
Abstract
Plasma electrolytic oxidation coating is obtained on AA6061 alloy using positive uni-polar pulsed DC in a sodium silicate-based electrolyte. The effect of process parameters such as solution concentration, process time, average current density, pulse frequency and positive on-time is investigated systematically and the corresponding voltage–time response is correlated with the coating growth rate. Surface morphology of the coatings is studied using scanning electron microscopy and the elemental distribution on the coating is investigated using energy dispersive X-ray spectroscopy. The concentration of sodium silicate in the solution is found to play a key role in determining the morphology and composition of the coating. X-ray diffraction studies indicate a transition from crystalline to amorphous nature of the coating with increase in silicate content of the electrolyte. Effect of pulse frequency on the voltage–time response and the corresponding coating growth rate is highly dependent on the solution concentration.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Plasma electrolytic oxidation (PEO) is a relatively novel electrochemical surface modification technique for obtaining thick oxide coatings on light metals such as aluminium, magnesium, titanium, etc [1,2,3,4,5,6]. PEO coatings, in general, exhibit superior mechanical properties such as high hardness and wear resistance as well as excellent corrosion resistance [7,8,9,10,11]. However, the properties of the coatings are highly dependent on the substrate, composition of electrolyte and the electrical regime used. Many electrolyte systems are reported for PEO process, of which the most commonly used are based on KOH/NaOH, silicate, phosphate or aluminate or other similar mild alkaline electrolytes or their combinations with several additives either to impart specific functional properties in the coating or to improve the coating growth process [12,13,14,15,16,17,18,19,20,21,22]. Among the various electrolytes, silicate based is reported to have better coating growth rate and are compatible with many additives [12,13,14,15,16,17,18,19,20,21,22,23,24].
In-spite of extensive research on PEO conducted on various metals and alloys, studies on AA 6061 are very limited. AA 6061 is one of the most commonly used alloys in space industry owing to its low density, high strength-to-weight ratio, high thermal conductivity and ease of fabrication [25]. To make its usage more versatile, suitable surface modifications are required to offer adequate protection against wear and corrosion and also to provide thermal or electrical insulation [26]. PEO coating on AA6061 is a promising technique to offer multifunctional properties to the substrate material. Further, its functional properties can be tuned with suitable modifications in the process parameters [27,28,29].
In order to fully utilize the capability of the PEO process for optimal functional properties, a systematic study of the influence of various parameters on the coating properties is required. The objective of the present work is to conduct systematic and in-depth study of PEO of AA 6061 alloy using a silicate-based electrolyte without any additives. The effects of electrolyte composition and various process parameters over a wide range and their interdependence on the coating growth rate and properties are investigated.
2 Experimental
Plasma electrolytic oxidation coating was developed on 4-mm-thick circular disc of AA 6061 samples with a diameter of about 5 cm. The samples were cleaned by ultrasonic degreasing in 2-propanol (isopropyl alcohol) at room temperature followed by rinsing with distilled water. A silicate-based electrolyte containing varying concentration of sodium silicate (20, 40, 60, 80 and 100 g l−1) was used for the process. A 25 kW PEO unit custom-built by Plasma Technology Ltd (Kowloon, Hong Kong) capable of providing bipolar or single positive or single negative pulsed DC output was used to produce the coatings. The positive terminal of the power supply was connected to the sample immersed in the electrolyte serving as anode and the negative terminal was connected to a stainless steel sheet immersed in the electrolyte acting as the cathode. PEO coatings were produced using positive uni-polar pulsed DC mode with a square waveform (a general schematic of which is shown in Supplementary Fig. 1).
The duty cycle is defined as
where Ton is the positive pulse on time and Toff is the positive pulse off time during a single cycle in a uni-polar pulsed DC output. In terms of frequency, duty cycle can be expressed as
Since frequency = 1/T, where T = Ton + Toff is the time period of a single pulse.
The effect of electrolyte composition and various process parameters such as process time, frequency, positive on-time, current density, etc on the coating growth and properties were studied in detail. The effect of process time in the different electrolytes was studied by conducting the process for 10, 20, 30, 40, 50 and 60 min at a constant average current density of 60 mA cm−2. Similarly, the influence of current density in the different electrolytes was elucidated by carrying out the process at different average current densities of 20, 40, 60, 80 and 100 mA cm−2 for 30 min at a frequency of 300 Hz and positive on-time of 500 µs. The influence of applied frequencies (50, 100, 300, 500 and 700 Hz), positive on-time (100, 300, 500 and 700 µs) and corresponding duty cycles (ranging between 2.5 and 49%) in the different electrolytes and their interdependence was studied systematically. The electrolyte temperature was maintained between 20 and 25 °C during the process using an air-cooled water chiller (Julabo). After completion of the process, the coating was immediately rinsed in running water and dried with hot air.
Voltage–time graph was plotted for every process by recording the output voltage at regular time interval. Coating thickness measurement was carried out with Fischer ISOSCOPE MP10E thickness metre (Germany) which works on the principle of eddy current. The reported thickness value is obtained by applying statistical average of 15–20 individual measurements taken on either surface of each sample to extract the mean data with minimum scatter. Surface morphology and elemental composition of the deposit were studied by scanning electron microscopy (Leica S 440 I, USA) equipped with energy dispersive X-ray spectroscopy facility (Oxford Instruments, INCA X-Max, UK). Since the coating is non-conducting, the test samples were sputter coated with gold prior to SEM imaging to minimize surface charging. SEM was operated at a voltage of 20 kV and a probe current of 100 pA was used. The detector used was secondary electron detector. EDX was operated at a voltage of 20 kV and a probe current of 890 pA was used. X-ray diffraction studies were carried out using a Philips X’pert-Pro instrument (PANalytical, The Netherlands) at 40 kV and 30 mA with Cu Kα radiation (λ = 1.5406 Å) in the range of 10°–100° and 0.008° step size.
3 Results and discussion
The various coating parameters such as concentration of sodium silicate, process time, average current density, frequency, positive on-time, etc are systematically varied and their influence on coating growth rate and properties are studied. The properties studied include coating thickness, surface morphology and microstructure, elemental and phase composition of coating, etc.
3.1 Effect of concentration of sodium silicate and process time
Samples were prepared using electrolytes containing varying amounts of sodium silicate (20, 40, 60, 80 and 100 g l−1) at a constant frequency of 300 Hz and a positive on-time of 500 µs (duty cycle = 15%). Corresponding to each concentration of electrolyte, different process times of 10, 20, 30, 40, 50 and 60 min were employed. Each process is carried out at a constant average current density of 60 mA cm−2.
Figure 1a shows the voltage–time response of sample processed in 40 g l−1 of sodium silicate solution for 60 min. The typical four-stage voltage–time response reported by many researchers [23, 24, 30] is clearly visible in the voltage–time graph where the first stage is marked by an abrupt linear increase in the voltage from around 288 V (at 15 s) to 415 V (at 1 min) within a short period of time, at the end of which dielectric breakdown of the oxide film occurs. After the dielectric breakdown, second stage commences which is marked by a decrease in the slope of the voltage–time curve. In the third stage, the rate of voltage increase becomes slightly faster compared to that in the second stage. The fourth stage is marked by a slight reduction in the rate of voltage increase and voltage fluctuations start appearing in the voltage time curve. These voltage fluctuations are reported to be due to high-intensity microdischarges originating from the metal–oxide interface which are termed as type B discharges by earlier researchers. Type A and type C discharges on the other hand are weaker than type B discharges and originate from the oxide–electrolyte interface [30,31,32]. Large and frequent voltage fluctuations are considered to be not suitable for coating growth as they are believed to be detrimental to the coating. Hence, by observing the voltage–time graph, it is possible to identify the onset of persistent voltage fluctuations for any process parameter under study and thereby choose the most suitable regime for PEO process where the detrimental type B discharges are absent. For a particular silicate concentration, the voltage–time response for various time intervals repeatedly followed similar pattern. (Supplementary Fig. 2 shows the voltage–time response of samples processed for various time intervals in a solution containing 40 g l−1 of sodium silicate and Supplementary Fig. 3 shows the difference in voltage for the various time intervals.)

Effect of sodium silicate concentration and process time on PEO process. a Voltage–time response of samples processed in 40 g l−1 of sodium silicate solution for 60 min, b voltage–time response of samples processed in electrolytes with different concentrations of sodium silicate (20, 40, 60, 80 and 100 g l−1) for 60 min, c coating thickness versus process time for different concentrations of sodium silicate (20, 40, 60, 80 and 100 g l−1) and d coating growth rate as a function of sodium silicate concentration
Figure 1b shows the voltage–time response of samples processed for 60 min using different concentrations of sodium silicate at a constant average current density of 60 mA cm−2. With increase in silicate concentration, a substantial reduction in the breakdown voltage is observed which is consistent with the results reported earlier by Liu et al. [12]. According to them this decrease in breakdown voltage is due to the increase in electrical conductivity of the solution with increased concentration of the electrolytes so that the energy required for arcing is reduced.
It has been observed that the voltage–time response shows a comparatively smooth curve up to a silicate concentration of 60 g l−1. However, when the concentration of sodium silicate is increased beyond 60 g l−1, lots of voltage fluctuations appear in the voltage–time graph which is characteristic of the strong high-intensity type B discharges typical of the fourth stage. The voltage fluctuations become persistent after about 14 min of process in the case of 80 g l−1 and after 6 min of process in the case of 100 g l−1 suggesting an early commencement of the fourth stage at higher silicate concentration.
Thickness of the coating versus process time is plotted for different concentrations of sodium silicate as shown in Fig. 1c. For any particular concentration of sodium silicate, the thickness of the coating exhibited a linear increase with respect to increasing process time although the rate at which it happened was different for different silicate concentrations. As the concentration of sodium silicate is increased, the coating growth rate initially increased up to 60 g l−1 and thereafter started decreasing and for 100 g l−1, the coating growth rate showed a drastic decrease. In order to understand the trend clearly, the coating growth rate (obtained as the values of slope of the linear fit of the respective thickness-process time plot) is plotted against the concentration of sodium silicate (Fig. 1d), where the drastic decrease in the coating growth rate beyond 60 g l−1 of sodium silicate is clearly visible. This may be attributed to the more intense type B discharges prevailing at higher silicate concentrations (as evident from the voltage–time plot) which are known to be detrimental to the coating.
3.2 Effect of average current density
Samples were prepared at different average current densities of 20, 40, 60, 80 and 100 mA cm−2 at constant pulse frequency of 300 Hz and positive on-time of 500 µs for 30 min in solutions containing 20, 40, 60, 80 and 100 g l−1 sodium silicate.
The voltage–time responses for 20 and 100 g l−1 solution at different average current densities are shown in Fig. 2a and b, respectively. Similarly, the voltage–time responses at average current densities of 20 and 100 mA cm−2 for different concentrations of sodium silicate are shown in Fig. 2c and d, respectively. For a given concentration of sodium silicate, as the average current density is increased from 20 to 100 mA cm−2, there is a substantial increase in the initial voltage (noted after 1 min of commencement of process); for example, from 104 to 513 V in the case of 20 g l−1 solution and from 78 to 328 V in the case of 100 g l−1 solution and also in the breakdown voltage. However, according to Liu et al. [12], for a particular concentration of electrolyte, there is no significant relationship between current density and breakdown voltage. A probable reason for this observation may be due to the fact that the range of current density studied by them is from 90 to 120 mA cm−2, where the variation of breakdown voltage was not markedly evident. On the other hand, Dehnavi et al. [24] studied the voltage–time response of samples coated at different current densities from 50 to 250 mA cm−2 and observed that on comparing the breakdown and maximum voltages achieved during the PEO of different samples, increasing the current density resulted in higher sparking and maximum voltages in each group of samples.

Voltage–time responses of samples processed at different average current densities (20, 40, 60, 80 and 100 mA cm−2) in solutions containing different concentrations of sodium silicate (20, 40, 60, 80 and 100 g l−1). a 20 g l−1 sodium silicate solution at different average current densities, b 100 g l−1 sodium silicate solution at different average current densities, c average current density of 20 mA cm−2 for different sodium silicate concentrations and d average current density of 100 mA cm−2 for different sodium silicate concentrations
Another interesting feature to be noted is that the duration of stage I of the process decreases steadily with increase in average current density and almost disappears when the average current density reaches the value of 100 mA cm−2. In the case of 100 g l−1 solution, even the stage II seems to disappear as the average current density reaches the value of 100 mA cm−2 which explains the early commencement of the third and fourth stage and appearance of the characteristic recurrent voltage fluctuations in the voltage–time graph. Stage I and II are, however, visible at lower current densities and the duration of the two stages increases as the average current density decreases. Moreover, the commencement of stage IV is delayed at lower current densities and hence at 20 mA cm−2 the commencement of stage IV is not observed within the time period under study. In general, the voltage–time graph at different average current density for a given solution concentration followed a similar pattern. A similar observation in terms of alteration in the duration and ratio of the different stages during PEO was reported by Dehnavi et al. [24]. However, complete disappearance of stage I or II was not reported probably due to the fact that the electrolyte used for the study consisted of only 2 g l−1 of sodium silicate and 2 g l−1 of potassium hydroxide.
On the other hand, when the voltage–time plots at a given current density for different solution concentrations are compared, it is observed that voltage–time response followed different trends for different solution concentrations. From Fig. 2c, it is evident that at a current density of 20 mA cm−2, the slope of stage II keeps on decreasing as the concentration of the solution increases and when the concentration of the solution becomes 100 g l−1, the voltage–time curve almost becomes parallel to the time axis indicating that for an appreciable time, the voltage remains almost constant. At higher current density of 100 mA cm−2, this trend is not visible as stage I and II are almost absent when the solution concentration is high as explained before. In general, it is understood that high-intensity type B discharges become prominent especially at higher concentrations and higher average current densities.
Figure 3a shows the coating thickness as a function of current density in electrolytes with different sodium silicate concentrations. It is evident from the plot that for any given concentration, the coating thickness increases with increase in average current density. However, the extent at which it increases depends on the solution concentration. For solution concentration of 20, 40 and 60 g l−1, the thickness of the coating increases linearly with increase in average current density, the slope of the linear curve increasing substantially with increase in concentration. For solution concentration of 80 g l−1, the coating thickness increases rapidly with increase in average current density up to 60 mA cm−2 and thereafter the slope of thickness versus current density plot decreases. Similarly, for solution concentration of 100 g l−1, the increase in coating thickness with increase in average current density is not very substantial. This observation can be attributed to the occurrence of high-intensity type B discharges at higher concentrations and higher average current densities as evident from the voltage–time response. This behaviour can be clearly understood when the coating thickness is plotted against concentration of sodium silicate at different average current densities as shown in Fig. 3b for 20 and 100 mA cm−2. Similarly, when the values of slope of the linear fit of the respective thickness-current density plot are plotted against the concentration of sodium silicate (Fig. 3c), the drastic decrease in the coating growth rate beyond 60 g l−1 of sodium silicate is clearly visible.

Effect of average current density on coating growth rate. a Coating thickness as a function of current density in electrolytes with different sodium silicate concentrations (20, 40, 60, 80 and 100 g l−1), b coating thickness as a function of concentration of sodium silicate at average current densities of 20 and 100 mA cm−2 and c slope of linear fit of thickness-current density plot versus concentration of sodium silicate
3.3 Effect of pulse frequency
Samples are processed at different pulse frequency of 50,100, 300, 500 and 700 Hz for 30 min using solutions containing sodium silicate of concentrations 20, 60 and 100 g l−1 at a constant average current density of 60 mA cm−2 and a positive on-time of 500 µs such that the corresponding duty cycle is 2.5, 5, 15, 25 and 35%, respectively.
Figure 4 shows the voltage–time response at different frequencies in electrolytes with different sodium silicate concentrations and the corresponding thickness of PEO coatings obtained. In general, it is observed that the voltage–time response at any particular frequency is dependent on the electrolyte concentration. For any given concentration, an increase in the applied frequency brings about a substantial reduction in the breakdown voltage and the final output voltage. In the case of 20 g l−1 solution, the output voltage at 50 Hz is so high that it crossed the limit of the power supply used (Max 700 V) and hence could not be processed. Even for a frequency of 100 Hz, the process had to be stopped at 25 min for the same reason. Hence, for a 20 g l−1 solution, higher applied frequency is better from the processing point of view. On the other hand, for 100 g l−1 solution, a lower applied frequency is beneficial since there are no major voltage fluctuations and corresponding coating growth rate is comparatively higher at low frequency. For 60 g l−1 solution, an intermediate frequency resulted in a smooth voltage–time response; higher frequency resulted in lots of voltage fluctuations and lower frequency resulted in high output voltage. Hence, in general, when studying the effect of applied pulse frequency, the solution concentration is an important parameter to be mentioned for completeness of observed data.

Effect of pulse frequency (50, 100, 300, 500 and 700 Hz) on PEO process carried out in electrolytes with different sodium silicate concentrations (20, 60 and 100 g l−1). a Voltage–time response at different frequencies in solution containing 100 g l−1 of sodium silicate, b voltage–time response at different frequencies in solution containing 60 g l−1 of sodium silicate, c voltage–time response at different frequencies in solution containing 20 g l−1 of sodium silicate and d thickness of PEO coatings obtained at different frequencies using electrolytes of different concentrations (20, 60 and 100 g l−1)
The coating thickness obtained in each case was in accordance with the voltage–time response (Fig. 4d). In the case of 20 and 60 g l−1 solution, there was no substantial variation in the coating thickness with variation in frequency. The smaller value of thickness of PEO coating in the case of 20 g l−1 solution at 100 Hz is due to the reduction in the process time from 30 to 25 min. However, for 100 g l−1 solution, the coating thickness at lower frequency was much higher may be due to the lesser B-type discharges as evident from the voltage–time response which are known to be detrimental to the coating. Hence, it is evident that solutions having higher concentration of sodium silicate (> 60 g l−1) can exhibit higher coating growth rate at lower applied frequency.
3.4 Effect of positive on-time
Samples are prepared using electrolyte containing 60 g l−1 of sodium silicate at a constant current density of 60 mA cm−2 and constant process time of 30 min. The positive on-time is maintained at 100, 300, 500 and 700 µs at different frequencies of 50,100, 300, 500 and 700 Hz.
Figure 5 shows the voltage–time response of the processes carried out at different positive on-time at different pulse frequencies. As a general trend, we can identify that at any given positive on-time, the output voltage decreases with increase in pulse frequency. Moreover, at higher frequencies, the stronger type B discharges become prominent especially at the later stages of the process. Since the power supply used for the present study is not capable of producing pulses of duty cycle < 2.5%, samples were not processed at frequency of 50 and 100 Hz at 100 µs and 50 Hz at 300 µs.

Voltage–time responses of PEO processes carried out at different positive on-time (100, 300, 500 and 700 µs) at different pulse frequencies (50, 100, 300, 500 and 700 Hz). a Positive on-time of 100 µs at different pulse frequencies, b positive on-time of 300 µs at different pulse frequencies, c positive on-time of 500 µs at different pulse frequencies and d positive on-time of 700 µs at different pulse frequencies
Even though the voltage–time response was different for different combinations of positive on-time and frequency, the coating thickness was found to be more or less similar around 40 ± 5 µm in every case.
3.5 Coating surface morphology and composition
In order to understand the effect of concentration of sodium silicate on the coating surface morphology, samples of same coating thickness obtained using different electrolytes at different process time are compared. Figure 6a–d are SEM images of samples of average coating thickness of 25 µm obtained using electrolytes containing sodium silicate of concentration 20, 40, 60 and 80 g l−1 with a corresponding process time of 60, 40, 20 and 10 min, respectively. In general, several literatures suggest the existence of two distinct regions in the SEM micrograph of PEO coating (30, [33,34,35,36]); a cratered region rich in aluminium formed by the rapid solidification of molten material ejected from the coating/substrate interface and a lighter nodular structure rich in Si suggesting the participation of silicate ions from the solution in the discharge process.

SEM images of samples of average coating thickness of 25 µm obtained using a 20 g l−1 solution for 60 min, b 40 g l−1 solution for 40 min, c 60 g l−1 solution for 20 min and d 80 g l−1 solution for 10 min
The SEM images of samples obtained at lower concentration of sodium silicate (say 20 and 40 g l−1) exhibit a similar morphology reported earlier, consisting of cratered and nodular regions distributed throughout the coating surface with the size of nodular region decreasing with increasing concentration of sodium silicate (Fig. 6a, b). However, as the concentration of sodium silicate is increased to 60 g l−1 and beyond, an altogether different morphology is observed wherein no distinct nodular structure is observed and the coating surface exhibited a more or less uniform morphology throughout (Fig. 6c, d). Moreover, the porosity of the coating also appeared to have increased with increase in sodium silicate concentration.
EDX analysis of the two distinct regions of the sample obtained using 20 g l−1 sodium silicate solution is shown in Fig. 7. Consistent with the earlier reports, the cratered region is aluminium rich and the nodular structure is silicon rich as shown in the respective spectra (Fig. 7a, b). Figure 7c shows the overall composition of the coating surface. As the concentration of the electrolyte increases, say at 60 g l−1, there is no distinct Si-rich or Al-rich regions consistent with observation from SEM images. Figure 8 shows the EDX analysis of different regions of the sample obtained using 60 g l−1 sodium silicate solution. Since there was no distinct cratered and nodular regions, EDX analysis was performed at a darker region and a lighter region. Throughout the coating surface, Si% is higher than Al% though their ratio is found to be different in different regions. Moreover, the Si/Al intensity ratio (wt%) in the coating is found to increase as the concentration of sodium silicate in the electrolyte increases (Supplementary Fig. 4). This can be attributed to the fact that as the silicate concentration increases, more silicate ions take part in the discharge events and get incorporated into the coating.

EDX analysis of sample obtained using 20 g l−1 sodium silicate solution a spectrum of cratered region, b spectrum of nodular structure, c overall spectrum

EDX analysis of sample obtained using 60 g l−1 sodium silicate solution a spectrum of darker region, b spectrum of lighter region, c overall spectrum
3.6 X-ray diffraction analysis
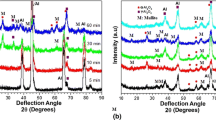
Figure 9 shows the X-ray diffraction pattern of samples of average coating thickness of ~ 25 µm prepared using electrolytes containing sodium silicate of concentration 20, 40, 60 and 80 g l−1 with a corresponding process time of 60, 40, 20 and 10 min, respectively. Coatings obtained using 20 and 40 g l−1 sodium silicate are found to be crystalline and shows the presence of aluminium oxide (Al2O3) and different mullite phases (Al5.65 Si0.35 O9.175, Al6 Si2 O13 and Al5 Si O9.5) along with the very intense peak of aluminium from the substrate. As the sodium silicate concentration is increased from 20 to 40 g l−1, the intensity of alumina phase decreased, whereas the intensity of mullite phase increased, which is consistent with the observation of increased Si/Al ratio in the EDX analysis.

X-ray diffraction pattern of samples of average coating thickness of 25 µm prepared using electrolytes containing sodium silicate of concentration 20, 40, 60 and 80 g l−1 with a corresponding process time of 60, 40, 20 and 10 min, respectively
When the concentration of sodium silicate is increased to 60 g l−1 and beyond, no peaks corresponding to the coating are obtained and only intense peaks of aluminium arising from the substrate are seen. This was initially thought to be due to the increased porosity in the coating (as observed in the SEM images). However, even when the thickness of the coating is increased to ~ 40 µm, XRD showed similar amorphous behaviour (Supplementary Fig. 5) for samples prepared using electrolytes with sodium silicate concentration of 60 g l−1 and more.
To further confirm the amorphous behaviour of the coating as discussed above, XRD of the samples is performed in a Bruker instrument equipped with glancing angle that restricts the X-rays only to the coating and not allowing to reach the substrate. A representative XRD with and without glancing angle of sample processed at 60 g l−1 sodium silicate is shown in Fig. 10. The diffraction pattern shows no evidence of peaks from the coating. It is therefore concluded that irrespective of coating thickness, coatings obtained using solution with sodium silicate concentration of 60 g l−1 and above in the range of parameters studied are X-ray amorphous, whereas those obtained at lower concentration of sodium silicate are crystalline in nature.

X-ray diffraction pattern of samples of average coating thickness of 40 µm obtained using instrument containing thin film attachment a without glancing angle, b with 5° glancing angle
This transition from crystalline to amorphous phase may be attributed to the fact that at higher silicate concentration, the temperature difference between the substrate and the solution is large due to intense microdischarges on the substrate. As a result, the ejected molten material undergoes rapid cooling when it comes in contact with the solution at much lower temperature. This rapid cooling enhances the formation of amorphous phase. This also explains the absence of distinct nodular and cratered regions in SEM images at higher silicate concentrations. The typical crater structure is formed when the molten material is allowed to solidify more slowly [24]. Moreover, since the solution contains higher concentration of silicate ions, incorporation of silicon in the coating as alumino-silicate phase is enhanced thereby reducing the concentration of alumina in the coating.
In general, the present study has undoubtedly established that the various process parameters are highly interdependent and the study of the effect of one of the parameters cannot be conducted independent of the others. Especially, the solution composition and concentration are strong deciding factors of the coating morphology and composition and the effect of other parameters on the coating growth behaviour and properties is highly dependent on the electrolyte used for the coating formation.
4 Conclusions
PEO coatings on AA6061 alloy are obtained using solution containing different concentrations of sodium silicate. The effect of various operating parameters such as average current density, process time, pulse frequency, positive on-time, etc is systematically investigated in the different solutions. The voltage–time response in each case is well correlated with the coating growth rate and resultant coating thickness. It is established that the various process parameters are highly interdependent and the solution composition plays a major role in determining the coating morphology, microstructure and composition.
-
1.
As the concentration of sodium silicate is increased, the coating growth rate initially increased up to 60 g l−1 and thereafter started decreasing and for 100 g l−1, the coating growth rate showed a drastic decrease most likely due to increased occurrence of the more intense microdischarges.
-
2.
At higher concentrations of sodium silicate, coating growth rate can be considerably improved if the process is carried out at low pulse frequency. On the other hand, a high pulse frequency is suitable for solutions having low concentration of sodium silicate since the output voltage can be brought down substantially and hence can extend the process time so as to obtain the required coating thickness.
-
3.
As the concentration of sodium silicate is increased, the coating microstructure shows a transition from crystalline to amorphous phase probably due to the rapid cooling rate of the ejected molten material. Accordingly, the morphology also shows a transition from a distinct crater and nodular structure to a more or less uniform morphology throughout. Moreover, the alumina content in the coating keeps on decreasing and the coating becomes almost composed completely of different phases of alumino-silicates (mullite).
References
Yerokhin AL, Nie X, Leyland A, Matthews A, Dowey SJ (1999) Plasma electrolysis for surface engineering. Surf Coat Technol 122:73–93
Walsh FC, Low CTJ, Wood RJK, Stevens KT, Archer J, Poeton AR, Ryder A (2009) Plasma electrolytic oxidation (PEO) for production of anodised coatings on lightweight metal (Al, Mg, Ti) alloys. Trans Inst Met Finish 87:122–135
Gupta P, Tenhundfeld G, Daigle EO, Ryabkov D (2007) Electrolytic plasma technology: science and engineering—an overview. Surf Coat Technol 201:8746–8760
Zhang Y, Fan W, Du HQ, Zhao YW (2017) Plasma electrolytic oxidation coatings for aluminum alloys. Mater Perform 56(9):38–41
Rizwan M, Alias R, Zaidi UZ, Mahmoodian R, Hamdi M (2018) Surface modification of valve metals using plasma electrolytic oxidation for antibacterial applications: a review. J Biomed Mater Res A 106(2):590–605
Matykina E, Arrabal R, Mohedano M, Mingo B, Gonzalez J, Pardo A, Merino MC (2017) Recent advances in energy efficient PEO processing of aluminium alloys. Trans Nonferrous Met Soc China 27(7):1439–1454
Tian J, Luo Z, Qi S, Sun X (2002) Structure and antiwear behavior of micro-arc oxidized coatings on aluminum alloy. Surf Coat Technol 154:1–7
Dearnley PA, Gummersbach J, Weiss H, Ogwu AA, Davies TJ (1999) The sliding wear resistance and frictional characteristics of surface modified aluminium alloys under extreme pressure. Wear 225–229:127–134
Voevodin AA, Yerokhin AL, Lyubimov VV, Donley MS, Zabinski JS (1996) Characterization of wear protective Al-Si-0 coatings formed on Al-based alloys by micro-arc discharge treatment. Surf Coat Technol 86–87:516–521
Wasekar NP, Jyothirmayi A, Krishna LR, Sundararajan G (2008) Effect of micro arc oxidation coatings on corrosion resistance of 6061-Al alloy. J Mater Eng Perform 17(5):708–713
Bajat JB, Vasili R, Stojadinovi S, Miškovi-Stankovi V (2013) Corrosion stability of oxide coatings formed by plasma electrolytic oxidation of aluminum: optimization of process time. Corrosion 69:693–702
Liu YJ, Xu JY, Lin W, Gao C, Zhang JC, Chen XH (2013) Effects of different electrolyte systems on the formation of micro-arc oxidation ceramic coatings of 6061 aluminum alloy. Rev Adv Mater Sci 33:126–130
Wang K, Koo B-H, Lee C-G, Kim Y-J, Lee S-H, Byon E (2009) Effects of electrolytes variation on formation of oxide layers of 6061 Al alloys by plasma electrolytic oxidation. Trans Nonferrous Met Soc China 19:866–870
Yerokhin AL, Voevodin AA, Lyubimov VV, Zabinski J, Donley M (1998) Plasma electrolytic fabrication of oxide ceramic surface layers for tribotechnical purposes on aluminium alloys. Surf Coat Technol 110:140–146
Gnedenkov SV, Khrisanfova OA, Zavidnaya AG, Sinebrukhov SL, Kovryanov AN, Scorobogatova TM, Gordienko PS (2000) Production of hard and heat-resistant coatings on aluminium using a plasma micro-discharge. Surf Coat Technol 123:24–28
Yerokhin AL, Nie X, Leyland A, Matthews A (2000) Characterisation of oxide films produced by plasma electrolytic oxidation of a Ti6Al4V alloy. Surf Coat Technol 130:195–206
Li H, Song R, Ji Z (2013) Effects of nano-additive TiO2 on performance of micro-arc oxidation coatings formed on 6063 aluminum alloy. Trans Nonferrous Met Soc China 23:406–411
Arrabal R, Matykina E, Viejo F, Skeldon P, Thompson GE, Merino MC (2008) AC plasma electrolytic oxidation of magnesium with zirconia nanoparticles. Appl Surf Sci 254:6937–6942
Stojadinovic S, Vasilic R, Belca I, Petkovic M, Kasalica B, Nedic Z, Zekovic Lj (2010) Characterization of the plasma electrolytic oxidation of aluminium in sodium tungstate. Corros Sci 52:3258–3265
Lukiyanchuk IV, Rudnev VS (2007) Tungsten oxide films on aluminum and titanium. Inorg Mater 43(3):264–267
Rudnev VS, Morozova VP, Lukiyanchuk IV, Adigamova MV, Tkachenko IA, Ustinov AYu, Kharitonskii PV, Frolov AM (2013) Oxide layers with ferro- and ferrimagnetic characteristics formed on aluminum via plasma electrolytic oxidation. Russ J Phys Chem A 87(6):1052–1056
Snizhko LO, Yerokhin AL, Pilkington A, Gurevina NL, Misnyankin DO, Leyland A, Matthews A (2004) Anodic processes in plasma electrolytic oxidation of aluminium in alkaline solutions. Electrochim Acta 49:2085–2095
Guan Y, Yuan X (2006) Correlation between discharging property and coatings microstructure during plasma electrolytic oxidation. Trans Nonferrous Met Soc China 16:1097–1102
Dehnavi V, Luan BL, Liu XY, Shoesmith DW, Rohani S (2015) Correlation between plasma electrolytic oxidation treatment stages and coating microstructure on aluminum under unipolar pulsed DC mode. Surf Coat Technol 269:91–99
Peters M, Leyens C (2009) Aerospace and space materials, materials science and engineering—vol. III. EOLSS Publications
Wernick S, Pinner R, Sheasby PG (1987) The surface treatment and finishing of aluminium and its alloys—vol 1, 5th edn. ASM International
Venugopal A, Srinath J, Krishna LR, Narayanan PR, Sharma SC, Venkitakrishnan PV (2016) Corrosion and nanomechanical behaviors of plasma electrolytic oxidation coated AA7020-T6 aluminum alloy. Mater Sci Eng A 660:39–46
Zhang Y, Fan W, Du HQ, Zhao YW (2018) Formation of corrosion-resistant MAO-treated aluminum alloy. Mater Perform 57(2):36–39
Ma D, Lu C, Fang Z, Yan W, Wei L, Ni Y, Xu Z (2016) Preparation of high absorbance and high emittance coatings on 6061 aluminum alloy with a pre-deposition method by plasma electrolytic oxidation. Appl Surf Sci 389:874–881
Dehnavi V, Surface modification of aluminum alloys by plasma electrolytic oxidation, Thesis from The University of Western Ontario, Canada, September 2014
Hussein RO, Nie X, Northwood DO, Yerokhin A, Matthews A (2010) Spectroscopic study of electrolytic plasma and discharging behaviour during the plasma electrolytic oxidation (PEO) process. J Phys D 43:105203 https://doi.org/10.1088/0022-3727/43/10/105203
Hussein RO, Northwood DO, Nie X (2010) Coating growth behavior during the plasma electrolytic oxidation process. J Vac Sci Technol A 28:766–773
Krishna LR, Purnima AS, Sundararajan G (2006) A comparative study of tribological behavior of microarc oxidation and hard-anodized coatings. Wear 261:1095–1101
Sundararajan G, Krishna LR (2003) Mechanisms underlying the formation of thick alumina coatings through the MAO coating technology. Surf Coat Technol 167:269–277
Jayaraj RK, Malarvizhi S, Balasubramanian V (2017) Optimizing the micro-arc oxidation (MAO) parameters to attain coatings with minimum porosity and maximum hardness on the friction stir welded AA6061 aluminium alloy welds. Def Technol 13(2):111–117
Sobolev A, Kossenko A, Zinigrad M, Borodianskiy K (2017) An investigation of oxide coating synthesized on an aluminum alloy by plasma electrolytic oxidation in molten salt. Appl Sci 7(9):889. https://doi.org/10.3390/app7090889
Acknowledgements
The authors wish to express their sincere gratitude to Prof. S. Sampath, Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore for his valuable guidance and technical assistance in conducting this research work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pillai, A.M., Rajendra, A. & Sharma, A.K. Influence of process parameters on growth behaviour and properties of coatings obtained by plasma electrolytic oxidation (PEO) on AA 6061. J Appl Electrochem 48, 543–557 (2018). https://doi.org/10.1007/s10800-018-1186-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1186-2