RG (2018) Single entity electrocatalysis: oxygen reduction mediated via methyl viologen doped Nafion nanoparticles. Phys Chem Chem Phys 20(23):15795–15806
Butt JB (2000) Reaction kinetics and reactor design. CRC Press, Boca Raton
Hoeger CD (2013) Foundational work in bioelectrochemical anaerobic reactor design with electron mediators
Zalc J, Löffler D (2002) Fuel processing for PEM fuel cells: transport and kinetic issues of system design. J Power Sources 111(1):58–64
Forster RJ, O’Kelly JP (2001) Protonation reactions of anthraquinone-2, 7-disulphonic acid in solution and within monolayers. J Electroanal Chem 498(1–2):127–135
Cook SK, Horrocks BR (2017) Heterogeneous electron-transfer rates for the reduction of viologen derivatives at platinum and bismuth electrodes in acetonitrile. ChemElectroChem 4(2):320
Wei Z, Shin W, Jiang H, Wu X, Stickle WF, Chen G et al (2019) Reversible intercalation of methyl viologen as a dicationic charge carrier in aqueous batteries. Nat Commun 10(1):1–10
Tichter T, Borah R, Nann T (2021) A theoretical framework for the electrochemical characterization of anisotropic micro-emulsions. ChemElectroChem 8(17):3397–3409
Yan J, Baird MA, Popple DC, Zettl A, Russell TP, Helms BA (2022) Structured-liquid batteries. J Am Chem Soc 144(9):3979–3988
Annibaldi V, Breslin CB (2019) Electrochemistry of viologens at polypyrrole doped with sulfonated β–cyclodextrin. J Electroanal Chem 832:399–407
Klein JM, Squire H, Dean W, Gurkan BE (2020) From salt in solution to solely ions: solvation of methyl viologen in deep eutectic solvents and ionic liquids. J Phys Chem B 124(29):6348–6357
Acknowledgements
The authors would like to thank the U.S. Department of Energy for primary financial support for this project (Grant No. DE-FE0031720). The authors would also like to thank Dr. Susan Odom (University of Kentucky) for providing additional insight and discussion on the charge carrier structures, and Dr. Chin Ng (University of Kentucky) for contributions to the modeling equations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moreno, D., Thompson, J., Omosebi, A. et al. Electrochemical analysis of charge mediator product composition through transient model and experimental validation. J Appl Electrochem 52, 1573–1584 (2022). https://doi.org/10.1007/s10800-022-01727-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-022-01727-5