
Abstract
Neuroinflammation is involved in a wide range of brain disorders, thus there is great interest in identifying novel anti-inflammatory agents to include in therapeutic strategies. Our previous in vitro studies revealed that beta-funaltrexamine (β-FNA), a well-characterized selective mu-opioid receptor (MOR) antagonist, inhibits inflammatory signaling in human astroglial cells, albeit through an apparent MOR-independent mechanism. We also previously determined that lipopolysaccharide (LPS)-induced sickness behavior and neuroinflammation in mice are prevented by pretreatment with β-FNA. Herein we investigated the temporal importance of β-FNA treatment in this pre-clinical model of LPS-induced neuroinflammation. Adult, male C57BL/6J mice were administered an i.p. injection of LPS followed by treatment (i.p. injection) with β-FNA immediately or 4 h post-LPS. Sickness behavior was assessed using an open-field test, followed by assessment of inflammatory signaling in the brain, spleen, and plasma. Levels of inflammatory chemokines/cytokines (interferon γ-induced protein, CXCL10; monocyte chemotactic protein 1, CCL2; and interleukin-6, IL-6) in tissues were measured using an enzyme-linked immunosorbent assay and nuclear factor-kappa B (NFκB), p38 mitogen activated kinase (p38 MAPK), and glial fibrillary acidic protein (GFAP) expression were measured by western blot. LPS-induced sickness behavior and chemokine expression were inhibited more effectively when β-FNA treatment occurred immediately after LPS administration, as opposed to 4 h post-LPS; and β-FNA-mediated effects were time-dependent as evidenced by inhibition at 24 h, but not at 8 h. The inhibitory effects of β-FNA on chemokine expression were more evident in the brain versus the spleen or plasma. LPS-induced NFκB-p65 and p38 MAPK expression in the brain and spleen were inhibited at 8 and 24 h post-LPS. These findings extend our understanding of the anti-inflammatory effects of β-FNA and warrant further investigation into its therapeutic potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anxiolytic, anti-depressant, and anti-psychotic medications generally target neurotransmitter signaling, yet often possess limited anti-inflammatory properties as well (Dantzer 2001; Davis et al. 2017; Salazar et al. 2012; Swiergiel and Dunn 2007). Neurological conditions, including mood disorders, often present with neuroinflammation (Uguccioni et al. 1999; Betteridge et al. 2013; Bai et al. 2015; Wei et al. 2022), thereby elevating interest in the therapeutic potential of anti-inflammatory agents. We previously discovered that β-funaltrexamine (β-FNA), a selective mu-opioid receptor (MOR) antagonist, inhibits cytokine-induced expression of cytokines/chemokines in vitro in human astroglial cells (Davis et al. 2007, 2013, 2020). Further investigation revealed that the anti-inflammatory actions were not dependent upon actions at the MOR (Davis et al. 2013, 2015). More specifically, when added (in threefold excess) prior to β-FNA, neither the general opioid receptor antagonist, naltrexone, the MOR selective (D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2, CTAP), nor the KOR selective (nor-binaltorphimine, Nor-BNI) antagonists reversed the inhibitory effects of β-FNA on proinflammatory-induced chemokine expression. Notably, unlike β-FNA, none of these opioid receptor antagonists inhibited inflammatory activation, even at a threefold higher concentration (Davis et al. 2013, 2015). While the exact mechanism by which β-FNA inhibits inflammatory signaling has yet to be fully elucidated, in vitro studies in astroglial cells suggest that disruption of key inflammatory signaling pathways are involved (Davis et al. 2008, 2007, 2013, 2015, 2020). More specifically, we demonstrated that β-FNA inhibited activation of both p38 mitogen activated kinase (p38 MAPK) and nuclear factor-kappa B (NFκB) in human astroglial cells (Davis et al. 2007, 2013, 2015). Investigations are ongoing to further define the anti-inflammatory mechanism of action for β-FNA. Importantly, we have also determined that β-FNA inhibits bacterial lipopolysaccharide (LPS)-induced neuroinflammation in adult male C57BL/6J mice (Davis et al. 2017). In this previous study, β-FNA was administered i.p. immediately prior to the i.p. injection of LPS and we limited our assessment of inflammatory signals to cytokines/chemokines in brain and plasma at 24 h post-LPS (Davis et al. 2017). In the present study, we extend and expand this line of investigation to assess the benefit of delayed β-FNA treatment, temporal effects of treatment, and additional inflammatory signaling molecules in both brain and spleen. Furthermore, we determined the effects of β-FNA on sickness behaviors. Together, these findings will advance our understanding of the neuroprotective actions and potential therapeutic benefits of β-FNA in treating neuroinflammation-associated conditions, including mood disorders.
Materials and methods
Animals
Seven-week-old male C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME) and maintained in USDA-approved facilities at Oklahoma State University-Center for Health Sciences (OSU-CHS). Mice were assigned three per plastic cage (dimensions of 10 cm × 17 cm × 28 cm). Each cage contained pine chip bedding, environmental stimulus was provided by cardboard tubes, and ad libitum access to food and water was provided. Room temperature was maintained at 21 °C with a 12:12 light:dark cycle. A total of 48 animals were approved for this study and were acclimated to housing conditions for 7 days prior to initiation of experiments and they were monitored daily. Animal manipulations and handling processes for all experiments were approved by the OSU-CHS Institutional Animal Care and Use Committee.
Experimental protocol
As previously described (Davis et al. 2017), mice (n = 5–8 per group) were administered LPS (Escherichia coli O55:B5; Sigma) i.p. at a dose of 0.83 mg/kg dissolved in saline. This LPS dose is routinely used in mice to induce neuroinflammation, and behavioral deficits, including anxiety-like and sickness behaviors (Salazar et al. 2012; Dantzer 2001; Swiergiel and Dunn 2007). After the LPS injection, mice were administered β-FNA (National Institute on Drug Abuse reagent supply program) at a dose of 50 mg/kg; i.p dissolved in saline or saline vehicle (200 µl) immediately or 4 h after LPS. The β-FNA dose was established based on effectiveness observed in our previous work (Davis et al. 2017; Ramachandra et al. 2011).
Behavioral measures
Behavioral analysis was done using an open-field test (OFT) where locomotor activity was used as an index of acute sickness behavior (Salazar et al. 2012; Dantzer 2001; Swiergiel and Dunn 2007). For this OFT, each mouse was individually placed in the open-field arena, measuring 40 cm × 40 cm, then monitored and recorded with Ethovision Software for 10 min. The dependent measures included distance moved (cm) and duration (s) in the center 35 cm × 35 cm region of the arena. Sickness behavior included decreased distance moved and decreased time spent in the center region (away from the walls) (Salazar et al. 2012; Dantzer 2001; Swiergiel and Dunn 2007).
Tissue collection
Immediately after behavioral testing, mice were euthanized by CO2 inhalation and subsequent decapitation. Trunk blood, brain, and spleen were collected into ice-cold tubes and immediately placed on ice. Plasma was collected after centrifugation (17,000×g, 15 min, 4 °C). All samples were then stored at − 80 °C until assays were performed. Tissue was homogenized in ice-cold triple detergent lysis buffer (Thomas Curtis et al. 2011) containing HALT Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) using a Sonic Dismembrator Model 100 (Fisher Scientific). The sample was then centrifuged (20,000×g, 20 min, 4 °C), followed by collection of the aqueous phase. Homogenized samples were used for the quantification of inflammatory factors. Total protein levels were measured using a bicinchoninic acid (BCA) protein assay as previously described (Davis et al. 2002) for subsequent determination of sample loading volume in western blot analyses and normalization of protein quantification data.
Measurement of inflammatory mediators
Standard dual-antibody solid-phase immunoassays were performed according to the manufacturer’s instructions (ELISA Development Kit, Peprotech) to quantify chemokine/cytokine [interferon gamma-induced protein 10, IP-10/CXCL10; monocyte chemoattractant protein-1, MCP-1/CCL2; interleukin-6, IL-6] levels in plasma and tissues. Absorbance of the final product was read on a BIOTEK Synergy 2 spectrophotometer at 450 nm using a Gen5 microplate reader and imager software by BIOTEK.
Western blot analysis was used to measure NFκB-p65, p38 MAPK, and glial fibrillary acidic protein (GFAP) levels in tissue homogenates. Beta-tubulin was also measured for normalization. Total protein (100 µg) was analyzed by SDS polyacrylamide gel electrophoresis (PAGE) and transferred to the PVDF membrane. The membrane was incubated in target-specific 1° antibody [p65 (1:1000), Cat# 4764S; p38 (1:1000), Cat# 9212S; GFAP (1:1000), Cat#3670S; β-tubulin (1:1000), Cat# 2146S; Cell Signaling Technology], washed 6 times in Tris-buffered saline with 0.1% Tween (TBST), then incubated in 2° antibody [Goat-anti-Rabbit IgG (1:10,000), Cat# 925-32211; Donkey-anti-Mouse IgG (1:10,000), CAT# 926-32212; Li-Cor). Licor-CLX Odyssey was used to image Western blots, and protein signals were analyzed using Image J for relative quantification.
Statistical analysis
Data were analyzed by a two-way ANOVA (treatment × time); select pairwise comparisons were further evaluated using Fisher’s LSD. Data are presented as mean ± SEM, with p values < 0.05 considered statistically significant. \({\mathrm{Prism}}^{\mathrm{TM}}\) Version 9 software (GraphPad Inc, San Diego, CA) was used to analyze data and prepare figures.
Results
Effects of β-FNA on LPS-induced sickness behavior
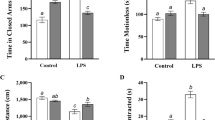
Two-way ANOVA for distanced moved (Fig. 1a) indicated significant main effects of treatment (F3,40 = 22.87, p < 0.0001) and time (F1,40 = 39.61, p < 0.0001), as well as a significant interaction (F3,40 = 6.273, p < 0.002) between main effects. Pairwise analyses using Fisher’s LSD revealed sickness behavior at the 8 and 24 h time points in the LPS groups as indicated by significantly reduced distance moved compared to the respective control group for each time point. At 8 h, the distance moved for the LPS + β-FNA and LPS + β-FNA 4 h groups were also significantly reduced relative to the control group. At the 24 h time point, sickness behavior was observed in the LPS and LPS + β-FNA 4 h groups as indicated by significantly reduced distance moved compared to the control group. Whereas protection was observed in the LPS + β-FNA group as indicated by the increase in distance moved at 24 h.

Effects of β-FNA on LPS-induced sickness behavior in male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 8 or 24 h post-LPS, mice were assessed in a 10 min open-field test (OFT). Endpoints measured include distance moved (a) and duration in the center area of the chamber (b). Data are reported as mean ± SEM. Two-way ANOVA (treatment × time) for distance moved revealed significant effects of both treatment (p < 0.0001) and time (p < 0.0001); and a significant interaction of treatment and time (p < 0.002). Two-way ANOVA for duration in the center area indicated significant effects of both treatment (p < 0.001) and time (p < 0.0001); and a significant interaction of treatment and time (p < 0.02). Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point
Further analysis of OFT data by two-way ANOVA of duration in the center region (Fig. 1b) revealed significant main effects of treatment (F3,38 = 7.754, p < 0.001) and time (F1,38 = 17.84, p < 0.0001), as well as a significant interaction (F3,38 = 3.716, p < 0.02) between main effects. Pairwise comparisons suggested sickness-like behavior 8 h after LPS administration as indicated by the reduced time spent in the center for the LPS group compared to the control group. Similarly, both β-FNA treatment groups (LPS + β-FNA and LPS + β-FNA 4 h) spent significantly less time in the center region at 8 h post-LPS compared to the control group. At 24 h, the LPS group seemed to spend less time in the center region compared to the control group, yet the difference was not significant (p = 0.09). The LPS + β-FNA 4 h group spent significantly less time in the center region compared to the control group.
Effects of β-FNA on LPS-induced cytokine and chemokine expression in brain, spleen, and plasma
In the brain, CXCL10 and CCL2 expression were significantly affected by treatment (F3,40 = 50.43, p < 0.001 and F3,40 = 17.31, p < 0.0001), whereas levels of CXCL10, CCL2, and IL-6 were significantly affected by time (F1,40 = 126.9, p < 0.001; F1,40 = 59.03, p < 0.0001; and F1,40 = 59.28, p < 0.0001) (Fig. 2). Furthermore, there were significant interactions between treatment and time for CXCL10, CCL2 and IL-6 in the brain (F3,40 = 14.44, p < 0.001; F3,40 = 11.74, p < 0.0001; and F3,40 = 6.392, p < 0.002). Pairwise analyses revealed CXCL10, CCL2, and IL-6 were increased at 8 h in the LPS group compared to the control group. At 8 h, levels of CXCL10, CCL2, and IL-6 in the brain of both the LPS + β-FNA and LPS + β-FNA 4 h groups were significantly higher than in the brain of the control groups. At 24 h, CXLC10 levels in the LPS and LPS + β-FNA 4 h groups were significantly higher than in the control group. The CXCL10 level in the of LPS + β-FNA group was significantly lower compared to the LPS group and approached the level in the control group. CCL2 levels were significantly higher in the LPS group at 24 h compared to controls. β-FNA was protective against CCL2 at 24 h in both the LPS + β-FNA and LPS + β-FNA 4 h group. The levels of IL-6 in the brain were similar among all groups at 24 h.

Effects of β-FNA on LPS-induced cytokine/chemokine expression in the brain of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of brain. Levels of CXCL10 (a), CCL2 (b), and IL-6 (c) in whole brain homogenates were measured by ELISA and normalized to total protein (pg/mg protein). Data (mean ± SEM) are presented as percent control. Two-way ANOVA (treatment × time) revealed a significant effect of treatment on CXCL10 (p < 0.001) and CCL2 (p < 0.0001); and time significantly affected CXCL10 (p < 0.001), CCL2 (p < 0.0001), and IL-6 (p < 0.0001). There was a significant interaction between treatment and time for all three inflammatory factors (p < 0.002). Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point
In the spleen, CXCL10, CCL2, and IL-6 expression were significantly affected by treatment (F3,40 = 43.20, p < 0.0001; F3,39 = 32.68, p < 0.0001; F3,40 = 31.56, p < 0.0001) and time (F1,40 = 335.2, p < 0.0001; F1,39 = 132.4, p < 0.0001; F1,40 = 301.4, p < 0.0001) and there was a significant interaction (F3,40 = 36.36, p < 0.0001; F3,39 = 17, p < 0.0001; F3,40 = 33.11, p < 0.0001) between these main effects (Fig. 3). Pairwise analyses indicated increased CXCL10, CCL2 and IL-6 at 8 h in the LPS group compared to controls. Similarly, CXCL10, CCL2, and IL6 levels in the spleen were significantly elevated in the spleen of both the LPS + β-FNA and LPS + β-FNA 4 h groups compared to controls. At the 24 h time point, only levels of CCL2 were significantly increased in the LPS group compared to the control group. Spleen CCL2 levels in the LPS + β-FNA and LPS + β-FNA 4 h treated mice were significantly decreased compared to the LPS group approached the level and were similar to levels in the control group.

Effects of β-FNA on LPS-induced cytokine/chemokine expression in the spleen of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of spleen. Levels of CXCL10 (a), CCL2 (b), and IL-6 (c) in whole spleen homogenates were measured by ELISA and normalized to total protein (pg/mg protein). Data (mean ± SEM) are presented as percent control. Two-way ANOVA (treatment × time) revealed a significant effect of both treatment and time on CXCL10, CCL2, and IL-6 (p < 0.0001 in all instances). There was a significant interaction between treatment and time for all three inflammatory factors (p < 0.0001). Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point
The levels of CXCL10 and CCL2 in the plasma were significantly affected by both treatment (F3,40 = 13.91 p < 0.0001; F3,38 = 4.964 p < 0.01) and time (F1,40 = 10.27, p < 0.003; F1,38 = 25.85, p < 0.001) and there were significant interactions (F3,40 = 7.020, p < 0.001; F3,38 = 4.985, p < 0.001) between these main effects (Fig. 4). Pairwise analyses of 8 h data revealed that compared to controls, CXCL10 levels in the plasma were only significantly elevated in the LPS + β-FNA group. Plasma CCL2 levels at 8 h were significantly increased in the LPS and LPS + β-FNA groups compared to the control group; while seemingly increased, CCL2 levels in the LPS + β-FNA 4 h mice were not statistically different from control levels (p = 0.0519). Plasma CXCL10 levels were significantly increased at 24 h in both the LPS and LPS + β-FNA 4 h groups compared to controls, whereas CXCL10 levels in the LPS + β-FNA group were statistically different from the LPS group. Plasma CCL2 levels at 24 h were not significantly different among the treatment groups. Due to limited sample volume, IL-6 levels were not assessed in the plasma.

Effects of β-FNA on LPS-induced chemokine expression in the plasma of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of plasma. Levels of CXCL10 (a), and CCL2 (b) in the plasma were measured by ELISA and normalized to total protein (pg/mg protein). Data (mean ± SEM) are presented as percent control. Two-way ANOVA (treatment × time) revealed a significant effect of treatment on CXCL10 (p < 0.0001) and CCL2 (p < 0.01) as well as a significant effect of time on these factors (p < 0.003). There was a significant interaction between treatment and time for both CXCL10 and CCL2 (p < 0.001). Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point
Effects of β-FNA on LPS-induced NFκB-p65, p38 MAPK, and GFAP expression in brain and spleen
The levels of NFκB-p65 and p38 MAPK in the brain were significantly affected by treatment (F3,40 = 4.970, p < 0.01; F3,40 = 3.115, p < 0.05) (Fig. 5). The expression of p38 MAPK in the brain was also significantly impacted by time (F1,40 = 4.124, p < 0.05). There were no significant interactions between treatment and time on expression of these signaling factors in the brain. Pairwise analyses determined that NFκB-p65 levels in the brain were significantly increased in the LPS group at 8 and 24 h relative to controls. At both time points, brain NFκB-p65 levels in the LPS + β-FNA and LPS + β-FNA 4 h groups were similar to control with LPS + β-FNA 4 h being significant different from the LPS group. The levels of p38 MAPK in the brain were increased significantly at 8 h in the LPS group compared to controls; whereas expression in the LPS + β-FNA 4 h group was not significantly different from the control group. At 24 h, p38 MAPK expression in the brain was similar among all four groups. The expression of GFAP in the brain was not significantly affected by either treatment (F3,40 = 1.177, p = 0.3305) or time (F1,40 = 1.941 p = 0.1713) (Fig. 6).

Effects of β-FNA on LPS-induced NFκB-p65 expression in the brain and spleen of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of brain and spleen. Levels of NFκB-p65 and beta-tubulin in the brain (a) and spleen (b) were determined by western blot analysis. NFκB-p65 levels are expressed relative to beta-tubulin levels and are reported as mean ± SEM. Two-way ANOVA (treatment × time) revealed a significant (p < 0.01) effect of treatment on NFκB-p65 in the brain; but no effect of time; and no significant interaction of treatment and time. Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point

Effects of β-FNA on LPS-induced GFAP expression in the brain of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of brain. Levels of GFAP and beta-tubulin in the brain were determined by western blot analysis. Levels of GFAP are expressed relative to beta-tubulin levels and are reported as mean ± SEM. Two-way ANOVA (treatment × time) did not reveal any significant main effects or interactions
The levels of NFκB-p65 and p38 MAPK in the spleen were significantly affected by treatment (F3,40 = 3.626, p < 0.05; F3,40 = 6.525, p < 0.01), but not by time (F1,40 = 0.01294, p = 0.91; F1,40 = 0.5655, p = 0.4565) (Fig. 7). Mice administered LPS had significantly greater levels of NFκB-p65 in the spleen at 8 and 24 h compared to control mice. The levels of NFκB-p65 in both the LPS + β-FNA and LPS + β-FNA 4 h groups were similar to controls at 8 and 24 h post-LPS. At 8 h, p38 MAPK levels in the LPS group were significantly elevated compared to the control mice, whereas the levels of p38 MAPK in the LPS + β-FNA and LPS + β-FNA 4 h groups were significantly different from levels in the LPS group. At 24 h, p38 MAPK expression in the spleen was similar among all four groups.

Effects of β-FNA on LPS-induced p38 MAPK expression in the brain and spleen of male C57BL/6J mice. Mice (n = 5–8/group) were injected (i.p.) with saline (control), LPS (0.83 mg/kg), LPS followed immediately by β-FNA treatment (50 mg/kg; i.p.; LPS + β-FNA), or LPS followed by β-FNA 4 h post-LPS (LPS + β-FNA 4 h). At 24 h post-LPS, mice were terminated followed by collection of brain and spleen. Levels of p38 MAPK and beta-tubulin in the brain (a) and spleen (b) were determined by western blot analysis. Levels of p38 MAPK are expressed relative to beta-tubulin levels and are reported as mean ± SEM. Two-way ANOVA (treatment × time) revealed a significant effect of both treatment (p < 0.05) and time (p < 0.05) on p38 MAPK in the brain; but no significant interaction of these main effects. Pairwise comparisons were asssed using a Fisher’s LSD test; *significantly different (p < 0.05) from the control group at the same time point
Discussion
Neurological conditions, including central nervous system infections, neurodegenerative diseases, and psychiatric disorders involve neuroinflammation (Da Ré et al. 2020; He et al. 2018; Na et al. 2021; Glass et al. 2010; Khansari and Sperlagh 2012; Kolb et al. 1999; Whitton 2010; Ziebell and Morganti-Kossmann 2010). Evidence of neuroinflammation in these conditions includes elevated expression of proinflammatory cytokines and chemokines (Bai et al. 2015; Blank et al. 2016; Ziebell and Morganti-Kossmann 2010). Peripheral (i.p.) LPS administration in mice is an established preclinical model of neuroinflammation in which proinflammatory cytokines/chemokines are elevated in the brain. For instance, we demonstrated that CXCL10 and CCL2 levels in the brain correlated with LPS-induced behavioral deficits (Davis et al. 2017). We and others have used this model to investigate the therapeutic potential of pharmacologic agents (Blank et al. 2016; Burton et al. 2011; Davis et al. 2015, 2017; Le Thuc et al. 2016). Indeed, we previously determined that LPS-induced CXCL10 and CCL2 expression in the brain of C57BL/6J mice was prevented by pretreatment with β-FNA (Davis et al. 2017). We now expand on these findings and show that β-FNA administration is also protective when administered after LPS injection.
Reduced locomotor activity and wall-hugging in the OFT are both reliable indicators of sickness behavior; and as expected, LPS-induced sickness behavior in both measures was more pronounced at 8 h than at 24 h post-LPS. Sickness behavior following a single LPS injection (i.p.) at 0.83 mg/kg often resolves by 24 h, however, in this study, decreased locomotor activity indicated that some level of sickness behavior remained at 24 h. Treatment with β-FNA immediately after the LPS injection prevented the sickness behavior at 24 h, whereas a 4 h delay in β-FNA treatment was not protective. The sickness behavior observed at 8 h was not prevented by β-FNA treatment regardless of the timing of administration, suggesting this dose of β-FNA is not sufficient to prevent this more severe LPS-induced sickness. It will be important in future studies to asses other behavior deficits to more fully appreciate the protective effects of β-FNA.
IL-6, CXCL10, and CCL2 are among the proinflammatory cytokines/chemokines that are increased in the CNS following brain injury, infection and in mood/behavior disorders (Burton et al. 2011; Kwon and Koh 2020; Lasselin et al. 2016; Patki et al. 2013; Ziebell and Morganti-Kossmann 2010) and are integral to neuropathogenesis (Stuart and Baune 2014; Ziebell and Morganti-Kossmann 2010). Therefore, we are particularly interested in the effects of β-FNA on the expression of these cytokines/chemokines in the brain in this pre-clinical model of LPS-induced neuroinflammation. LPS-induced expression of IL-6, CXCL10, and CCL2 in the brain was more pronounced at 8 h compared to 24 h post injection which is consistent with the relative severity of sickness behavior observed at these two time points. β-FNA failed to inhibit LPS-induced cytokine/chemokine expression in the brain at this early time point, which is consistent with the lack of an effect on sickness behavior at 8 h. The anti-inflammatory effects of β-FNA were most pronounced when administered immediately following the LPS injection, as opposed to delaying treatment by 4 h. Although, β-FNA treatment 4 h post-LPS was also sufficient to inhibit LPS-induced CCL2 expression in the brain. It remains unclear why CCL2, but not CXCL10, is sensitive to delayed β-FNA but it may be related to differences in transcription/translational control of these two chemokines, yet further investigation is needed. It is also interesting to note that IL-6 is not elevated at 24 h and likely plays a minimal role in the sickness behavior.
Thus far these findings suggest that β-FNA is likely inhibiting a relatively early event in the LPS-induced inflammatory pathway. Indeed, β-FNA inhibited LPS-induced NFκB-p65 expression in the brain and spleen, early on at 8 h and at 24 h post-LPS. Inhibition of p38 MAPK expression in both the brain and spleen by β-FNA at 8 h, but not 24 h post-LPS provides additional evidence that inflammatory signaling is being disrupted.
The differential effects of β-FNA among tissues and dosing regimen is interesting. As in our previous report, β-FNA effects on LPS-induced chemokine expression are generally more pronounced in the brain than in the periphery. However, in our previous study, peripheral tissue was limited to plasma, and in the current study we also assessed levels in the spleen (Davis et al. 2017). In the spleen, only LPS-induced CCL2 expression (not CXCL10 or IL-6) was sensitive to the inhibitory effects of β-FNA and only when treatment occurred immediately after LPS administration. Also, in the present study LPS-induced plasma CXCL10 levels, but not CCL2 levels, were decreased by immediate β-FNA (and not by β-FNA at 4 h post LPS). Altogether, it appears that treatment with β-FNA immediately after LPS administration is more effective than treatment prior to LPS or delayed several hours. However, one limitation to this conclusion is the fact that we did not specifically test β-FNA pre-treatment versus post-LPS treatment in the same study. It will be interesting in future studies to investigate additional β-FNA doses and treatment regimens. It will also be critical to determine the effects of β-FNA on neuroimmune signaling in specific brain regions (i.e., hippocampus and frontal cortex).
We expected that LPS would activate astroglia, as indicated by increased GFAP expression (Ifuku et al. 2012; Shah et al. 2020; Wang et al. 2019). However, while there was an apparent increase in GFAP expression 24 h following LPS administration (but not in β-FNA-treated mice), the increase did not reach the level of statistical significance (p = 0.08). One explanation for the modest LPS-induced increase in GFAP expression is that whole brain tissue was assessed rather than hippocampus or cortex as reported by others (Shah et al. 2020; Wang et al. 2019). It will be crucial in future studies to determine the extent to which β-FNA differentially affects LPS-induced astrocyte activation across select brain regions.
This study was not designed to specifically assess mechanism of action for β-FNA-mediated inhibition of inflammatory signaling. However, we have substantial information from in vitro studies in human astrocytes that suggest β-FNA inhibits NFκB-p65 and p38 MAPK signaling pathways resulting in decreased chemokine expression (Davis et al. 2007, 2013, 2015). β-FNA seems to be impacting the early signaling events at 8 h whereas the effects on secreted factors (i.e., cytokines/chemokines) were observed at a later time-point when expression typically peaks. Importantly, investigations are ongoing to fully elucidate the molecular mechanism of action for the anti-inflammatory effects.
Overall, it is important to note that the field’s understanding of β-FNA’s anti-inflammatory and neuroprotective effects in vivo is still very limited, and in a relatively early stage of discovery. To our knowledge, we were the first to report the anti-inflammatory effects of β-FNA (Davis et al. 2007). This initial report and several others to follow all used an in vitro approach to demonstrate the anti-inflammatory effects of β-FNA in human astroglial cells. We later demonstrated for the first time that β-FNA is also anti-inflammatory in mice (Davis et al. 2015). Compared to the current study, our initial in vivo investigation utilized a relatively high dose of LPS (5 mg/kg; i.p.), a relatively lower dose of β-FNA (28 mg/kg; i.p.) and assessed inflammatory and behavioral outcomes at 6 h post-treatment (Davis et al. 2015). We found that β-FNA inhibited LPS-induced CXCL10 expression in the brain (but not in the plasma) and failed to prevent sickness-like behavior. Thereafter, we expanded upon these studies and assessed the effects of β-FNA (12.5–50 mg/kg; i.p.) on LPS (0.83 mg/kg, i.p.)-induced inflammation and sickness-like behavior at 24 h post-treatment (Davis et al. 2017). We found that β-FNA inhibited LPS-induced CXCL10 and CCL2 expression in the brain as well as sickness-like behavior when administered concurrently with LPS. To our knowledge, the only other report on the anti-inflammatory and neuroprotective effects of β-FNA in vivo is by Wu et al. (2020) in which they determined that β-FNA (intraventricular infusion) protects against cerebral ischemia/reperfusion injury in rats. Interestingly, β-FNA reduced expression of several inflammatory factors that were induced in this pre-clinical stroke model (Wu et al. 2020).
The current findings are critical in our efforts to advance β-FNA as potential therapeutic option for neuroinflammatory conditions. This advancement comes with our focus on the importance of (1) the timing of β-FNA administration, (2) the temporal effects of β-FNA, (3) expanded panel of inflammatory factors (IL-6, NF-κB, p38 MAPK), and glial activation in vivo.
Conclusion
In summary, we have advanced this line of investigation by further demonstrating the anti-inflammatory and neuroprotective effects of β-FNA in a pre-clinical model of LPS-induced neuroinflammation. β-FNA treatment reduces LPS-induced sickness behavior and inflammatory signaling. The beneficial effects of β-FNA are greatest when treatment occurs immediately after LPS administration compared to treatment 4 h post-LPS, and the effects seem to be more pronounced in the brain compared to spleen and plasma. These findings are expected to advance therapeutic strategies to brain disorders involving neuroinflammation.
Data availability
Data will be made available for review as needed upon legitimate request.
References
Bai YM, Su TP, Li CT, Tsai SJ, Chen MH, Tu PC, Chiou WF (2015) Comparison of pro-inflammatory cytokines among patients with bipolar disorder and unipolar depression and normal controls. Bipolar Disord 17:269–277
Betteridge JD et al (2013) Inflammatory bowel disease prevalence by age, gender, race, and geographic location in the US military health care population. Inflamm Bowel Dis 19(7):1421–1427
Blank T, Detje CN, Spieß A, Hagemeyer N, Brendecke SM, Wolfart J, Staszewski O, Zöller T, Papageorgiou I, Schneider J, Paricio-Montesinos R, Eisel UL, Manahan-Vaughan D, Jansen S, Lienenklaus S, Lu B, Imai Y, Müller M, Goelz SE, Baker DP, Schwaninger M, Kann O, Heikenwalder M, Kalinke U, Prinz M (2016) Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity 44:901–912
Burton MD, Sparkman NL, Johnson RW (2011) Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J Neuroinflamm 8:54
Da Ré C, Souza JM, Fróes F, Taday J, Dos Santos JP, Rodrigues L, Sesterheim P, Gonçalves CA, Leite MC (2020) Neuroinflammation induced by lipopolysaccharide leads to memory impairment and alterations in hippocampal leptin signaling. Behav Brain Res 379:112360
Dantzer R (2001) Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci 933:222–234
Davis RL, Dertien J, Syapin PJ (2002) Ethanol-induced modulation of inducible nitric-oxide synthase activity in human A172 astrocytoma cells. Alcohol Clin Exp Res 26:1404–1411
Davis RL, Buck DJ, Saffarian N, Stevens CW (2007) The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol 186:141–149
Davis RL, Buck DJ, Saffarian N, Mohan S, DeSilva U, Fernando SC, Stevens CW (2008) Beta-funaltrexamine inhibits inducible nitric-oxide synthase expression in human astroglial cells. J Neuroimmune Pharmacol 3:150–153
Davis RL, Das S, Buck DJ, Stevens CW (2013) β-funaltrexamine inhibits chemokine (CXCL10) expression in normal human astrocytes. Neurochem Int 62:478–485
Davis RL, Das S, Thomas Curtis J, Stevens CW (2015) The opioid antagonist, β-funaltrexamine, inhibits NF-κB signaling and chemokine expression in human astrocytes and in mice. Eur J Pharmacol 762:193–201
Davis RL, Stevens CW, Thomas Curtis J (2017) The opioid antagonist, beta-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice. Physiol Behav 173:52–60
Davis RL, McCracken K, Buck DJ (2020) β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells. Neuroimmunol Neuroinflamm 7:300–310
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
He P, Yan S, Zheng J, Gao Y, Zhang S, Liu Z, Liu X, Xiao C (2018) Eriodictyol attenuates LPS-induced neuroinflammation, amyloidogenesis, and cognitive impairments via the inhibition of NF-κB in male C57BL/6J mice and BV2 microglial cells. J Agric Food Chem 66:10205–10214
Ifuku M, Katafuchi T, Mawatari S, Noda M, Miake K, Sugiyama M, Fujino T (2012) Anti-inflammatory/anti-amyloidogenic effects of plasmalogens in lipopolysaccharide-induced neuroinflammation in adult mice. J Neuroinflamm 9:197
Khansari PS, Sperlagh B (2012) Inflammation in neurological and psychiatric diseases. Inflammopharmacology 20:103–107
Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A (1999) Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J Neuroimmunol 93:172–181
Kwon HS, Koh SH (2020) Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 9:42
Lasselin J, Elsenbruch S, Lekander M, Axelsson J, Karshikoff B, Grigoleit JS, Engler H, Schedlowski M, Benson S (2016) Mood disturbance during experimental endotoxemia: predictors of state anxiety as a psychological component of sickness behavior. Brain Behav Immun 57:30–37
Le Thuc O, Cansell C, Bourourou M, Denis RG, Stobbe K, Devaux N, Guyon A, Cazareth J, Heurteaux C, Rostène W, Luquet S, Blondeau N, Nahon JL, Rovère C (2016) Central CCL2 signaling onto MCH neurons mediates metabolic and behavioral adaptation to inflammation. EMBO Rep 17:1738–1752
Na S, Duan X, Wang R, Fan Y, Xue K, Tian S, Yang Z, Li K, Yue J (2021) Chronic neuroinflammation induced by lipopolysaccharide injection into the third ventricle induces behavioral changes. J Mol Neurosci 71:1306–1319
Patki G, Solanki N, Atrooz F, Allam F, Salim S (2013) Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res 1539:73–86
Ramachandra V, Kang F, Kim C, Nova AS, Bajaj A, Hall FS, Uhl GR, Gonzales RA (2011) The μ opioid receptor is not involved in ethanol-stimulated dopamine release in the ventral striatum of C57BL/6J mice. Alcohol Clin Exp Res 35:929–938
Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O’Connor JC (2012) Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav 62:202–209
Shah MA, Park DJ, Kang JB, Kim MO, Koh PO (2020) Baicalin alleviates lipopolysaccharide-induced neuroglial activation and inflammatory factors activation in hippocampus of adult mice. Lab Anim Res 36:32
Stuart MJ, Baune BT (2014) Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: a systematic review of biomarker studies. Neurosci Biobehav Rev 42:93–115
Swiergiel AH, Dunn AJ (2007) Effects of interleukin-1beta and lipopolysaccharide on behavior of mice in the elevated plus-maze and open field tests. Pharmacol Biochem Behav 86:651–659
Thomas Curtis J, Chen Y, Buck DJ, Davis RL (2011) Chronic inorganic mercury exposure induces sex-specific changes in central TNFα expression: importance in autism? Neurosci Lett 504:40–44
Uguccioni M et al (1999) Increased expression of IP-10, IL-8, MCP-1, and MCP-3 in ulcerative colitis. Am J Pathol 155(2):331–336
Wang Y, Ni J, Zhai L, Gao C, Xie L, Zhao L, Yin X (2019) Inhibition of activated astrocyte ameliorates lipopolysaccharide-induced depressive-like behaviors. J Affect Disord 242:52–59
Wei Y et al (2022) Brain-spleen axis in health and diseases: a review and future perspective. Brain Res Bull 182:130–140
Whitton PS (2010) Neuroinflammation and the prospects for anti-inflammatory treatment of Parkinson’s disease. Curr Opin Investig Drugs 11:788–794
Wu CC, Chang CY, Shih KC, Hung CJ, Wang YY, Lin SY, Chen WY, Kuan YH, Liao SL, Wang WY, Chen CJ (2020) β-Funaltrexamine displayed anti-inflammatory and neuroprotective effects in cells and rat model of stroke. Int J Mol Sci 21:3866
Ziebell JM, Morganti-Kossmann MC (2010) Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7:22–30
Funding
This work was supported in part by Oklahoma Health Research Program (Oklahoma Center for the Advancement of Science and Technology, HR 18-033) (RLD), the Office of the Vice President of Research at Oklahoma State University Center for Health Sciences (RLD), and Graduate and Professional Student Association (GPSGA) Research Materials Awards (SM).
Author information
Authors and Affiliations
Contributions
SM: performed experiments and assays, analyzed data, prepared manuscript. KM: performed experiments and assays, interpreted data, edited manuscript. DB: performed experiments and assays, interpreted data, edited manuscript. JTC: conceptualization and design of study, assisted with statistical analyses, interpreted data. RLD: conceptualization and design of study, assisted with statistical analyses, interpreted data, troubleshooted assays, edited manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or no-financial interests to disclose.
Ethical approval
Animal manipulations and handling processes for all experiments were approved by the OSU-CHS Institutional Animal Care and Use Committee, protocol # 2020-1236.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Myers, S., McCracken, K., Buck, D.J. et al. Anti-inflammatory actions of β-funaltrexamine in a mouse model of lipopolysaccharide-induced inflammation. Inflammopharmacol 31, 349–358 (2023). https://doi.org/10.1007/s10787-022-01113-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-022-01113-9