
Abstract
The different configurations of the photopyroelectric calorimetry which can be adopted for the evaluations of the thermophysical parameters in liquids and in solids, and the advantages, with respect to other calorimetric techniques, are briefly described. A review of the recent results obtained by the photopyroelectric back-detection configuration for the determination of the different thermophysical properties in various materials is presented with particular attention to the cases where the temperature dependence of such parameters is studied. The results concerning the studies carried out by performing simultaneous calorimetric and imaging evaluations are also described. Finally, the possibility of performing “absolute” measurements of the thermal parameters, eliminating the influence of eventual coupling fluid, is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The evaluation of the thermal parameters of materials is of great importance. In fact quantities like the thermal diffusivity and the thermal effusivity are important from the technical point of view. The thermal diffusivity D = k/ρc (k is thermal conductivity, c the specific heat, and ρ the density), in non steady-state situations, constitutes a measure of how rapidly heat can propagate throughout the material, and the thermal effusivity, e = (kρc)1/2, characterizes the ability the material has to exchange thermal energy with the surroundings [1]. From a more fundamental point of view, the knowledge of both D and e enables the determination of c and k which are very important basic physical parameters. When measured with large enough a temperature resolution over a phase transition, the behavior of c and k can allow the study of, respectively, the static [2] and the dynamic critical phenomena [3] associated with the thermal parameters, for a more complete characterization of the transition [4,5,6,7,8,9].
The evaluation of c and k generally involves the use of separate technique. However, an ac calorimetric technique had been proposed to determine simultaneously both parameters but requiring a frequency scan to evaluate k [10]. Such a procedure has proven very time-consuming when measuring the temperature dependence of k, such as in the cases of its analysis over phase transitions. The photopyroelectric (PPE) technique can also be used for the characterization of the thermophysical parameters in liquids and solids, like other photothermal techniques [11,12,13,14,15,16,17,18,19]. In fact, PPE in the so-called back-detection (BD) configuration [20] was also shown to be capable of measuring simultaneously both c and k in solids [4] and in liquids [21,22,23,24] operating at a single frequency value and, therefore, in a much more practical way with respect to the previously proposed technique.
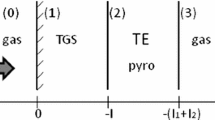
Another advantage of the PPE technique is its versatility, since it can be used in different configurations, each with its peculiar capabilities. Figure 1 shows the schemes of the various PPE configurations, namely the front detection (FD), the inverse detection (ID) and the back-detection (BD) which differ depending on the relative positions of the illuminated surface and that where the detection occurs. The specific peculiarities of each configuration have been summarized in a recent review [7]. Specifically, FD is the only one which allows a very large frequency range of operation, nearly 5 decades. This proved very useful when investigating the frequency dependence spectroscopy of the specific heat over glassy transitions, obtained from the analysis of the thermal effusivity [25,26,27,28,29,30]. On the other hand, it was shown that ID could be fruitfully applied, among others, for the direct determination of the specific heat and of the thermal conductivity by operating in corresponding specific frequency ranges [31, 32]. Recently, frequency scans of the ID signal enabled the determination of the thermal effusivity in different samples characterized by a wide range of values of such a parameter [33,34,35].

Sketch of the illumination conditions for various PPE configurations, namely the front detection (FD), the inverse detection (ID) and the back-detection (BD) ones
The capability of the BD configuration to measure simultaneously c and k operating at a single modulation frequency has made it very useful to monitor, in a straightforward fashion, the temperature dependence of such parameters particularly over temperature ranges where phase changes of the analyzed material may occur. Finally, the BD configuration has been shown to be able to detect also latent heat over first-ordered phase transitions [36, 37]. In this work we present relatively recent results, obtained by the BD configuration, not reported in the previous review work and concerning the evaluation of the temperature dependence the thermal parameters of solid and liquid samples, with particular attention to the cases where the parameters change because of variations occurring in the mesophases of the analyzed compounds.
2 Experimental
The most commonly adopted pyroelectric transducer consists of a LiTaO3 crystal typically 100–500 μm thick, though polyvinylidene di-fluoride (PVDF) films of various thicknesses (9–100 μm) are also employed. LiTaO3 sensors allow a more sensitive ac temperature detection over a wider temperature range, while PVDF is significantly cheaper. The electrodes on the pyroelectric transducer surfaces are typically optically opaque thin metallic (Au, Ag or Ti) films, while indium tin oxide (ITO) electrodes can be used when optical transparency is required. The ac sample heating can be provided by modulated laser beams. When the sample is solid, the thermal contact with the pyroelectric transducer is obtained by applying very thin layer (a few microns) of coupling fluid between them, while liquid samples are usually contained in glass/quartz cells of calibrated thickness (typically 10–100 μm). For transparent liquids, the visible laser light absorption is ensured by applying a thin opaque metallic layer on the appropriate cell/transducer surface. The electric signal from the transducer is fed to a dual-channel lock-in amplifier which measures both the amplitude and the phase of the PPE signal.
In some instances the PPE calorimeter has been used in an upgraded configuration where the setup was integrated with a polarization microscopy imaging system, operating in the reflection mode, as sketched in Fig. 2 [38]. Light from a lamp source was first passed through a polarizer and then driven trough the sample. The light reflected from the metallic layer at the cell bottom was passed through an analyzer and then collected by a long working distance zoom lens onto a CCD camera.

Sketch of setup for integrated calorimetric and polarization microscopy imaging evaluations
3 Results and Discussion
3.1 Measurements as a Function of Temperature
For the back-detection configuration, the expressions for the amplitude and phase when the sample is thermally thick are [39]:
where the subscripts s, p and b refer, respectively, to the sample, the pyroelectric transducer and the backing medium, P is the transducer pyroelectric coefficient, A the transducer area, I the absorbed light intensity, Ls the sample thickness, ω the angular frequency, τd= RC, and R and C, respectively, the effective transducer detection electronics resistance and capacitance. It can be observed that the signal amplitude and phase depend on such parameters like I and τd which are not easy to estimate accurately, so that a calibration procedure is required. One possibility would be to divide the signal obtained with the sample by that obtained by illuminating only the bare transducer, so that the factor between square brackets in the amplitude and the second term in the phase would not appear in the two expressions. Consequently, the phase would depend only on Ds, while the amplitude also on es, thus enabling the determination of cs and ks. In particular, the value of Ds can be obtained from the slope of the frequency scan of the phase data [9], or from thickness scans of the phase values at a single frequency as performed in liquid samples [40, 41].
When the temperature dependence of the thermal parameters is studied, a very practical normalization procedure consists in dividing the signal at the various temperatures T by that obtained at a reference temperature To where the values of the thermal parameters are known, yielding [7]:
so that Ds(T) and es(T) can both be determined.
3.2 Results in Liquids
Concerning the liquid samples, the studies over the phase transitions were carried out mostly in liquid crystals (LC) which present a great variety of transitions, but also in linear alkanes compounds. In LC samples, with respect to the isotropic (I) liquid state, the nematic (N) phase is characterized by an average common orientation of the molecules. At lower temperature, in the smectic A (A) phase the molecules are aligned parallel to each other and they are disposed in regularly spaced parallel planes, which are perpendicular to the molecular orientation direction, but there is no correlation in the molecular positions within the planes. At lower temperature still, the material eventually turns to the crystalline phase.
The various mesophases usually show up in multidomain samples, with the average molecular orientation being different in the various domains. The possibility to analyze single-domain samples would be of great usefulness particularly for those aspects related to molecular orientational order, molecular tilt angles and translational correlation length [42]. In samples exhibiting the nematic phase, a permanent uniform molecular alignment can be achieved by inducing an aligning agent on the cell walls [43]. For the samples with no N phase between the isotropic phase and the more ordered A phase, such a procedure proves useless and a new method was tested based on the application of a uniform strain field in the sample volume [LC] in order to produce an effect similar to that taking place in the case of the flow-induced alignment in nematics [44]. The strain perpendicular to the cell walls can be induced on the LC molecules by a small vertical displacement of the pyroelectric transducer [LC] wetting the LC sample. The achievement of the homeotropic single-domain alignment produced in the A phase can be verified, in the upgraded PPE calorimetric setup, both by polarization microscopy observations and by monitoring the thermal diffusivity which is known to show larger values for homeotropic samples than for non aligned focal conics ones [45]. This is related to the fact that the thermal transport parameters are largest when measured with the heat flow direction parallel to the molecular long axis [46].
The results of such an attempt are reported in Fig. 3 which displays the polarization microscopy patterns, obtained between crossed polarizers, when a 10 µm thick sample of 10CB LC, which changes from isotropic directly to the A phase, was first cooled from the isotropic to the A phase. Here it formed a multidomain focal conics texture [45], whose pattern is reported in Fig. 3a. Then, the vertical displacement is applied, in the smectic A phase, to the pyroelectric transducer achieving the non-birefringent homeotropic single domain (Fig. 3b). The successful outcome of the attempt is also certified by the thermal diffusivity values displayed in Fig. 3c. When the sample is cooled from the I to the A phase, the thermal diffusivity values decrease over the transition because of the very defective poly-domain focal conics structure [47]. When the sample is aligned into a homeotropic single domain, by the application of the “vertical” strain in the A phase near 45 °C, the thermal diffusivity increases up to its maximum values. The sample is then heated up to the isotropic phase where the thermal diffusivity decreases again. The described aligning procedure enabled to study the transition between the smectic A and the hexatic B (B) (where molecules have also hexagonal positional coordination within the smectic planes) phases in a single-domain sample of 65OBC LC for the first time [48]. It was thus possible to study of the critical behavior of the specific heat over the B-A transition in presence of different amount of disorder, namely maximum in a focal conic textured sample and minimum in a homeotropic single-domain one [48].

(a) Focal conic texture in the smectic A phase of 10CB liquid crystal; (b) non-birefringent polarization microscopy pattern obtained in homeotropically aligned smectic A phase; (c) thermal diffusivity values corresponding to situation in (a) (black symbols) and in (b) (gray symbols)
Another very interesting study which could be carried out in the upgraded PPE setup was that concerning the effect of the inclusion of photochromic molecules on the properties of the phases, and of the transitions between them, in 8CB LC [49]. The optical access to the sample enables the calorimetric and imaging evaluations to be performed, while the sample is irradiated with UV light to induce the structure change of the photochromic (naphthopyran) molecules. This is something which cannot be achieved by any of the existing calorimetric techniques. With the UV light off, the effect of the naphthopyran molecules hosted in the LC is to induce a homeotropic alignment in the LC volume in the A and N phases yielding relatively large values of the thermal conductivity as shown in Fig. 4b. The UV light is then switched on after the sample is taken in the isotropic phase from where it is cooled again down to the A phase. The effect of the change in the photochromic molecules conformation (more planar structure) upon UV light illumination is, in the first place, to shift the various phase transition temperatures to larger values as shown in the specific heat data in Fig. 4a. However, what it can also be observed is that the thermal conductivity decreases substantially as the 8CB/naphthopyran mixture is cooled from the nematic to the smectic phase. This can be associated with the loss of the homeotropic alignment of the LC molecules which could be readily be confirmed by the “focal conics” texture detected in the polarization microscopy observations (Fig. 4c). The loss of the alignment in the smectic phase can be attributed, upon their structure change induced by the UV light illumination, to the progressive inclusion of the photochromic molecules within the smectic planes, from an initial position between the planes when the UV light is off [50, 51]. This causes a disruptive effect on the LC alignment because the size and shape of the naphthopyran molecules are incompatible with those of the liquid crystal molecules to maintain a homeotropic single-domain smectic structure. The single-domain sample thus breaks up into a multidomain focal conics structure.

Specific heat (a) and thermal conductivity (b) in 8CB LC/naphthopyran mixture with UV light off (gray symbols) and on (black symbols). (c) Focal conics texture in the smectic phase with UV light on
A similar effect, but with very different timescale, occurs when the UV light is switched on when the sample is initially in the homeotropic A phase. The thermal conductivity then also decreases because, due to the disruptive effect by the illuminated naphthopyran molecules, but only about 5 h after UV switch-on time (Fig. 5a), this being due to the smectic material being much more viscous than the nematic one. This causes, with respect to the previous case, a much slower migration of the naphthopyran molecules into the smectic planes. Another important difference with respect to the previous case consists in the marked difference of the focal conics texture. In fact, when the focal conics texture nucleates from an initially aligned smectic phase, like the latter case, where the parallel plane orientation is uniform in all the samples, the so-called parabolic textures is obtained [52], as shown in Fig. 5b [49]. Different is the case where such a nucleation occurs as the sample is cooled over the NA transition. In fact, since no initial plane disposition can exist in the N phase, as the smectic phase is entered, the focal conic domains can nucleate with any orientation of the smectic planes giving rise to a texture like that shown in Fig. 4c.

(a) Thermal conductivity time dependence after UV light is switched on in the homeotropic smectic phase of the 8CB LC/naphthopyran mixture. (b) Parabolic focal conics texture after the loss of homeotropic alignment occurs
A final very recent example of the thermal parameters analysis by the upgraded PPE setup is that concerning the study of the transitions between the so-called rotator phases [53] in linear alkane sample. These are solid phases where the molecules possess three-dimensional crystalline order for their positions but no long range rotational order about the molecular long axis [54, 55]. The various rotator phases differ from one another in terms of side packing, molecular tilt, layer stacking and azimuthal ordering, and calorimetric data have been reported for samples with a number of carbon atoms in the molecules ranging between 23 and 30 (C23 to C30) [56, 57].
The particular aspect under investigation has been the hysteretic behavior, between the cooling and heating measuring runs of the sample, of the transition temperature between the various phases to help establish the order of the involved transitions. As an example, Fig. 6a, b reports the specific heat and thermal conductivity obtained in a heptacosane (C27) sample. Beside the liquid phase, three rotator phases, the so-called RIV, RIII and RV [58], were detected at lower temperatures, as well as two crystalline phases, the so-called Mdci and ODci ones [59]. A substantial hysteresis can be observed for the transition temperature between some of the occurring phase transitions. In the figures, the included vertical dotted lines, between the various phases, and the corresponding phases labels refer only to the cooling measurement.

(a) Specific heat and (b) thermal conductivity profiles for C27 in the range crystal phase to liquid phase. The black symbols correspond to the cooling runs and the gray symbols to the heating runs. The vertical dashed lines indicate the boundaries between the rotator phases during the cooling of the sample
An important issue is raised when the hysteretic behavior between the rotator phases is to be analyzed. In fact, from the polarization microscopy observations, performed in the upgraded PPE setup, it can be observed that the texture obtained at 56 °C in the RIV phase during the cooling run from the liquid phase (Fig. 7a) is markedly different from that obtained at the same temperature during the subsequent heating run from the crystalline phase (Fig. 7c). Specifically, the texture in the latter case contains elements which resemble those observed in the texture of the Mdci crystalline phase at 47 °C (Fig. 7b), with the possibility that it maintains memory of the grain structure of the crystalline phase. This may give rise to distortions in the RIV phase obtained in the heating run due to such grain constrains. An indication in this direction is provided by the smaller thermal conductivity values obtained during the heating of the sample than during the cooling, in the temperature range of the RV to RIV phases (Fig. 6b), since such a parameter is known to decrease with the presence of defects and strain in the material. Finally, when the heating measurements was performed by inverting the previous cooling run before reaching the crystalline phase, then the texture patterns in the RIV phase were shown to be the same as in the cooling runs. Hence, to prevent distortion in the material overshadowing the true characteristics of the transitions involved between the various rotator phases, the comparison between the results obtained during the heating and cooling measurements was obtained by cycling the sample only in the RV–RIV temperature range, without ever entering the crystalline phase. It could finally be established that the RIV-RIII and the RIII-RV transitions were, respectively, of first and of second order [60].

Polarization microscopy images performed in C27 obtained: (a) in the RIV phase during the first cooling run; (b) in the Mdci crystalline phase during the first cooling run; (c) in the RIV phase during the subsequent heating run
3.3 Results in Solids
Concerning solid samples, PPE calorimetric studies were mostly carried out over samples showing various magnetic phases, where the transitions between them occur over a very large temperature range. For this reason, a setup was developed and calibrated, able to perform measurements between 15 K and 380 K [61]. Using this kind of approach many investigations have been carried out in solids [62,63,64,65,66]. Of particular relevance is the systematic study over the paramagnetic–antiferromagnetic transition in a wide variety of manganite compounds, RMnO3, where R is a variable element, an investigation carried out over a very wide temperature range, as shown by the thermal diffusivity profiles reported in Fig. 8 [67]. There, the minima reflect the corresponding peaks in the heat capacity and therefore they identify the temperatures at which the transition occurs for the various compounds The rate with temperature with which the peaks in c diverge characterizes the analyzed transition [7]. The universality class [2] of the transition in the various compounds could thus be determined and compared with the predictions by theoretical models [67].

Thermal diffusivity profiles over the paramagnetic to antiferromagnetic phase transitions in several manganite compounds (RMnO3), where R represents all the elements displayed in the figure (from Ref. [67])
4 Absolute Measurements in Solids and Liquids
One of the challenges posed by the use of the PPE calorimetry is to find a way where the measurements of the thermal parameters can be determined in an “absolute” way, and that is to say, without the need to make any calibration of the setup other than that which could be performed during the actual measurements. In this context it must be remarked that, with solid samples, the thermal contact between the sample and the transducer is ensured by the application of a thin layer of coupling fluid between the two, the presence of which is not envisaged in the model leading to Eqs. 1 and 2. The effect of this layer on the evaluation of the sample thermal parameters is minimized by reducing as much as possible its thickness. Finding a way to eliminate completely such an effect would also be very useful. The first successful attempt to perform absolute measurements of the sample thermal diffusivity in solids, eliminating the coupling fluid influence, was obtained by performing the ratio of the PPE signals obtained in the BD and FD configurations [13]. According to a model, which takes into account also the coupling fluid layer, the expression for the phase of the BD/FD signal ratio is:
which coincides with the phase expression obtained by employing the BD only (the first term of Eq. 2), and it does not depend on any parameter associated with the coupling fluid or the pyroelectric transducer. The absolute determination of Ds can then be obtained by performing frequency scans of the signal phase obtained in this way. By doing so, it was found that the thermal diffusivity values were larger than the ones obtained previously for corresponding samples by the BD technique only, where the effect of the coupling fluid could be minimized but not eliminated altogether. This confirms the success in eliminating the effect of the coupling fluid by the BD/FD signal combination procedure. As for the absolute determination of the sample thermal effusivity, it was first obtained in liquid samples only, by combining the PPE signals obtained in the BD and FD configurations, where the sample was sandwiched between two transducer separated by a spacer of known thickness [23]. Very recently, a method was presented where the absolute measurements of thermal diffusivity and effusivity could be simultaneously performed by using an approach similar to that used in Ref. [35], but where the sample side, opposite that in contact with the transducer, was kept in contact with a medium (backing) of known, non-negligible thermal effusivity. In this way the expression of the phase of the ratio of the BD to FD signals yields [68]:
It can be seen that such a phase is independent from any parameter of the coupling fluid. It is also independent from the laser intensity and from the transducer thermal, electric and geometrical parameters. This implies that provided the backing material thermal effusivity is known, absolute measurement of the thermal diffusivity and effusivity of both liquids and solids is possible. In the large frequency range, the second term in Eq. 6 disappears, so that Ds can be separately evaluated in that range. In the low frequency range, from such a second term, es can also be determined. With this procedure absolute measurements of the thermal effusivity and thermal diffusivity were successfully performed for several solid and liquid samples [68].
5 Conclusions
In this work, the principles of the various photopyroelectric calorimetric configurations for the evaluations of the thermophysical parameters in liquids and in solids were briefly described. It was shown how the photopyroelectric calorimetry, in the back-detection configuration, enables the determination of the thermal diffusivity, the thermal conductivity and of the specific heat in a single measurement, thus making PPE very useful to characterize phase transitions over which the thermal parameters may show very sharp changes. A review of some recent results obtained over phase transitions to analyze the temperature dependence of c and k were illustrated, where both the static and the dynamic critical behaviors associated with the thermal parameters could be analyzed. Particular attention was placed on the results where simultaneous calorimetric and imaging evaluations, in an upgraded photopyroelectric setup calorimeter, were performed. Finally, the possibility of performing “absolute” measurements of the thermal parameters eliminating the influence of eventual coupling fluid has also been discussed.
References
B.K. Bein, J. Pelzl, Plasma Diagn. 6, 211 (1989)
L.P. Kadanoff, W. Goetze, D. Hamblen, R. Hecht, E.A.S. Lewis, V.V. Palciauskas, M. Rayl, J. Swift, D. Aspnes, J. Kane, Rev. Mod. Phys. 39, 395 (1967)
P.C. Hohenberg, B.I. Halperin, Rev. Mod. Phys. 49, 435 (1977)
M. Marinelli, F. Murtas, M.G. Mecozzi, U. Zammit, R. Pizzoferrato, F. Scudieri, S. Martellucci, M. Marinelli, Appl. Phys. A 51, 387 (1990)
M. Marinelli, F. Mercuri, U. Zammit, R. Pizzoferrato, F. Scudieri, D. Dadarlat, Phys. Rev. B 49, 9523 (1994)
M. Marinelli, F. Mercuri, U. Zammit, F. Scudieri, Phys. Rev. E 53, 701 (1996)
U. Zammit, M. Marinelli, F. Mercuri, S. Paoloni, F. Scudieri, Rev. Sci. Instrum. 82, 121101 (2011)
A. Oleaga, A. Salazar, E.H. Bocanegra, Phys. Rev. B 80, 024426 (2009)
A. Oleaga, A. Salazar, D. Prabhakaran, J.-G. Cheng, J.-S. Zhou, Phys. Rev. B 85, 184425 (2012)
C.C. Huang, J.M. Viner, J.C. Novack, Rev. Sci. Instrum. 56, 1390 (1985)
J.A. Balderas-Lòpez, A. Mandelis, Rev. Sci. Instrum. 74, 5219 (2003)
M. Kuriakose, M. Depriester, D. Dadarlat, A. Hadj, Sahraoui. Meas. Sci. Technol. 24, 025603 (2013)
A. Salazar, A. Oleaga, Rev. Sci. Instrum. 83, 014903 (2012)
F. Mercuri, S. Paoloni, N. Orazi, C. Cicero, U. Zammit, Appl. Phys. A 123, 317 (2017)
F. Mercuri, R. Gnoli, S. Paoloni, N. Orazi, U. Zammit, C. Cicero, M. Marinelli, F. Scudieri, Restaurator 34, 195 (2013)
N. Orazi, F. Mercuri, U. Zammit, S. Paoloni, M. Marinelli, A. Giuffredi, C.S. Salerno, Stud. Conserv. 61(4), 236–244 (2016)
F. Mercuri, N. Orazi, S. Paoloni, C. Cicero, U. Zammit, Appl. Sci. 7, 1010 (2017)
F. Mercuri, C. Cicero, N. Orazi, S. Paoloni, M. Marinelli, U. Zammit, Int. J. Thermophys. 36, 1189 (2015)
F. Mercuri, N. Orazi, U. Zammit, A. Giuffredi, S.C. Salerno, C. Cicero, S. Paoloni, J. Archaeol. Sci. Rep. 14, 199 (2017)
A. Mandelis, F. Care, K.K. Chan, L.C.M. Miranda, Appl. Phys. A 38, 117 (1985)
M. Marinelli, U. Zammit, F. Scudieri and S. Martellucci, in Proceedings of Nato Advanced Study Institute Phase Transitions in Liquid Crystals, Erice 2-12 May 1991, ed. by S. Martellucci and A.N. Chester, (NATO ASI Ser. B 290, Plenum, 1992), p. 189
M. Marinelli, U. Zammit, F. Mercuri, R. Pizzoferrato, J. Appl. Phys. 72, 1096 (1992)
U. Zammit, S. Paoloni, F. Mercuri, M. Marinelli, F. Scudieri, AIP Adv. 2, 012135 (2012)
S. Paoloni, F. Mercuri, U. Zammit, J. Chem. Phys. 145, 124506 (2016)
E.H. Bentefour, C. Glorieux, M. Chirtoc, J. Thoen, J. Chem. Phys. 120, 3726 (2004)
M. Marinelli, F. Mercuri, S. Paoloni, U. Zammit, Phys. Rev. Lett. 95, 237801 (2005)
N.O. Birge, Phys. Rev. B 34, 1631 (1986)
E.H. Bentefour, C. Glorieux, M. Chirtoc, J. Thoen, J. Appl. Phys. 93, 9610 (2003)
E.H. Bentefour, C. Glorieux, M. Chirtoc, J. Thoen, J. Chem. Phys. 120, 3726 (2004)
S. Paoloni, U. Zammit, M. Marinelli, F. Mercuri, R. Pizzoferrato, Rev. Sci. Instrum. 84, 054904 (2013)
J. Caerels, C. Glorieux, J. Thoen, Phys. Rev. E 65, 031704 (2002)
S. Pittois, B. Van Roie, C. Glorieux, J. Thoen, J. Chem. Phys. 121, 1866 (2004)
D. Dadarlat, C. Neamtu, Meas. Sci. Technol. 17, 3250 (2006)
D. Dadarlat, C. Neamtu, M. Streza, R. Turcu, I. Craciunescu, D. Bica, L. Vekas, J. Nanopart. Res. 10, 1329 (2008)
A. Salazar, A. Oleaga, V. Shvalya, E. Apiñaniz, Int. J. Therm. Sci. 100, 60 (2016)
F. Mercuri, M. Marinelli, S. Paoloni, U. Zammit, F. Scudieri, Appl. Phys. Lett. 92, 251911 (2008)
F. Mercuri, S. Paoloni, U. Zammit, F. Scudieri, M. Marinelli, Phys. Rev. E 74, 041707 (2006)
F. Mercuri, U. Zammit, F. Scudieri, M. Marinelli, Phys. Rev. E 68, 417081 (2003)
S. Delenclos, M. Chirtoc, A. Hadj Saharaoui, C. Kolinsky, J.M. Buisine, Rev. Sci. Instrum. 73, 2773 (2002)
S. Delenclos, D. Dadarlat, N. Houriez, S. Longuemart, C. Kolinsky, A. Hadj, Sahraoui. Rev. Sci. Instrum. 73, 2773 (2002)
D. Dadarlat, C. Neamtu, R. Pop, M. Marinelli, F. Mercuri, J. Optoelctron. Adv. Mater. 9, 2847 (2007)
J.M. Seddon, Structural studies of liquid crystals by X-Ray diffraction, in Handbook of Liquid Crystals, vol. 1, Fundamentals, ed. by J. Goodby, G.W. Gray, H.W. Spiess, V. Vill (Wiley, Weinheim, 1998), pp. 635–679
B.-Y. Kim, W.-K. Lee, Y.-H. Kim, H.-G. Park, P. Tae-Kyu, D.-S. Seo, Liq. Cryst. 38, 349 (2011)
K. Hongladarom, W.R. Burghardt, S.G. Baek, S. Cementwala, J. Magda, Macromolecules 26, 772 (1993)
Y. Bouligand, Defect and textures, in Handbook of Liquid Crystals, vol. 1, Fundamentals, ed. by J. Goodby, G.W. Gray, H.W. Spiess, V. Vill (Wiley, Weinheim, 1998), pp. 406–453
M. Marinelli, F. Mercuri, S. Foglietta, U. Zammit, F. Scudieri, Phys. Rev. E 54, 1604 (1996)
S. Paoloni, F. Mercuri, M. Marinelli, R. Pizzoferrato, U. Zammit, Liq. Cryst. 40, 1535 (2013)
F. Mercuri, S. Paoloni, M. Marinelli, R. Pizzoferrato, U. Zammit, J. Chem. Phys. 138, 074903 (2013)
S. Paoloni, F. Mercuri, M. Marinelli, R. Pizzoferrato, U. Zammit, T. Kosa, L. Sukhomlinova, B. Taheri, J. Chem. Phys. 143, 134901 (2015)
Y. Lansac, M.A. Glaser, N.A. Clark, O.D. Lavrentovich, Nature 398, 54 (1999)
B. Zalar, O.D. Lavrentovich, H. Zeng, D. Finotello, Phys. Rev. E 62, 2252 (2000)
C.S. Rosenblatt, R. Pindak, N.A. Clark, R.B. Meyer, J. Phys. 38, 1105 (1977)
D.M. Small, The Physics of Chemistry of Lipids: From Alkanes to Phospholipids (Plenum, New York, 1986)
G.R. Strobl, B. Ewen, E.W. Fisher, W.J. Piesczek, J. Chem. Phys. 61, 5257 (1974)
G. Ungar, N. Masic, J. Phys. Chem. 89, 1036 (1985)
E.B. Sirota, D.M. Singer, J. Chem. Phys. 101, 10873 (1994)
E.B. Sirota, H.E. King Jr., D.M. Singer, H.H. Shao, J. Chem. Phys. 98, 5809 (1993)
U. Zammit, M. Marinelli, F. Mercuri, S. Paoloni, J. Phys. Chem. B 114, 8134 (2010)
F. Rajabalee, V. Metivaud, D. Mondieig, Y. Haget, M.A. Cuevas-Diarte, J. Mater. Res. 14, 2644 (1999)
S. Paoloni, F. Mercuri, U. Zammit, J. Leys, C. Glorieux, J. Thoen, J. Chem. Phys. 148, 094503 (2018)
A. Oleaga, V. Shvalya, A.S. Sefat, A. Salazar, Int. J. Thermophys. 37, 4 (2016)
M. Marinelli, F. Mercuri, U. Zammit, R. Pizzoferrato, F. Scudieri, D. Dadarlat, Phys. Rev. B 49, 9523 (1994)
M. Marinelli, F. Mercuri, D.P. Belanger, Phys. Rev. B 51, 8897 (1995)
M. Marinelli, F. Mercuri, S. Foglietta, D.P. Belanger, Phys. Rev. B 54, 4087 (1996)
A. Salazar, M. Massot, A. Oleaga, A. Pawlak, W. Schranz, Phys. Rev. B 75, 224428 (2007)
M. Massot, A. Oleaga, A. Salazar, D. Prabhakaran, M. Martin, P. Berthet, G. Dhalenne, Phys. Rev. B 77, 1324438 (2008)
A. Oleaga, A. Salazar, D. Prabhakaran, J.-G. Cheng, J.-S. Zhou, Phys. Rev. B 85, 184425 (2012)
U. Zammit, F. Mercuri, S. Paoloni, M. Marinelli, R. Pizzoferrato, J. Appl. Phys. 117, 105104 (2015)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zammit, U., Mercuri, F., Paoloni, S. et al. Thermophysical Parameters Evaluation by Pyroelectric Detection. Int J Thermophys 39, 122 (2018). https://doi.org/10.1007/s10765-018-2443-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-018-2443-7