
Abstract
Objectives
Chronic neuroinflammation has become one of the important causes of common neurodegeneration disease. Therefore, the target of this study was to explore the protective action of glabridin on lipopolysaccharide (LPS)-induced neuroinflammation in vivo and in vitro and its mechanism.
Methods
The neuroinflammation model was established by LPS-induced BV2 cells. The cell viability with various concentrations of glabridin was determined by MTT assay, and the content of NO in each group was detected. A neuroinflammatory model was established in male C57BL/6J mice for a water maze test. Subsequently, NF-κB and SOD indices were measured by ELISA, GFAP and IBA-1 indices were measured by immunofluorescence, and Nissl staining was used to explore the Nissl bodies in the hippocampus of mice.
Results
In vitro experiments, our results expressed that glabridin could markedly increase the cell activity of LPS-induced BV2 cells and reduce the NO expression in cells. It indicated that glabridin had a remarkable impact on the neuroinflammation of LPS-induced BV2 cell protection. In vivo neuroinflammation experiments, mice treated with different doses of glabridin showed significantly improved ability of memory compared with the LPS group in the Morris water maze test. The levels of NF-κB, GFAP, and the number of positive cells in Nissl staining were decreased. High-dose glabridin significantly increased the SOD content in the brain tissue and decreased the IBA-1 levels.
Conclusion
Glabridin can significantly reduce or even reverse LPS-induced neuroinflammation, which may be related to the fact that glabridin can reduce the NO expression, NF-κB, IBA-1, GFAP, and other inflammatory mediators, upregulate the expression of SOD to relieve oxidative stress of brain and inhibit the activation of gliocyte in brain tissue.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation, which is more common in the central nervous system (CNS), is a complicated response that encompasses multiple stages and is brought on by the loss of nerve cells in the brain. These responses can include autoimmune activity, trauma, infections, poisons, and processes that change the activity of neurons. (Carro et al. 2017; Tan et al. 2020). Studies have noted that neuroinflammation refers to the pathogenesis of Alzheimer's disease (AD), Parkinson's disease (PD), Depressive disorder (DD), and other common neurodegenerative diseases. The impairment of learning and memorial function and emotional dysfunction ascribed these neurodegenerative diseases have serious harm to patients and society. Neuroinflammation is caused mainly by active forms of microglia and astrocytes, both of which are attached to CNS immune glia with a dynamic biochemical cascade of inflammatory factors that alter the CNS microenvironment (Giovannoni and Quintana 2020; Kwon and Koh 2020; Silver et al. 2014).
People with Alzheimer's disease may have high levels of endotoxin lipopolysaccharide (LPS) in their brains, and this substance can cause neuroinflammation and cognitive decline. (Leng and Edison 2021; Liu et al. 2018; Zhan et al. 2018). The cholinergic drug galantamine is effective in mitigating the neuroinflammation and cognitive abilities decline induced by LPS (El-Saber Batiha et al. 2020; Li et al. 2016). Additionally, cannabinoids may mediate some of the effects of anti-inflammatory by altering immunocyte metabolism ability and inducing autophagy (van Niekerk et al. 2019), and can also reduce inflammation by silencing small-diameter neurons (McKenna and McDougall 2020; Rajesh et al. 2008). Although studies have demonstrated that non-steroidal anti-inflammatory drugs (NSAIDs) can remarkably mitigate the relative risk of dementia, long-term use of NSAIDs may lead to side effects such as gastrointestinal reactions and liver function damage (Bindu et al. 2020). Therefore, the treatment of neuroinflammation is still a very challenging task.
Traditional herbal remedies are potential alternative therapies for treating a wide range of diseases (Yang et al. 2017). Some natural products are considered to have less side effects and better effects in the treatment of neuroinflammation than synthetic drugs. Consequently, medicinal plants have become increasingly widely accepted (Rao et al. 2019). Licorice glabridin (1,3-Benzenediol-,4-(3,4-dihydro-8,8-dimethyl-2h,8h-benzo[1,2-b:3,4-b'] dipyran-3-yl)-,(R)-), a licorice flavonoid, is a chief active substance extracted from licorice. Glabridin has been used on food and diet and demonstrated significant anti-inflammatory and anti-atherosclerotic activities, estrogen-like effects, and the ability of energy metabolism regulation (Markina et al. 2022; Wang et al. 2015).
Numerous studies have demonstrated that glabridin has neuroprotective and other beneficial effects on degenerative neurological diseases (Karthikkeyan et al. 2021; Li et al. 2015; Simmler et al. 2013). Kang JS et al. found that Glabridin inhibited inducible nitric oxide synthase (iNOS) expression and NO production in RAW 264.7 cells and peritoneal macrophages, and in an animal model, glabridin attenuated LPS-induced septic shock, resulting in decreased amounts of NO and TNF-α in plasma (Kang et al. 2005). Jee Youn Kim et al. found that DCs as inflammatory target cells of glabridin, which blocked NF-κB and MAPK signalings to inhibit dendritic cell maturation, and inhibited the production of pro-inflammatory cytokines, including IL-12p40, TNF-α, and IL-1β (Kim et al. 2010). Yehuda et al. found that glabridin inhibited LPS-induced NO production, iNOS gene expression, and nitrotyrosine formation in macrophages, which exhibited potential antiinflammatory effects under hyperglycemic conditions (Yehuda et al. 2015). However, the bioavailability of glabridin is low, whether oral formulations can improve memory and the mechanism is still dimness. This study we used LPS-induced mouse microglial cell lines BV2 and C57BL/6J mice to construct in vitro and in vivo models of neuroinflammation to study the anti-neuroinflammatory effect and mechanism of different doses of glabridin. Our research results indicate the ability of glabridin to reduce neuroinflammation and contribute ideas to this mechanism of action, revealing the latent of glabridin as therapeutic substances for neuroinflammation.
Material and methods
Materials
Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), Penicillin–Streptomycin Solution, tryptase, phosphate-buffered saline (PBS) were purchased from Gibco (Carlsbad, CA, USA). Gamma-aminobutyric acid receptor (GABAAR) antibody (No. ab109364) Cyanine3 (CY3) (No. ab6939) and Fluorescein Isothiocyanate (FITC) (No. ab6717) was purchased from Abcam (Cambridge, UK). Methyl-thiazolyl-tetrazolium (MTT) was purchased from Beijing Solarbio Science and Technology Company (Beijing, China). Dimethyl sulfoxide (DMSO) was purchased from Damao (Tianjin, China). LPS was purchased from Thermo Fisher Scientific (Suzhou, China). T75 cell culture flasks and 96-well plates were purchased from NEST (Wuxi, China). Glabridin powder was purchased from Aladdin (Shanghai, China). Azeliragon (TTP488) was purchased from InvivoChem. A nitric oxide (NO) kit (No. S0021) was purchased from Beyotime (Shanghai, China). The ELISA kit was purchased from the Enzyme Label Biotechnology Company (Jiangsu, China). Adult (18–22 g) male C57BL/6J specific pathogen-free (SPF) mice were obtained from the Guangdong Medical Laboratory Animal Center from Sun Yat-Sen University (SCXK2011-0029, Guangzhou, China). The animal experimental protocol of this project adheres to the principles of animal protection, animal welfare and ethics, and all the test steps were in strict accordance with the Guidelines for the Care and Use of Laboratory Animals. (The 7th edition, USA). Tissue homogenizer was purchased from Tiangen Biotech Co., Ltd., Beijing, China.
Preparation of glabridin
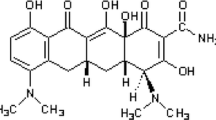
Glabridin (molecular weight 324.37) powders (C20H20O4, purity ≥ 99%) were dissolved into 10 mM stock solution with Sodium Carboxymethyl Cellulose (CMC), solution, filtered using a microporous membrane with 0.22 μm, and kept aseptically in a freezer at –20 °C (Fig. 1).

Chemical structure of glabridin (C20H20O4)
Cell culture
Complete medium was obtained by adding 1% penicillin–streptomycin and 10% FBS to DMEM. Cells were grown in a T75 culture flask, cells were added to the 12 mL of complete culture medium and cultured in a cell incubator (Unity Lab Services, USA) at a stable temperature of 37 °C in a 5% CO2 atmosphere. The nutrient solution was changed once a day.
The culture flask was observed under a microscope (Chongqing Optec Instrument Company, LTD), and BV2 cells were digested by 0.25% trypsin for counting after they were fully grown. Further, cells were added to 96-well plates with a density of 1 × 105 cells/well. 100 μg/ml LPS powder was dissolved in PBS (Nam et al. 2018; Wang et al. 2021), and 100 ng/mL DMEM was treated in each well. The cells were then reinserted into an incubator for an additional 24 h under the same conditions as previously stated.
Cell viability assay
Methyl-thiazolyl-tetrazolium (MTT) was used as a target of cell activity (Pascua-Maestro et al. 2018). Inoculated BV2 cells were placed in 96-well plates with a density of 5 × 103 cells in each well and classified into a blank group, a control group, and five administration groups for the cytotoxicity detection of glabridin at five concentrations. A 96-well plate was placed for 24 h in a constant-temperature incubator. Remove sterile glabridin from the 4 °C and dilute with DMEM to concentrations gradient of 0.05, 0.1, 0.2, 0.4 and 0.8 μM. After culturing for 24 h, the culture medium was taken away from each well, drugs at five different concentrations (100 μL) were added to each well according to the groups. The equal amount of DMEM was treated in the control group; cells were not cultured and no reagents were added to the blank group.
The 96-well plate was placed in a constant-temperature incubator for further culturing for 24 h. Subsequently the cell supernatant was removed and each well was added with 100 μL 0.5 mg/mL MTT in DMEM without FBS under dark conditions. After the cell culture plate was incubated for 4 h at 37 °C, then MTT solution was took away. The cell supernatant was removed, and 150 μL DMSO was treated to each well. After even shaking, each well of the absorbance was estimate at 570 nm using a microplate analyzer (Thermo Scientific Company, USA) (Zhang et al. 2019).
Measurement of nitric oxide production
The BV2 cells were cultured in 96-well plate (100 µl per well) at a concentration of 5 × 105 cells/ml and classified into a blank group, control group, LPS group, and four administration groups. The model and administration groups were treated with LPS, and the control group was cultured normally with DMEM. The 96-well plates were placed in a constant-temperature incubator for 24 h, and the medium was removed from the wells. After the sterile glabridin was diluted to 0.05, 0.1, 0.15 and 0.2 μM concentrations with DMEM, the solution was added to the four administration groups. DMEM was added to the model and control groups. The blank group did not plant cells, and no reagents were added. The plate was then put into an incubator for 24 h. The plates were removed from the incubator and, and 50 μL of the supernatant from each group was absorbed by a new 96-well plate and added to the corresponding position of the new plate. After culturing in an incubator for 10 min, the absorbancy of each group was measured by a microplate analyzer (Thermo Scientific Company, USA) at 562 nm.
Animals and experimental design
Twenty-five adult male C57BL/6J mice with body weight of 20 ± 2 and no specific pathogenic bacteria were separated into five groups according to mass and the free degree (E): (1) no treatment control group for 3 mice, (2) LPS group for 5 mice, (3) TTP488 group for 5 mice, (4) high-dose glabridin group for 5 mice, and (5) low-dose glabridin group for 5 mice. To maintain the possibility of the significance level result, the value of E is between 10 and 20, the formula is as follows:
E = Sum of experimental animals in each group− Number of groups
The mice were allowed free access to water and food and were adapted to the environment of 23 ~ 25 ℃ for one week before entering the experiment. The administration regimen is based on existing research (Bluthé et al. 1999; Kwon et al. 2008), the control group was intraperitoneally injected with saline and the LPS group was intraperitoneally injected with LPS at 250 μg/kg. The TTP488 group was administered 5 mg/kg TTP488 and LPS, the high-dose glabridin group received 100 mg/kg glabridin and LPS, and the low-dose glabridin group 50 mg/kg and LPS. Treatments were administered intragastrically for 14 days. Based on the weight data recorded on the mice before intragastric administration, the daily volume of intragastric administration was estimated, and the standard was 0.2 mL per 10 g (weight of mice). The experimental protocol was approved by the Experimental Animal Center of Guangdong Province (approval documents: SCXK/20130002), and the source of purchase is Guangdong Experimental Animal Center.
Morris water maze test
The Morris water maze test experiment lasted for seven days and included a learning and memory stage (D1-D5), directional navigation stage (D6), and spatial exploration stage (D7). We conducted the experiment as previously described (Fotakis and Timbrell 2006). Briefly, mice during the learning and memory phases were lay in quadrants of the water maze test system, and the water escape time was recorded as the incubation period. For memory training, the mice were kept on a platform for 20 s. During the directional navigation phase, the mice were placed in the quadrant farthest from the platform, and both the incubation period and time to escape on the platform reflected their memory ability. In the spatial exploration phase, the survival platform on D7 was taken away. The mice were laid up in the same quadrant of water, and over the next 60 s, the repetition of passing through the former platform area and their movement tracks were recorded (Dinel et al. 2020; Vorhees and Williams 2006).
Immunofluorescence assays
After the water maze experiment, the blood of the mice was collected by enucleation. Then the mice were killed by cervical vertebrae removal. Blood samples were coagulated at room temperature for 30 min and centrifuged at 2000 rpm for 10 min to obtain serum, which was stored at 4 ℃. The brain tissues of the mice were collected and the samples of the hippocampus were removed. Part of the whole brain and hippocampus of mice were fixed with 4% PFA tissue fixation solution, and then 4 µm thick paraffin sections were cut into. Sections were immersed in xylene I for 20 min and xylene II for 20 min for dewaxing and rehydration. Put slices into the repair box full of citrate antigen retrieval solution (PH6.0), heat it in a microwave for 8 min to boil, then paused heating for 8 min to hold it warm and then turn it to a medium–low temperature for 7 min. After natural cooling at room temperature, place the slide into PBS (PH7.4), shake then wash it by using the decolorization shaker (3 times, 5 min each time). The tissue of mice was incubated at 4 ℃ overnight with GABAAR antibody diluted at 1:60 after staining. After douching with phosphate-buffered saline (PBS) for 3 times, sections were incubated at 25 ℃ for 1 h with a secondary antibody diluted at 1:800 and protected from light. IgG (H + L) (CY3 conjugated Goat Anti-Rabbit IgG (H + L)) and IgG (H + L) [FITC conjugated Goat Anti-Rabbit IgG (H + L)] were added to stain for 10 min and then rinsed three times with PBS.
Nissl standing
Brain tissue samples were washed with the PBS (twice, 5–10 min), paraffin embedded, prepared 5 μM thick sections, centrifuged at 12000 rpm for 10 min. The tissues were immersed completely in xylene I for 20 min and xylene II for 20 min for dewaxing, heated to 60 ℃ and stained with 1% toluidine blue for 40 min. After rinsing with distilled water, dehydrate with graded ethanol (70, 95, 100%). The sections were then washed with xylene for 5 min. Tissue sections were placed in a repair box full of citrate antigen retrieval solution (PH 6.0) in a microwave for antigen repair, with a medium heat of 8 min to boiling, a hold fire of 8 min followed by a medium–low temperature of 7 min. After naturally lowering the temperature, the slides were situated in PBS (PH 7.4) and washed three times with a decolorizing shaking bed for 5 min each time.
We focused on the neural cone cells found in the cortex and hippocampus to assess neuronal damage. After staining these cells, we used an inverted fluorescent microscope to note the staining depth, cell membrane integrity, and nuclear clarity. Images of normal neurons were collected and observed under a microscope (magnification × 400). Three non-overlapping and intact areas were irregularly selected for each tissue section. Reckoned the number of normal neurons and took their average value.
ELISA assay
The remaining mouse brain tissue was collected, and nine times the weight of the protein lysate was added to make the brain homogenate by RIPA buffer. After centrifugation at 5000 rpm for 3 min, the supernatant was collected and stored at – 20 ℃ in a freezer. Manufacturer instructions were followed to determine total protein content by using the Beyotime BCA Protein Concentration Assay Kit. Afterward, we ensured that the protein contents of all samples were diluted with RIPA buffer to the same level. We determined superoxide dismutase (SOD) and NF-κB levels using an HRP-ELISA kit (EnzymeLabel Biotechnology Company, Jiangsu) following the company’s protocol.
Statistical analysis
One-way analysis of variance (ANOVA) was followed by Dennett’s multiple comparison test. Zen (Carl Zeiss AG, Oberkochen, Baden-Württemberg, Germany) was used to quantify the dynamic change process of phenotypic parameters of microglia and astrocytes and add scales. GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) was used to compare data. Image-J (National Institutes of Health, Bethesda, MD, USA) was used to measure the number of stained positive cells and calculate the integral optical density of the slice. Data are shown as mean ± standard error. p-values < 0.05 were considered to have statistical significance.
Results
Assessing glabridin cell viability
Drug cytotoxicity testing can reflect whether the drug itself has toxic effects on cells and the strength of these effects, which was convenient to eliminate interference and analyze the effects of drugs on LPS models. The results of Fig. 2A showed that 0.2 μM glabridin had obvious toxicity to cells and the IC50 value of glabridine was 0.399 μM.

Effects of glabridin in the BV2 cell viability and nitric oxide production. A MTT assay was used to evaluate the cell viability of glabridin at different concentrations. B NO assay was used to evaluate the production of nitric oxide in the treatment group (*p < 0.05 and **p < 0.01)
NO is a pro-inflammatory signaling molecule in the brain system that is regulated by BV2 cells and is released in great quantities during neuroinflammation. As such, it is a crucial marker for determining the extent of neuroinflammation. The NO content in the LPS group was significantly higher than that in the control group (p < 0.05) and demonstrating the modeling was effective. As shown in Fig. 2B, all four doses of glabridin significantly inhibited NO activity when compared with the LPS group. In the selected concentration range, the inhibitory effect of NO activity was proportional to the concentration of glabridin. These results indicated that glabridin had a good protective effect on LPS-induced neuroinflammation in the selected concentration range. When the concentration of glabridin was 0.2 μM, the NO content was reduced to close to the control group. However, due to the significant toxicity of the 0.2 μM concentration of glabridin to cells, it was not considered as the concentration in this study.
Water Maze experiment
The movement trajectory of LPS mice in the directed cruise stage was very confusing, whereas TTP488 and glabridin-treated mice had a shorter path to the platform (Fig. 3A). In addition, the time required for TTP488 and glabridin group to reach the platform was significantly shortened by fivefold and 2.5-fold, respectively, compared with LPS group (Fig. 3C) (p < 0.01). During the space exploration stage, high-dose glabridin-treated mice took most of their routes through the target quadrant (Fig. 3B). Mice that spent a long time in the target quadrant or crossed the removed platform multiple times had greater memory abilities. The mice in the TTP488 and high-dose groups took longer time (3.5-fold) to pass the removed target platform than the mice in the LPS group (Fig. 3D) (p < 0.05), while the glabridine group took longer time (2.4-fold) to stay in the target quadrant (Fig. 3E) (p < 0.05). These results indicate that LPS had a significant effect on the memory ability of mice. In this model, the TTP488 group performed similarly to the control group when contrasted to the LPS group (p < 0.01), indicating that the TTP488 almost completely prevented LPS-induced memory impairment. There was no obvious difference between the data of high-dose and low-dose groups, but the mice in both groups were obviously faster than those in the LPS treatment control (p < 0.05).

Effects of glabridin on LPS-induced memory deficits and related brain functions. A Motion trails of mice in the directional cruise stage. B Motion trails of mice in the spatial exploration stage. C The escape incubation period in the directional navigation stage. D The number of times the mice passed through the former area of platform in the spatial exploration stage. E The percentage of time the mice spent searching for the removed platform in the spatial exploration stage. (*p < 0.05 and **p < 0.01)
Effect of glabridin on reducing NF-κB content and protecting nerve cells from oxidative stress
NF-κB is a fundamental cellular transcription factor that mediates the inflammatory response. It can activate the transcriptional function of genes and specifically bind to inflammatory factor genes. Therefore, NF-κB is a crucial index that reflects the extent of inflammation, and the severity of inflammation is proportional to the level of NF-κB. In contrast to the control group, NF-κB activity in the LPS group was obviously increased by 1.13-fold (p < 0.05). Contrast with that in the LPS group, the activity of NF-κB in the TTP488 control and high-dose glabridin groups was decreased by 1.14-fold obviously (p < 0.05), which showed a similar decline range (Fig. 4A). These results indicated that the inhibitory effect of glabridin at these two concentrations on NF-κB secretion was similar to that of the TTP488.

Effect of glabridin on NF-κB and SOD production induced by LPS. The levels of NF-κB (A) and SOD (B) in brain homogenates were examined by the ELISA assay. (*p < 0.05)
Neuroinflammation may lead to oxidative stress injury in nerve cells, and SOD can effectively protect nerve cells from oxidative stress injury during inflammation. Therefore, the SOD content can reflect the effectiveness of neuroinflammation in protecting nerve cells (Wang et al. 2020). Within a certain range, the protective effect of neuroinflammation was proportional to the SOD content. In Fig. 4B, the SOD content in the brain tissue of the LPS group was significantly decreased by 1.25-fold (p < 0.05) compared to the control group. The SOD content significantly increased in the TTP488 and high-dose glabridin groups compared to the LPS group, by 1.15 and 1.28 times (p < 0.05), respectively. The positive effects of glabridine and TTP488 were similar, indicating that glabridine can achieve a good protective effect on nerve cells in neuroinflammation.
Glabridin restored the number of Nissl bodies in nerve cells
There are many Nissl bodies in the body of neurons, which are composed of basophilic clumps and particles. They are mainly used for protein synthesis in neurons and analyzing their number can reflect the functional activity of nerve cells. In the LPS group, the number of Nissl bodies was decreased, the color depth was shallow, the cell membrane was porous, the nucleus was poorly defined, and the conical cells were sparsely arranged. However, opposite results were observed in the control group. Following the application of glabridin and TTP488, the staining became more intense, the organization became more pronounced, and the nucleus was obviously visible (Fig. 5A). A certain amount of the nerve cells were recovered (Fig. 5B). Among these, the TTP488 group and the high-dose glabridin group increased by 1.29 and 1.26 times, respectively, in comparison to the LPS group, which was comparable to the control group (p < 0.05). The implication of these results is that glabridin and TTP488s could reduce the degree of neuronal damage by inhibiting neuroinflammation.

Effects of glabridin on LPS-induced neuronal injury (A) Nissl staining images of the dentate gyrus (DG) and glutamine 1 (CA1) in the hippocampus, and cortex of all treatment groups. (B) Normal neuronal counts in the mouse cortex and hippocampus. (*p < 0.05)
Glabridin plays an anti-inflammatory role by inhibiting the activation of astrocytes and microglia
Fluorescein was labeled on the corresponding antibody. The antibody was directly bound to the antigen and the content of the detected index was reflected by the fluorescence intensity. The indices of glial fibrillary acidic protein (GFAP) and Ionized calcium-binding adapter molecule 1 (IBA-1) were detected. The elevated levels of both factors are markers of microglia and astrocytes activation (Lafrenaye et al. 2020). The darker the CY3 (IBA-1) or FITC (GFAP), the more severe the inflammation (Norden et al. 2016) The fluorescence effect of high-dose glabridin group was similar to that of the TTP488 group, and the fluorescence intensity and quantity were decreased compared with LPS group (Fig. 6A). Figure 6B and C demonstrate that the LPS group had higher protein expressions of IBA-1 (0.7-fold) and GFAP (1.5-fold) than the control group (p < 0.05). Conversely, the high-dose glabridin group had significantly lower protein expressions of IBA-1 (1.7-fold) and GFAP (1.4-fold) (p < 0.05). It is suggested that brain inflammation was more serious in the LPS group and glabridin may play an anti-inflammatory role by the inhibition of activated astrocytes and microglia, indicating that the TTP488 drug and high- and low-dose glabridin groups could effectively reverse neuroinflammation.

LPS-induced inflammatory reaction in the cerebral cortex and hippocampus A Immunofluorescent cell images of microglia (IBA-1) and astrocytes (GFAP) in all experimental groups were stained with fluorophore-linked antibodies to detect the expression of DG and CA1 in the hippocampus of mice. B The quantified measurement of GFAP in the immunofluorescence assay. C The quantified measurement of IBA-1 in the immunofluorescence assay. (*p < 0.05 and **p < 0.01)
Discussion
The anti-inflammatory and neuroprotective effects of glabridin have long been verified (Liu et al. 2017), but the protective effectiveness and mechanism of glabridin on neuroinflammation are not comprehensive (Simmler et al. 2013). In this study, LPS was used to induce neuroinflammatory response in BV2 cells. To investigate the potential mechanism underlying the efficient protective action of varying doses of glabridine on BV2 cells, an intraperitoneal injection model of learning and memory impairment was created. Results showed that the production of NO was effectually inhibited and the surviving rate of BV2 cells under the influence of LPS was increased by glabridin. In mice with learning and memory impairment, glabridin also significantly reversed LPS-induced memory impairment and restored memory ability (Cui et al. 2008). Simultaneously, the changes in SOD and NF-κB content in mouse brain indicated that the protective mechanism of glabridine on nerve cells may be related to regulating SOD content, alleviating oxidative stress damage or regulating NF-κB and other inflammatory factors to restore brain microenvironment (Griciuc et al. 2013). The fluorescence intensity of DG and CA1 areas was reduced and the levels of IBA-1 and GFAP were decreased after glabridin treatment, suggesting that glabridin could effectively reverse neuroinflammation by inhibiting activated astrocytes and microglia.
The absorption of glabridin in the human body will combine with certain proteins in the stomach and loses its original effect, resulting in very low bioavailability. Regarding the reports on glabridin, its safety has covered a wide range of doses. These results only discussed its therapeutic effect on neuroinflammation at a certain safe dose, but further investigation about the specific action process of glabridin after entering the human body needs to be clarified.
In summary, glabridin has a protective effect on LPS-induced neuroinflammation and has great potential in the development of therapeutic drugs for neuroinflammation (Park et al. 2010). However, the target sites and molecular mechanisms of the protective effect of glabridin on neuroinflammation still need to be further investigated.
Abbreviations
- LPS:
-
Lipopolysaccharide
- MTT:
-
Methyl-thiazolyl-tetrazolium
- NO:
-
Nitric oxide
- NF-κB:
-
Nuclear factor kappa-B
- SOD:
-
Superoxide dismutase
- ELISA:
-
Enzyme-linked immunosorbent assay
- TTP488:
-
Azeliragon
- GFAP:
-
Glial fibrillary acidic protein
- IBA-1:
-
Ionized calcium-binding adapter molecule 1
- DG:
-
Dentate gyrus
- CA1:
-
Glutamine 1
- CY3:
-
Cyanine3
- FITC:
-
Fluorescein Isothiocyanate
References
Bindu S, Mazumder S, Bandyopadhyay U (2020) Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol 180:114147
Bluthé R-M, Castanon N, Pousset F, Bristow A, Ball C, Lestage J, Dantzer R (1999) Central injection of IL-10 antagonizes the behavioural effects of lipopolysaccharide in rats. Psychoneuroendocrinology 24(3):301–311
Carro E, Bartolome F, Bermejo-Pareja F, Villarejo-Galende A, Molina JA, Ortiz P, Orive G (2017) Early diagnosis of mild cognitive impairment and Alzheimer’s disease based on salivary lactoferrin. Alzheimers Dement (amst) 8:131–138. https://doi.org/10.1016/j.dadm.2017.04.002
Cui YM, Ao MZ, Li W, Yu LJ (2008) Effect of glabridin from Glycyrrhiza glabra on learning and memory in mice. Planta Med 74(4):377–380. https://doi.org/10.1055/s-2008-1034319
Dinel AL, Lucas C, Guillemet D, Laye S, Pallet V, Joffre C (2020) Chronic supplementation with a mix of Salvia officinalis and Salvia lavandulaefolia improves Morris water maze learning in normal adult C57Bl/6J mice. Nutrients 12(6):1777. https://doi.org/10.3390/nu12061777
El-Saber Batiha G, Magdy Beshbishy A, El-Mleeh A, Abdel-Daim MM, Prasad Devkota H (2020) Traditional uses, bioactive chemical constituents, and pharmacological and toxicological activities of Glycyrrhiza glabra L. (Fabaceae). Biomolecules 10(3):352. https://doi.org/10.3390/biom10030352
Fotakis G, Timbrell JA (2006) In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett 160(2):171–177. https://doi.org/10.1016/j.toxlet.2005.07.001
Giovannoni F, Quintana FJ (2020) The role of astrocytes in CNS inflammation. Trends Immunol 41(9):805–819. https://doi.org/10.1016/j.it.2020.07.007
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Tanzil RE (2013) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78(4):631–643. https://doi.org/10.1016/j.neuron.2013.04.014
Kang JS, Yoon YD, Cho IJ, Han MH, Lee CW, Park S-K, Kim HM (2005) Glabridin, an isoflavan from licorice root, inhibits inducible nitric-oxide synthase expression and improves survival of mice in experimental model of septic shock. J Pharmacol Exp Ther 312(3):1187–1194
Karthikkeyan G, Pervaje R, Pervaje SK, Prasad TSK, Modi PK (2021) Prevention of MEK-ERK-1/2 hyper-activation underlines the neuroprotective effect of Glycyrrhiza glabra L. (Yashtimadhu) against rotenone-induced cellular and molecular aberrations. J Ethnopharmacol 274:114025. https://doi.org/10.1016/j.jep.2021.114025
Kim JY, Kang JS, Kim HM, Ryu HS, Kim HS, Lee HK, Han S-B (2010) Inhibition of bone marrow-derived dendritic cell maturation by glabridin. Int Immunopharmacol 10(10):1185–1193
Kwon HS, Koh SH (2020) Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 9(1):42. https://doi.org/10.1186/s40035-020-00221-2
Kwon HS, Oh SM, Kim JK (2008) Glabridin, a functional compound of liquorice, attenuates colonic inflammation in mice with dextran sulphate sodium-induced colitis. Clin Exp Immunol 151(1):165–173. https://doi.org/10.1111/j.1365-2249.2007.03539.x
Lafrenaye AD, Mondello S, Wang KK, Yang Z, Povlishock JT, Gorse K, Kochanek PM (2020) Circulating GFAP and Iba-1 levels are associated with pathophysiological sequelae in the thalamus in a pig model of mild TBI. Sci Rep 10(1):13369. https://doi.org/10.1038/s41598-020-70266-w
Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17(3):157–172. https://doi.org/10.1038/s41582-020-00435-y
Li C, Eom T, Jeong Y (2015) Glycyrrhiza glabra L. extract inhibits LPS-induced inflammation in RAW macrophages. J Nutr Sci Vitaminol (tokyo) 61(5):375–381. https://doi.org/10.3177/jnsv.61.375
Li G, Nikolic D, van Breemen RB (2016) Identification and chemical standardization of Licorice raw materials and dietary supplements using UHPLC-MS/MS. J Agric Food Chem 64(42):8062–8070. https://doi.org/10.1021/acs.jafc.6b02954
Liu K, Pi F, Zhang H, Ji J, Xia S, Cui F, Sun X (2017) Metabolomics analysis to evaluate the anti-inflammatory effects of polyphenols: Glabridin reversed metabolism change caused by LPS in RAW 264.7 cells. J Agric Food Chem 65(29):6070–6079. https://doi.org/10.1021/acs.jafc.7b01692
Liu Y, Zhang Y, Zheng X, Fang T, Yang X, Luo X, Yu Y (2018) Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J Neuroinflammation 15(1):112. https://doi.org/10.1186/s12974-018-1141-5
Markina YV, Kirichenko TV, Markin AM, Yudina IY, Starodubova AV, Sobenin IA, Orekhov AN (2022) Atheroprotective effects of glycyrrhiza glabra L. Molecules 27(15):4697. https://doi.org/10.3390/molecules27154697
McKenna M, McDougall JJ (2020) Cannabinoid control of neurogenic inflammation. Br J Pharmacol 177(19):4386–4399. https://doi.org/10.1111/bph.15208
Nam HY, Nam JH, Yoon G, Lee JY, Nam Y, Kang HJ, Hoe HS (2018) Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J Neuroinflammation 15(1):271. https://doi.org/10.1186/s12974-018-1308-0
Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP (2016) Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 64(2):300–316. https://doi.org/10.1002/glia.22930
Park SH, Kang JS, Yoon YD, Lee K, Kim KJ, Lee KH, Park SK (2010) Glabridin inhibits lipopolysaccharide-induced activation of a microglial cell line, BV-2, by blocking NF-kappaB and AP-1. Phytother Res 24(Suppl 1):S29-34. https://doi.org/10.1002/ptr.2872
Pascua-Maestro R, Corraliza-Gomez M, Diez-Hermano S, Perez-Segurado C, Ganfornina MD, Sanchez D (2018) The MTT-formazan assay: Complementary technical approaches and in vivo validation in Drosophila larvae. Acta Histochem 120(3):179–186. https://doi.org/10.1016/j.acthis.2018.01.006
Rajesh M, Mukhopadhyay P, Hasko G, Pacher P (2008) Cannabinoid CB1 receptor inhibition decreases vascular smooth muscle migration and proliferation. Biochem Biophys Res Commun 377(4):1248–1252. https://doi.org/10.1016/j.bbrc.2008.10.159
Rao T, Tan Z, Peng J, Guo Y, Chen Y, Zhou H, Ouyang D (2019) The pharmacogenetics of natural products: a pharmacokinetic and pharmacodynamic perspective. Pharmacol Res 146:104283. https://doi.org/10.1016/j.phrs.2019.104283
Silver J, Schwab ME, Popovich PG (2014) Central nervous system regenerative failure: role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harb Perspect Biol 7(3):a020602. https://doi.org/10.1101/cshperspect.a020602
Simmler C, Pauli GF, Chen SN (2013) Phytochemistry and biological properties of glabridin. Fitoterapia 90:160–184. https://doi.org/10.1016/j.fitote.2013.07.003
Tan W, Zhang Q, Dong Z, Yan Y, Fu Y, Liu X, Duan X (2020) Phosphatidylcholine Ameliorates LPS-induced systemic inflammation and cognitive impairments via mediating the gut-brain axis balance. J Agric Food Chem 68(50):14884–14895. https://doi.org/10.1021/acs.jafc.0c06383
van Niekerk G, Mabin T, Engelbrecht AM (2019) Anti-inflammatory mechanisms of cannabinoids: an immunometabolic perspective. Inflammopharmacology 27(1):39–46. https://doi.org/10.1007/s10787-018-00560-7
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1(2):848–858. https://doi.org/10.1038/nprot.2006.116
Wang L, Ding J, Zhu C, Guo B, Yang W, He W, Sun T (2021) Semaglutide attenuates seizure severity and ameliorates cognitive dysfunction by blocking the NLR family pyrin domain containing 3 inflammasome in pentylenetetrazole-kindled mice. Int J Mol Med 48(6):219. https://doi.org/10.3892/ijmm.2021.5052
Wang L, Yang R, Yuan B, Liu Y, Liu C (2015) The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharm Sin B 5(4):310–315. https://doi.org/10.1016/j.apsb.2015.05.005
Wang Z, Wu J, Hu Z, Luo C, Wang P, Zhang Y, Li H (2020) Dexmedetomidine alleviates lipopolysaccharide-induced acute kidney injury by inhibiting p75NTR-mediated oxidative stress and apoptosis. Oxid Med Cell Longev 2020:5454210. https://doi.org/10.1155/2020/5454210
Yang WT, Zheng XW, Chen S, Shan CS, Xu QQ, Zhu JZ, Wang Y (2017) Chinese herbal medicine for Alzheimer’s disease: clinical evidence and possible mechanism of neurogenesis. Biochem Pharmacol 141:143–155. https://doi.org/10.1016/j.bcp.2017.07.002
Yehuda I, Madar Z, Leikin-Frenkel A, Tamir S (2015) Glabridin, an isoflavan from licorice root, downregulates iNOS expression and activity under high-glucose stress and inflammation. Mol Nutr Food Res 59(6):1041–1052
Zhan X, Stamova B, Sharp FR (2018) Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in alzheimer’s disease brain: a review. Front Aging Neurosci 10:42. https://doi.org/10.3389/fnagi.2018.00042
Zhang Y, Naguro I, Herr AE (2019) In situ single-cell western blot on adherent cell culture. Angew Chem Int Ed Engl 58(39):13929–13934. https://doi.org/10.1002/anie.201906920
Acknowledgements
This work was funded by the Guangdong Provincial Key Laboratory of Plant Resources Biorefinery (No. 2021GDKLPRB02) and the Basic and Applied Basic Research Foundation of Guangdong Province (No. 2020A1515110715).
Funding
Guangdong Provincial Key Laboratory of Plant Resources Biorefinery, 2021GDKLPRB02, Lanyue Zhang, the Basic and Applied Basic Research Foundation of Guangdong Province, 2020A1515110715, Lanyue Zhang
Author information
Authors and Affiliations
Contributions
JW: Research, digitize and write the first draft. YW: Research, digitize and write the first draft. ZT: Digitize, investigation and verification. YY: Data collation. SH: Investigation. ZL: Formal analysis. YL: Data collation and modification. LZ: Conceptualization, supervision, resources and capital acquisition. ZD: Conceptualization, methodology, supervision, review and proofreading.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest in the publication of this paper.
Ethical approval
All animal experiments are conducted in accordance with relevant government and institutional regulations on the ethical use of animals and are approved by the Experimental Animal Center of Guangdong Province (approval documents: SCXK/20130002).
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Weng, J., Wang, Y., Tan, Z. et al. Glabridin reduces neuroinflammation by modulating inflammatory signals in LPS-induced in vitro and in vivo models. Inflammopharmacol 32, 1159–1169 (2024). https://doi.org/10.1007/s10787-023-01424-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-023-01424-5