
Abstract
Purpose
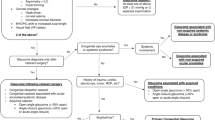
To correlate the features of certain types of infantile glaucoma with the progression and the prognosis of the disease, highlighting probable risk factors.
Methods
Seventy-six patients with pediatric glaucoma were recruited in this retrospective study. All patients underwent ophthalmological examination in the Department of Ophthalmology of the Saarland University Medical Center from January 2001 to December 2012. Our pediatric patients were classified into four different categories of glaucoma: (1) primary congenital glaucoma (presenting buphthalmus), (2) aniridia-related glaucoma, (3) Peters/Rieger’s anomaly-related glaucoma and (4) congenital cataract-related glaucoma. Personal data comprised age, sex, nationality, systemic diseases and gestational age. The best-corrected visual acuity (BCVA), the cup–disk ratio (CDR), the intraocular pressure (IOP), the corneal diameter and thickness, along with the Haab striae and corneal haze, were recorded.
Results
The majority of the children were male (58%) and suffered from aniridia-related glaucoma (38%). Children with aniridia exhibited the worst BCVA. The CDR and IOP were significantly higher in children with primary congenital glaucoma, compared to the other groups, at the first visit. Those children also were with the largest corneal diameter and prevalence of Haab striae compared to the rest groups, whereas corneal haze was found more often and was more pronounced in children with Peters/Rieger’s syndrome.
Conclusions
We concluded that glaucoma was earlier detected in children with primary congenital glaucoma, who exhibited increased corneal diameter and high percentage of Haab striae comparing to the other groups. However, these children responded successfully to any therapeutic intervention, exhibiting better BCVA and IOP values than the rest groups at the second visit.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glaucoma is the second leading cause of vision loss around the world. The percentage of blind children with glaucoma ranges from 1.2% in Great Britain, 3% in northern India to 7% in southern India [1,2,3]. In Toronto of Canada, congenital glaucoma was found to be the most common type (38% of all patients), followed by aphakic glaucoma (20%) and Sturge–Weber syndrome-associated glaucoma (10%) [4]. A recent population-based study in Olmstead County (MN, USA) estimated that the incidence of childhood glaucoma (including patients under 20 years old) was 2.29 out of 100,000 residents [5]. In Tongren Hospital in Beijing (Republic of China), 1.055 patients with pediatric glaucoma were seen from 2002 to 2008. Congenital glaucoma was the most common type seen in 46% of the patients, followed by traumatic (12%) and aphakic glaucoma (9%) [6].
Congenital glaucoma forms a heterogeneous group of diseases, leading to optic neuropathy and visual field changes. It can be categorized into primary, secondary, being related to other pathologies (cataract, aniridia, Peters’ anomaly, Axenfeld–Rieger anomaly, trauma or uveitis) and acquired subtypes [7,9,9]. The dysgenesis of trabecular meshwork seems to be responsible for the elevated resistance to aqueous humor outflow and the subsequent increase in intraocular pressure. This dysgenesis has been associated with the migration of the cells of the neural crest to the iridocorneal angle, during the embryonic and fetal development of the eye. The correct morphogenesis of trabecular pathways requires the differentiation of the cells to a porous and lamellate meshwork, as well as the ingrowth of Schlemm’s canal and the posterior movement of the iris root. The primary congenital or infantile glaucoma is usually sporadic, whereas hereditary cases are often caused by mutations in the CYP1B1 gene, which encodes for the enzyme cytochrome P450 1B1. Mutations in the latent transforming growth factor beta binding protein 2 (LTBP2) or in the transcription factor FOXC1 have been rarely implicated in primary congenital glaucoma [10]. The diagnosis of glaucoma is based on tonometry combined with corneal pachymetry, fundoscopy and gonioscopy, especially in cases of secondary glaucoma [7].
The aim of this study is to correlate the features of the pediatric glaucoma with the progression and the prognosis of the disease. This correlation could reveal some risk factors, which contribute to the development of pediatric glaucoma.
Patients and methods
Study design
This is retrospective study, regarding patients of Homburger visual school, who underwent ophthalmological examination in the Department of Ophthalmology of the Saarland University Medical Center during the period 2001–2012. The diagnosis of infantile glaucoma was made from 1992 to 2011, based on elevated intraocular pressure (over 21 mmHg), central corneal thickness and increased cup–disk ratio (CDR).
The purpose of this retrospective study was to evaluate visual acuity, ocular findings, including intraocular pressure and cup–disk ratio, as well as ocular and systemic comorbidities in children with various types of infantile glaucoma. These results could draw conclusions, capable of predicting the progression of the disease.
Patient population
The patients were classified into four groups according to the etiology/type of the glaucoma, as follows: the first group included patients with primary congenital glaucoma (buphthalmus), and the second group involved patients with aniridia-related glaucoma, whereas the third and fourth group recruited patients with Peters/Rieger’s anomaly-related and congenital cataract-related glaucoma, respectively. Only the affected eye was included in the study, except for children with bilateral involvement, where both eyes were included. Patients with glaucoma secondary to trauma or uveitis and patients presenting more than one subtypes of glaucoma were excluded from the study.
Data collection
Personal data comprised age, sex, nationality, systemic diseases and gestational age. The ocular history included questions regarding congenital cataract, Rieger and Peters’ anomaly, buphthalmus and aniridia. The slit lamp findings provided information about Haab striae and corneal opacity. Measurements of corneal diameter and thickness were also performed. Intraocular pressure (IOP) was calculated for the evaluation of glaucoma at presentation, and the type of measurement (Goldmann, iCare, etc.) was recorded. The best-corrected visual acuity (BCVA), objective refraction and the cup–disk ratio (CDR) were evaluated. BCVA, CDR and IOP were recorded twice, at first and last visit of the patients. All the ophthalmological examinations were conducted in the Department of Ophthalmology of Saarland University Medical Center (Homburg, Saar, Germany). The anti-glaucomatous medication and its modifications were also noted.
Statistical analysis
All data were entered into a Microsoft Excel sheet and were analyzed, using the statistical program IBM SPSS Statistics 22.0. Descriptive analysis of all parameters, including the age, the gender, the nationality, the prematurity, BCVA, CDR, IOP and the slit lamp findings (corneal diameter and thickness, Haab striae and corneal opacity) were first carried out. Nonparametric analysis Kolmogorov–Smirnov was used to check the distribution of the variables. Statistical hypothesis was performed to verify any statistical differences.
The possible differences in the prevalence of Haab striae and corneal haze among the four groups were estimated using the Chi-squared test. The nonparametric Wilcoxon matched-pairs signed-rank test was used to calculate the significance of differences in BCVA, CDR and IOP, between 1st and l2th (last visit) values, within each group. Independent samples t tests and Mann–Whitney tests were applied to identify the possible differences in BCVA, CDR and IOP among the four groups. Mann–Whitney test was used when neither Kolmogorov–Smirnov analysis nor Levene’s test for equality of variances indicated the normal distribution of the variables. A p value less than 0.05 was considered to indicate statistical significance.
Results
Demographics–personal data
Seventy-six patients were recruited in this study, including 13 children with primary congenital glaucoma (presenting buphthalmus), 29 with aniridia-related glaucoma, 19 children with Peters/Rieger’s anomaly-related glaucoma and 15 with congenital cataract-related glaucoma. Males (58%, 44) predominated over females (42%, 32). In fact, there were 7 males and 6 females in primary congenital glaucoma group, while the group of aniridia included 16 males and 13 females. Ten males and 9 females consisted in the group of Peters/Rieger’s syndrome, whereas the group of congenital cataract included 11 males and 4 females. The age of the patients at the time of diagnosis was 11, 117, 73 and 36 months in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively.
Germans outnumbered among participants, who also came from Albany, USA, France, Italy, Luxembourg, Poland and Turkey (Fig. 1). Nineteen (19%) children among participants were premature at birth time. The percentages of premature children were 15, 10, 32 and 21% in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively.

The distribution of nationalities among four groups
Slit lamp findings and visual acuity
BCVA at the first visit was 0.3, 0.2, 0.3 and 0.3 decimal in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively. The correspondent values at the second visit were recorded as follows: 0.5, 0.2, 0.2 and 0.4 decimals, respectively. The BCVA exhibited statistically significant increase between the two visits within primary congenital glaucoma group (Wilcoxon test, p = 0.021), whereas the changes were not significant within the other three groups (Wilcoxon test, aniridia group p = 0.269, Peters/Rieger’s syndrome group p = 0.211, congenital cataract group p = 0.833). Evaluating the differences in BCVA among the four groups, we noted that BCVA at the second visit was better in patients with primary congenital glaucoma comparing to aniridia and Peters/Rieger’s syndrome groups (Mann–Whitney test, p < 0.001 and p = 0.001, respectively). Furthermore, patients with aniridia exhibited lower BCVA at first and second visits, being compared to patients with congenital cataract (Mann–Whitney test, 1st visit p = 0.027, 2nd visit p = 0.002).
CDR at the first visit was 0.5, 0.2, 0.4 and 0.2 in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively. The correspondent values at the second visit were recorded as follows: 0.3, 0.3, 0.3 and 0.4 decimals. The increase in CDR observed at the second visit was significant in aniridia group (Wilcoxon test, p = 0.029), whereas no statistically significant changes were found in the CDR of the other groups. On the other hand, significant higher CDR was noted in primary congenital glaucoma group compared to aniridia one at the first visit (Mann–Whitney test, p = 0.013). Moreover, the alteration of CDR at the second visit was estimated to be significant both between the abovementioned groups (Mann–Whitney test, p = 0.030), as well as between primary congenital glaucoma and congenital cataract groups (Mann–Whitney test, p = 0.006). Similarly, significant difference was observed in the fluctuations of CDR between patients Peters/Rieger’s syndrome and congenital cataract (Mann–Whitney test, p = 0.043).
Corneal diameter was 12.6 ± 1.4, 10.4 ± 2.2, 10.5 ± 1.4 and 9.7 ± 1.7 mm in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively. Corneal thickness was as follows: 609.9 ± 52.8 μm in primary congenital glaucoma group, 669.1 ± 146.0 μm in aniridia group, 655.4 ± 148.5 μm in Peters/Rieger’s syndrome group and 584.0 ± 69.2 μm in congenital cataract group. The distribution of corneal diameter and thickness among participants for the right and left eye is presented in Figs. 2 and 3.

The distribution of corneal diameter (in mm) in the right and left eye among participants

The distribution of corneal thickness (in μm) in the right and left eye among participants
The differences in corneal diameter of both eyes exhibit statistical significant differences between primary congenital glaucoma and both Peters/Rieger’s syndrome (Mann–Whitney test, p = 0.004) and congenital cataract (Mann–Whitney test, p = 0.037) groups. Corneal diameter was found significant higher in patients with primary congenital glaucoma compared to those with aniridia, Peters/Rieger’s syndrome and congenital cataract (Mann–Whitney test, p = 0.001, p < 0.001 and p < 0.001, respectively). No statistically significant differences in corneal thickness were found among the four groups.
Seventy (92%) children appeared no Haab striae during the slit lamp examination. The distribution of Haab striae among the rest children was as follows: 3 (4%) children exhibited Haab striae at the right eye and 2 (3%) at the left one, and 1 child (1%) was found to have Haab striae at both eyes. No Haab striae were detected in aniridia and Peters/Rieger’s syndrome groups, while the percentages of Haab striae in the rest groups are depicted in Fig. 4. Corneal haze was found in 29% of children, including 3 (4%) children with corneal haze at the right eye, 2 (3%) with corneal haze at the left eye and 1 (1%) child exhibiting corneal haze at both eyes. The distribution of cornel haze among four groups is presented in Figs. 5 and 6.

The distribution of Haab striae in primary congenital glaucoma and congenital cataract groups

The distribution of corneal haze in primary congenital glaucoma and aniridia-related glaucoma groups

The distribution of corneal haze in Peters/Rieger’s syndrome and congenital cataract groups
The differences in prevalence of Haab striae between primary congenital glaucoma and both aniridia (p < 0.001) and Peters/Rieger’s syndrome (p = 0.004) groups were statistically significant, given that no Haab striae were detected in children with aniridia or Peters/Rieger’s anomaly. Significant differences in prevalence of Haab striae were also detected between children with primary congenital glaucoma and congenital cataract (p = 0.037). Statistically significant differences were also found in prevalence of corneal haze between Peters/Rieger’s syndrome and both primary congenital glaucoma (p = 0.013) and aniridia (p = 0.026) groups. Similarly significant differences in corneal haze were found between Peters/Rieger’s syndrome and congenital cataract group (p = 0.010).
IOP management
The patients’ IOP at the first visit was found to be 21.5, 19.2, 19.7 and 17.1 mm Hg in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively. The IOP at the last visit was 14.9, 18.5, 17.1 and 16.9 mmHg in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively. Although IOP was decreased in all groups by the time of the last visit, a statistically significant reduction was only noted in buphthalmus group (Wilcoxon test, p = 0.003). The IOP in primary congenital glaucoma and congenital cataract group exhibited statistically significant difference both at first and last visit (Mann–Whitney test, p = 0.040 and p = 0.013, respectively). The decrease in IOP, which was recorded at the second visit, was also differed significantly in these groups (Mann–Whitney test, p = 0.013). Significant differences in IOP were also found between primary congenital glaucoma and aniridia groups at the last visit of the patients Mann–Whitney test, p = 0.007 and p = 0.005, respectively).
Goniotomy, trabeculectomy, cyclophotocoagulation and Ahmed valve implantation were performed to reduce IOP. All patients with primary congenital glaucoma underwent anti-glaucomatous surgeries, including goniotomy in 1 patient, trabeculectomy in 7 patients, cyclophotocoagulation in 3 patients and Ahmed valve implantation in 2 patients. In aniridia group, goniotomy, trabeculectomy, cyclophotocoagulation and Ahmed valve implantation were performed in 1, 4, 15 and 5 patients, respectively. Only seven patients in Peters/Rieger’s syndrome group underwent surgical interventions, including trabeculectomy in 2 patients, cyclophotocoagulation in 3 patients and Ahmed valve implantation in 2 patients. Similarly, no goniotomy was carried out in congenital cataract group, where trabeculectomy, cyclophotocoagulation and Ahmed valve implantation were performed in 1, 7 and 3 patients, respectively. Details about surgical interventions in each group are displayed in Table 1.
Ten patients with buphthalmus received medical treatment at the presentation, whereas this number was declined in 4 patients at the latest follow-up. In aniridia group, 25 patients were under anti-glaucomatous drops at presentation, while 12 patients kept the medication at the latest follow-up. Seven patients with Peters/Rieger’s syndrome received anti-glaucomatous medication at presentation, but only 2 patients maintained the treatment at the latest follow-up. All patients with congenital cataract group were under anti-glaucomatous drops at presentation, but this number was reduced in 11 patients at the latest follow-up. The medical treatment used in presentation and at the latest follow-up in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups is presented in Tables 2, 3, 4 and 5.
Systemic diseases and chromosomal abnormalities
The systemic diseases and chromosomal abnormalities recorded in the four groups of children with glaucoma are presented in Table 6.
Primary congenital glaucoma (Buphthalmus)
Children recruited in this group had already glaucoma at the time of birth, or the disease was developed during the first months of their life, without exhibiting ocular comorbidities. A small percentage of these children suffered from specials syndromes (Stickler syndrome type 2 and Marc Hall syndrome, Sturge–Weber–Krabbe, pontocerebellar hypoplasia syndrome, type PCH3 with optic atrophy). Some patients suffered also from a systemic disease, including idiopathic juvenile arthritis, asthma and epilepsy.
Aniridia-related glaucoma
A group of children with congenital aniridia also suffered from genetically assured WAGR syndrome (Willms tumor, Aniridia, Genitourinary abnormalities, mental retardation). Several comorbidities were also detected, including microcephaly and hip dysplasia, heart failure, hypothyroidism and eczema, obesity, clubfoot, language impairment, Marinesco–Sjögren syndrome and Perthes disease, epilepsy, and oculocutaneous albinism.
Peter’s anomaly/Rieger syndrome-related glaucoma
Axenfeld–Rieger syndrome (maxillary hypoplasia, microdontia, hypodontia, congenital hip dislocation) coexisted with Peters-plus syndrome (short-limbed dwarfism, cleft lip/palate and learning difficulties) in several cases. The comorbidities which were observed included achondroplasia and seborrheic eczema, phokomelie and deformity of the face, hydrocephalus and mental retard, Haller–Streiff syndrome, facial asymmetry, cardiomyopathy with ventricular septal defect, atrioventricular block III with pacemaker, developmental delay and Balken dystrophy with cerebellar vermis atrophy (genetic investigation was initiated). Congenital cataract was also diagnosed in these children.
Congenital cataract-related glaucoma
This is a group of patients who underwent surgery for congenital cataract and experienced glaucoma symptoms in the later stages. The refractive errors were corrected with contact lenses, intraocular lenses or glasses. Other comorbidities included multiply disabled visual impaired (MDVI), ANC syndrome and clubfoot, Haller–Streiff syndrome, attention deficit hyperactivity disorder (ADHD), galactose epimerase deficiency, fructose malabsorption, obesity (without endocrine cause), Lowe syndrome, Trisomy 21, oculocutaneous albinism and galactosemia.
Multiple ocular abnormalities
Some children could not be clearly classified into a group due to coexisting malformations. In these cases aniridia was combined with congenital cataract or an anterior chamber disorder and cataract or buphthalmus. For statistical reasons, one primary diagnosis was chosen and these patients were classified into the above four groups with respect to their age and sex.
Discussion
There are few studies dealing with pediatric glaucoma, including usually a small number of children. In our study, the male gender predominated in the general population and in all subgroups of pediatric glaucoma. Although there are few studies about gender distribution in the literature so far, our results are in compliance with other studies. Tatham et al. [11] studied the incidence of glaucoma in children undergoing cataract surgery, recruiting 38 boys and 36 girls. Aponte et al. [5] investigated the incidence and clinical characteristics of childhood glaucoma, finally including 16 boys and 14 girls. Twice as many boys as girls participated in the study of MacKinnon, which assessed the efficacy of timolol in pediatric glaucoma [12]. 57% of patients with aniridia were men in the study of Eden [13]. No X-chromosomal glaucoma genes have been found to explain the numerical advantage of male gender in glaucoma patients.
We noted that glaucoma was detected early in children with primary congenital glaucoma (average age 11 months) compared to the rest groups with other ocular abnormalities (average age 117 months in aniridia group, 73 months in Peters/Rieger’s anomaly group and 36 months in congenital cataract group). Cramer et al. observed that the relative frequency of patients’ age, recorded in aniridia group at time of glaucoma diagnosis, was as follows: 15% from birth to 9 years, 15% 10–19 years, 15% 20–29 years, 15% 30–39 years, 35% 40–49 and 5% 50–59 years [14]. According to Bussières and colleagues, the mean age at the time of diagnosis of pediatric glaucoma diagnosis was 5.7 years old [15]. In our study the rate of premature births in the total population was 18.7%, reaching up to 32 and 21% in children with Peters/Rieger’s anomaly and congenital cataract, respectively. The prevalence of prematurity in primary congenital glaucoma and aniridia groups was estimated to be 15 and 10%, respectively. Similar reference in the literature has been made by Tonoki et al. [16]. Although 82% of affected children are born with glaucoma, control studies on premature infants could possibly eliminate the development of glaucoma.
Haab striae occur most frequently in children with congenital glaucoma, an observation which are confirmed by our study (38% in buphthalmus group). We also noted that 7% of children with congenital cataract had Haab striae, whereas the latter were not noted among the rest children. Mendes et al. noted that the mean corneal diameter was significantly higher in children with buphthalmus (11–15.5 mm, mean 14.13 mm, SD 1.28) compared to control group (11.5–12.5 mm, mean 12.01, SD 0.09). The same study group observed that the mean central corneal thickness was decreased in patients with congenital glaucoma and Haab striae (539 ± 46 µm) compared to glaucomatous children without Haab striae (571 ± 56 µm) and to healthy individuals (559 ± 28 µm). However, the differences in corneal thickness among the three groups were not statistically significant [17]. The corneal diameter of both eyes was 11 mm in our study. Moreover, children with primary congenital glaucoma had the largest values, accounting for 12.5 mm in the right and 12.75 mm in the left eye. On the other hand, children with congenital cataract had the smallest corneal diameter both in the right (9.5 mm) and left eye (9.63 mm). The values of corneal diameter in aniridia and Peters/Rieger’s anomaly groups were 11.5 and 10.5 mm for the right eye and 11.0 and 11.0 mm for the left eye, respectively.
Values of the central corneal thickness in the right and left eye were noted to be 605.5 and 621.5 nm, respectively. In primary congenital glaucoma (right eye: 599.0 nm and left eye: 642 nm) and cataract (right eye: 597.0 nm and left eye: 596.0 nm) groups, the corresponding values of corneal thickness were slightly lower. Another study revealed that patients with primary congenital glaucoma exhibited corneal thickness of 534 ± 72.3 μm (extreme values 430–610 μm) [18]. In this study, the thickest corneas were found in aniridia group (right eye: 631.5 μm, left eye 629.0 μm). Lopez et al. [19] reported that patients with aniridia had corneal thickness 639 μm, whereas Lee et al. [20] measured thicker corneas (by 100 μm) in similar groups. The Peters/Rieger’s anomaly group behaved like aniridia one, exhibiting slightly lower values (right eye: 588.5 µm, left eye 591.0 µm).
Corneal opacities appeared most frequently in children with Peters/Rieger’s anomaly (58%), followed by aniridia patients (24%), in our study. Furthermore, we noticed that in primary congenital glaucoma, the one eye was always affected (total 16%), while in the congenital cataract group, only 13% of the children had corneal opacities. Khan revealed that corneal opacities may also occur along with congenital glaucoma in children with anterior chamber anomalies. Nevertheless, these features are not specific for glaucoma [21]. Corneal haze was detected with variable prevalence in 67 patients with aniridia and buphthalmus from Saudi Arabia [22].
Systemic diseases were observed in all groups (Table 6). The relationship of Sturge–Weber syndrome with glaucoma has already been shown in the literature. It has been estimated that 30% of patients with Sturge–Weber syndrome are diagnosed with glaucoma; 60% of the latter develop the disease before the second year of life [23]. One case was only detected in our patient sample. Furthermore, one patient with Lowe syndrome was diagnosed. Usually, 50% of patients with Lowe syndrome suffer from glaucoma, which is either congenital or developed within the first year of life [24]. Glaucoma related to Haller–Streiff syndrome can be a consequence of dysgenesis of iridocorneal angle [25].
Patients with Stickler syndrome are usually introduced with the autosomal dominant type 1 at ophthalmologists, whereas Type 3 exhibits no abnormalities in the eyes. Types 1 and 2 have a high risk of developing cataract and glaucoma, along with retinal detachment in 50% of the cases [26]. 20% (14–27%) of patients with juvenile idiopathic arthritis also develop uveitic glaucoma subsequent to uveitis or steroid treatment [27, 28]. Pontocerebellar hypoplasia (PCH) type 3, in contrast to type 1 and 2, has been associated with ophthalmologic abnormalities, such as optic atrophy, glaucoma and megalocornea [29]. Only one case report of hydrocephalus associated with congenital glaucoma has mentioned in the literature [30]. In a Croatian study, recruiting 153 children with trisomy 21 (Down syndrome), the incidence of various ophthalmic diseases was determined as follows: 98.5% refractive errors, 1.3% cataracts and 1.9% glaucoma [31]. Although no further cases of phokomelia, achondroplasia and glaucoma have been described in the literature, we detected a mother and his son with aniridia. Galactosemia has been mentioned in the literature only in the context of Lowe syndrome. Fructose malabsorption has not been associated with glaucoma in the literature. Seborrheic and atopic eczema have been only referred as atopic reaction subsequently to anti-glaucoma medication [32].
In our study, although the highest values of IOP at the first visit were measured in children with primary congenital glaucoma, these values were significantly reduced only in this group at the second visit (14.9 from 21.5 mmHg, p = 0.003). Bussière et al. [15] supported that the mean IOP calculated in children with glaucoma at the time of diagnosis was 29.1 mmHg. Another study in Ethiopia estimated that the average initial IOP was 54 ± 2 mmHg and reduced by 43% after surgical intervention [33]. We found that the CDR at the first visit was 0.5, 0.2, 0.4 and 0.2 in primary congenital glaucoma, aniridia, Peters/Rieger’s syndrome and congenital cataract groups, respectively, whereas the correspondent values at the second visit were recorded as follows; 0.3, 0.3, 0.3 and 0.4 decimals. CDR in our study was 0.3 at both visits, whereas Bussière et al. [15] measured an average value of 0.5 at the time of diagnosis. Although the CDR at the first visit was significantly elevated in children with primary congenital glaucoma compared to the other groups, the subsequent decrease at the second visit was also significant in our study. The buphthalmus study group reported that the CDR was ranged between 0.3 and 0.8, exhibiting an average of 0.6 [34].
On the other hand, in our study children with aniridia exhibited the worst visual acuity (0.2 decimals at both visits) compared to primary congenital glaucoma (0.3 and 0.5 decimal at 1st and 2nd visits, respectively), Peters/Rieger’s syndrome (0.3 and 0.2 decimal at 1st and 2nd visits, respectively) and congenital cataract groups (0.3 and 0.4 decimal at 1st and 2nd visits, respectively). Evaluating the differences in BCVA among the four groups, we noted that BCVA measured at the second visit was better in patients with primary congenital glaucoma, being compared to aniridia and Peters/Rieger’s syndrome group (Mann–Whitney test, p < 0.001 and p = 0.001, respectively). The BCVA was 0.3 decimals at both visits in our study, whereas Beck estimated that 30–50% of children with secondary glaucomas have visual acuity of 20/50 or better [35]. In the study of Gramer et al. [14], 60% of the recruited children appeared a visual acuity of 20/100 or worse.
In this study we investigated the features of various types of pediatric glaucoma, looking for possible risk factors, which should worsen the course of the disease and lead to faster progression. Systemic diseases were found to be related to glaucoma development, although they were detected more often in children with aniridia. We observed that patients with glaucoma due to aniridia prevailed over the other groups, followed by children with Peters/Rieger’s syndrome and congenital cataract. The earliest diagnosis of glaucoma was observed in children with primary congenital glaucoma (11 months), whereas the disease was developed later in children with aniridia (117 months). Furthermore, the values of corneal diameter were found significant higher in patients with primary congenital glaucoma compared to aniridia, Peters/Rieger’s syndrome and congenital cataract groups, whereas no statistically significant differences in central corneal thickness were found among the four groups. Haab striae occurred most frequently in children with primary congenital glaucoma (38%) and only in 7% of children with congenital cataract, whereas this feature was not detected in the rest groups.
However, BCVA and IOP in children with primary congenital glaucoma were significantly improved after any therapeutic intervention (at the second visit), in contrast to the other three groups values. The CDR values were elevated at the second visit in children with aniridia. The highest percentage of premature children was noted in children with Peters/Rieger’s syndrome (32%), followed by children with congenital cataract (21%) and aniridia (10%). Corneal opacities presented in higher frequency among children with Peters/Rieger’s anomaly (58%), followed by aniridia (24%), primary congenital glaucoma (16%) and congenital cataract (13%) groups.
We concluded that glaucoma was detected earlier in children with primary congenital glaucoma, who exhibited increased corneal diameter and high percentage of Haab striae, compared to the other groups. Although these clinical features were more intense in children with primary congenital glaucoma, these children responded successfully to any therapeutic intervention. On the other hand, the absence of significant improvement in BCVA and IOP (at the last visit) in children with aniridia-related glaucoma, Peters/Rieger’s syndrome-related glaucoma and congenital cataract-related glaucoma indicates that these patients do not respond well in treatment.
References
Durnian JM, Cheeseman R, Kumar A, Raja V, Newman W, Chandna A (2010) Childhood sight impairment: a 10-year picture. Eye (Lond) 24(1):112–117
Bhattacharjee H, Das K, Borah RR, Guha K, Gogate P, Purukayastha S, Gilbert C (2008) Causes of childhood blindness in the northeastern states of India. Indian J Ophthalmol 56(6):495–499
Dorairaj SK, Bandrakalli P, Shetty C, Misquith D, Ritch R (2008) Childhood blindness in a rural population of southern India: prevalence and etiology. Ophthalmic Epidemiol 15(3):176–182
Taylor RH, Ainsworth JR, Evans AR, Levin AV (1993) The epidemiology of pediatric glaucoma: the Toronto experience. J AAPOS 3(5):308–315
Aponte EP, Diehl N, Mohney BG (2010) Incidence and clinical characteristics of childhood glaucoma: a population-based study. Arch Ophthalmol 128(4):478–482
Qiao CY, Wang LH, Tang X, Wang T, Yang DY, Wang NL (2009) Epidemiology of hospitalized pediatric glaucoma patients in Beijing Tongren Hospital. Chin Med J 122(10):1162–1166
Beck AD (2001) Diagnosis and Management of pediatric glaucoma. Pediatr Ophthalmol 14(3):501–512
Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT (2006) A review of anterior segment dysgeneses. Surv Ophthalmol 51(3):213–231
Moore W, Nischal KK (2007) Pharmacologic management of glaucoma in childhood. Paediatr Drugs 9(2):71–79
Tamm ER (2011) Development of the iridocorneal angle and congenital glaucoma Ophthalmologe 108(7):610-4, 616-7. (Review)
Tatham A, Odedra N, Tayebjee S, Anwar S, Woodruff G (2010) The incidence of glaucoma following paediatric cataract surgery: a 20-year retrospective study. Eye (Lond) 24(8):1366–1375
McMahon CD, Hetherington J Jr, Hoskins HD Jr, Shaffer RN (1981) Timolol and pediatric glaucomas. Ophthalmology 88(3):249–252
Edén U, Beijar C, Riise R, Tornqvist K (2008) Aniridia among children and teenagers in Sweden and Norway. Acta Ophthalmol 86(7):730–734
Gramer E, Reiter C, Gramer G (2012) Glaucoma and frequency of ocular and general diseases in 30 patients with aniridia: a clinical study. Eur J Ophthalmol 22(1):104–110
Bussières JF, Therrien R, Hamel P, Barret P, Prot-Labarthe S (2009) Retrospective cohort study of 163 pediatric glaucoma patients. Can J Ophthalmol 44(3):323–327
Tonoki H, Harada N, Shimokawa O, Yosozumi A, Monzaki K, Satoh K, Kosaki R, Sato A, Matsumoto N, Iizuka S (2011) Axenfeld-Rieger anomaly and Axenfeld-Rieger syndrome: clinical, molecular-cytogenetic, and DNA array analyses of three patients with chromosomal defects at 6p25. Am J Med Genet A 155A(12):2925–2932
Mendes MH, Sakata L, Betinjane AJ (2011) Central corneal thickness and its correlations with other ocular biometric data in patients with congenital glaucoma. Arq Bras Oftalmol 74(2):85–87
Jordão ML, Costa VP, Rodrigues Mde L, Paula JS (2013) Comparison of dynamic contour tonometry and Goldmann applanation tonometry in relation to central corneal thickness in primary congenital glaucoma. Graefes Arch Clin Exp Ophthalmol 251(1):117–121
Lopez JP, Freedman SF, Muir K, Duncan L, Stephens D, Atenafu E, Levin AV (2011) Central corneal thickness in children and adolescents with pediatric glaucoma and eye disorders at risk of developing glaucoma. J Pediatr Ophthalmol Strabismus 48(2):108–116
Lee H, Khan R, O’Keefe M (2008) Aniridia: current pathology and management. Acta Ophthalmol 86(7):708–715
Khan AO (2011) Conditions that can be mistaken as early childhood glaucoma. Ophthalmic Genet 32(3):129–137
Khan AO, Aldahmesh MA, Al-Abdi L, Mohamed JY, Hashem M, Al-Ghamdi I, Alkuraya FS (2011) Molecular characterization of newborn glaucoma including a distinct aniridic phenotype. Ophthalmic Genet 32(3):138–142
Brémond-Gignac D (2002) Sturge–Weber–Krabbe syndrome. J Fr Ophtalmol 25(6):655–656
Loi M (2006) Lowe syndrome. Orphanet J Rare Dis 1:16
Roulez FM, Schuil J, Meire FM (2008) Corneal opacities in the Hallermann–Streiff syndrome. Ophthalmic Genet 29(2):61–66
Snead MP, McNinch AM, Poulson AV, Bearcroft P, Silverman B, Gomersall P, Parfect V, Richards AJ (2011) Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (Lond) 25(11):1389–1400. https://doi.org/10.1038/eye.2011.201
Kotaniemi K, Sihto-Kauppi K (2007) Occurrence and management of ocular hypertension and secondary glaucoma in juvenile idiopathic arthritis-associated uveitis: an observational series of 104 patients. Clin Ophthalmol 1(4):455–459
Tejada P, Enríquez E, Del Valle JMR, Barceló A, Gutiérrez E, de Inocencio J (2012) Juvenile idiopathic arthritis-associated uveitis complicated by glaucoma and Brown’s syndrome. Rheumatology (Oxf) 51(9):1729–1730
Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, Petit F, Afenjar A, Goizet C, Barresi S, Coussement A, Ioos C, Lazaro L, Joriot S, Desguerre I, Lacombe D, des Portes V, Bertini E, Siffroi JP, de Villemeur TB, Rodriguez D (2012) Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis 7:18
Mandal AK, Hornby SJ, Jones RB (2002) Congenital hydrocephalus associated with congenital glaucoma and natal teeth. Indian J Ophthalmol 50(4):322–323
Karlica D, Skelin S, Culic V, Galetović D, Znaor L, Karlica H, Pavelić J (2011) The ophthalmic anomalies in children with Down syndrome in Split-Dalmatian County. Coll Antropol 35(4):1115–1118
Rojo-España R, Tomas-Mallebrera L, Gimeno-Clemente N, Marquina-Vila A, Morales-Suárez-Varela M (2011) Epidemiological study of periocular dermatitis in a specialised hospital department. Iran J Allergy Asthma Immunol 10(3):195–205
Ben-Zion I, Tomkins O, Moore DB, Helveston EM (2011) Surgical results in the management of advanced primary congenital glaucoma in a rural pediatric population. Ophthalmology 118(2):231–235
Tchabi S, Sounouvou I, Yehouessi L, Doutetien C, Bassabi SK (2007) Congenital glaucoma at CNHU in Cotonou: a review of 27 cases. Mali Med 22(4):14–17
Beck AD (2001) Diagnosis and management of pediatric glaucoma. Ophthalmol Clin N Am 14(3):501–512
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflict of interest to declare, and no financial support was offered for the present study.
Rights and permissions
About this article

Cite this article
Moschos, M.M., Nitoda, E., Fenzel, I. et al. Prognostic factors of pediatric glaucoma: a retrospective study. Int Ophthalmol 39, 359–373 (2019). https://doi.org/10.1007/s10792-018-0819-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-018-0819-0