
Abstract
Apigenin, a flavonoid found in many plants, has various biological properties. We aimed to investigate the anti-inflammatory and anti-oxidative activity of apigenin against carbon tetrachloride (CCl4)-induced acute liver injury in mice and hydrogen peroxide (H2O2)-induced oxidative stress in HepG2 cells and possible mechanism. In vivo, apigenin significantly reduced alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity in serum of mice challenged by CCl4 and markedly alleviated the lipid peroxidation as indicated by the increased level of superoxide dismutase (SOD), reduced glutathione (GSH), glutathione peroxidases (GSH-Px) and catalase (CAT), and the decreased malondialdehyde (MDA) in liver tissue. Apigenin also ameliorated inflammation by downregulating tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and upregulating IL-10. Consistently, the elevated ALT and AST level; the impaired balance between SOD, GSH activity, and excessive ROS; and the increased gene expression of TNF-α and IL-6 resulting from H2O2-induced oxidative stress were restored by apigenin. Moreover, the results from Western blot, real-time qPCR, and immunofluorescence assay indicated that apigenin enhanced the activity of TNF receptor-associated factor (TRAF) 2/3 and cellular inhibitor of apoptosis protein (c-IAP) 1, ameliorated NF-κB-inducing kinase (NIK), and mediated the nuclear translocation of NF-κB2, therefore had an inhibitory effect on the non-canonical NF-κB pathway which was activated in both models. siNIK canceled the protective effect of apigenin on H2O2-induced HepG2 cells. Altogether, our results demonstrated that apigenin mitigated liver injury by ameliorating inflammation and oxidative stress through suppression of the non-canonical NF-κB pathway, indicating the potential of apigenin for treatment of the liver injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The liver is the largest parenchymatous organ and one of the most important organs in the human body [1]. It plays a vital role in many metabolic and clearance functions in order to maintain homeostasis and health, including the metabolism of carbohydrates, proteins and lipids, the uptake and detoxification of toxins and pathogens, and so on [2]. It is therefore foreseeable that the liver can be vulnerable to many xenobiotics or endogenously generated harmful substances, and liver diseases can be life-threatening [3]. Acute liver injury has been considered as an initiating factor and a common pathway to numerous liver diseases [4], and it is associated with a high degree of morbidity and mortality [5]. Thus, it is essential to explore a novel agent in terms of acute liver injury.
Carbon tetrachloride (CCl4) is one of the most widely used toxicant for inducing experimental acute liver injury in animals [6]. After ingestion, CCl4 is activated by cytochrome P450 (CYP450) in the liver into trichloromethyl radical (CCl3·), which readily reacts with oxygen to form trichloromethylperoxy radical (CCl3OO·). CCl3OO· initiates lipid peroxidation by oxidizing membrane polyunsaturated fatty acids and covalently binds with proteins and lipids to generate other reactive oxygen species (ROS) such as the superoxide radical [7]. Meanwhile, CCl4 inducing the expression of pro-inflammatory cytokines mediates the process of inflammation, which is also a contributing factor of CCl4-induced acute liver injury [8].
Initially thought to be mainly related to lymphocyte development and adaptive immunity, the non-canonical NF-κB pathway has recently been shown to also contribute to regulating liver inflammation, integrity, and function [9, 10]. NF-κB-inducing kinase (NIK), a central signaling component, triggers the phosphorylation of p100 (also known as NF-κB2) and causes its inducible processing to p52. Under quiescent conditions, the steady level of NIK is extremely low due to constant ubiquitylation and degradation facilitated by an ubiquitin ligase complex consists of TNF receptor-associated factor (TRAF) 2/3 and cellular inhibitor of apoptosis protein (c-IAP) 1/2 [11]. Upon activation through a select group of TNFR superfamily members [12,13,14], degradation of any component of this ubiquitin ligase complex will lead to accumulation of NIK, constitutive p100 to p52 processing and eventually nuclear translocation of RelB-p52 NF-κB complex. Hepatic NIK is reported to be a regulator of liver inflammation, oxidative stress, and liver injury [9, 15, 16]. The non-canonical NF-κB pathway can be considered a plausible drug target for alleviating CCl4-induced acute liver injury and H2O2-induced cell injury where oxidative stress and inflammation are implicated.
Apigenin is a flavonoid found at a significant level in a wide range of fruits, vegetables, and herbs [17]. Numerous efforts have been made to investigate apigenin’s anti-inflammatory [18], anti-oxidative [19], anti-apoptotic [20], and anti-tumor [10] effects. However, the effect of apigenin on CCl4-induced acute liver injury is yet to be discovered. Here, we explored the role of apigenin in CCl4-induced acute liver injury focusing on its effect on inflammation and oxidative stress, and the role of the non-canonical NF-κB pathway was investigated to elucidate the mechanism by which apigenin exerts its activity.
MATERIAL AND METHOD
Materials
Apigenin (> 98.0%) and bifendate (> 97%) were obtained from ALADDIN-E. COM. (Shanghai, P.R.C.). Alanine aminotransferase (ALT), aspartate aminotransferase (AST), superoxide dismutase (SOD), reduced glutathione (GSH), glutathione peroxidases (GSH-Px), catalase (CAT), and malondialdehyde (MDA) reagent kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, P.R.C.). BCA protein assay kit, ROS assay, radio immunoprecipitation assay (RIPA) lysis buffer, Trizol, and phenylmethanesulfonyl fluoride (PMSF) were obtained from Beyotime Biotechnology (Shanghai, P.R.C.). Mouse tumor necrosis factor-alpha (TNF-α) ELISA kit, mouse interleukin-6 (IL-6), and mouse IL-10 ELISA kit were provided by Neobioscience (Shenzhen, P.R.C.). Primary antibodies for TRAF2/3, c-IAP1, NIK, and RelB were purchased from Cell Signaling Technology, Inc. (Danvers, MA, US), NF-κB2 p100/p52 from Santa Cruz Biotechnology (Santa Cruz, CA, US), and β-actin from Bioworld Technology, Inc. (Nanjing, P.R.C.). PrimeScript™ RT Master Mix (Perfect Real Time) and TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) were provided by Takara Biomedical Technology (Beijing) Co., Ltd. (Dalian, P.R.C.).
Animal Experiment
Male ICR mice (18–22 g) were purchased from Weitong Lihua Experimental Animal Co. Ltd. (Beijing, P. R. C). Mice were bred with room temperature at 24 ± 2 °C, relative humidity 50–60%, and unlimited access to fodder and water. After 7 days of adaptation to the environment, the mice were randomly divided into 6 groups (n = 6), among which were the control group; model group; apigenin 50, 100, and 200 mg/kg groups; and bifendate group. Control and model groups were orally administrated with the vehicle (0.5% CMC-Na, 10 mL/kg), apigenin groups were administrated with the corresponding drugs by gavage (50, 100, and 200 mg/kg, dissolved in 0.5% CMC-Na, 10 mL/kg), and the bifendate group was administrated with bifendate (100 mg/kg, dissolved in 0.5% CMC-Na, 10 mL/kg). After 7 days of administration, the CCl4-induced acute liver injury model was established 2 h after the last administration. The control group was intraperitoneally treated with the vehicle (olive oil, 25 mL/kg), and the rest of the groups with CCl4 (2.5 mL/kg, 10% in olive oil). All mice were sacrificed 12 h after the exposure to CCl4, and blood samples and liver tissue were collected for further investigation. All animal experiment was approved by the Institutional Animal Care and Use Committee of China Pharmaceutical University.
Cell Culture
The human liver carcinoma cell line HepG2 (obtained from Shanghai Institute of Biochemistry and Cell Biology) were cultured in Dulbecco’s modified eagle medium (DMEM) with 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 μg/mL). The cells were incubated under 37 °C with 5% CO2 and 100% relative humidity.
ROS Assay
The level of ROS in HepG2 cells was identified using 2,7-dichloroflurescein diacetate (DCFH/DA) probe. In brief, HepG2 cells were seeded in 6-well plate at a density of 4 × 105 cells/well. The control and model groups (n = 3) were added DMEM with 10% FBS, and the apigenin groups (10, 20, and 40 μM) and the bifendate group (10 μM) were pretreated with respective drugs (diluted in DMEM with 10% FBS). After 24-h incubation, the medium of all cells other than the control group was replaced by 800 μM H2O2 (diluted in DMEM) and the control group by DMEM. After being exposed to H2O2 for 24 h, the medium was aspirated and the cells were rinsed once with phosphate-buffered saline (PBS). DCFH/DA (10 μM, diluted in DMEM) was added to each well and the cells were incubated at 37 °C for 30 min shielded from light. The probe was washed off with PBS 3 times, and the fluorescence intensity of each well was observed with the inverted fluorescence microscope (Nikon, Tokyo, Japan).
Measurement of ALT and AST
The quantities of ALT and AST in both serum and cells’ medium supernatant were identified using commercial chemical assay kits according to the manufacturers’ directions.
Measurement of MDA, SOD, GSH, GSH-Px, and CAT
The quantities of MDA, SOD, GSH, GSH-Px, and CAT in livers and SOD and GSH in cells were detected using commercial chemical assay kits. In brief, livers and cells were homogenized with normal saline or RIPA lysis buffer respectively and then centrifuged for 10 min (3500×g) at 4 °C. The indexes above were detected following the manufacturers’ directions.
Histopathological Analysis
Liver tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sliced into 5-μm sections. Then, the sections were dewaxed with xylene and rehydrated in a graded series of ethanol, followed by staining with hematoxylin and eosin. To evaluate the severity of CCl4-induced liver injury, histological examination under a microscope and analysis of necrotic area were performed in a double-blinded manner by independent investigators who was unaware of the treatment conditions made histological assessments.
ELISA Assay
The quantities of TNF-α and IL-6 in the liver were determined by ELISA assay. Fifty microliters of liver lysate and positive control was added to the 96-well plate and incubated at 37 °C for 90 min. The lysate was aspirated and the wells were rinsed 5 times. One hundred microliters of detection antibody was added in each well, and the plate was sealed and incubated at 37 °C for 60 min. The washing procedure was repeated. One hundred microliters of HRP-linked secondary antibody was added in each well, and the plate was sealed and incubated at 37 °C for 30 min. The washing procedure was repeated. One hundred microliters of TMB substrate was added in each well and the plate was sealed and incubated at 37 °C for 15 min. One hundred microliters of STOP solution was added in each well and the plate was gently shook for a few seconds. The absorbance was read at 450 nm.
Western Blot
The expression of TRAF2/3, c-IAP1, NIK, p100, and p52 in the liver was estimated by Western blot. The liver samples of all groups (n = 3) were homogenized in RIPA lysis buffer with 1 mM PMSF and phosphatase inhibitor cocktail. The homogenate was centrifuged for 10 min (12,000×g) at 4 °C, and the supernatant where the protein existed was kept. The loading buffer was added and the samples were heated at 99 °C for 5 min. The protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride (PVDF). After the transfer, the membranes were incubated in 5% w/v bovine serum albumin (BSA) for 2 h at room temperature and then in primary antibodies (diluted as recommended) overnight at 4 °C. When the incubation with primary antibodies was done, the membranes were washed with TBST 3 times for 10 min each. The membranes were incubated with horseradish peroxidase (HRP)-linked antibody for 2 h at room temperature and then, the secondary antibody was washed off as before. ECL plus was added on the membrane and the expression of protein was identified by ChemiDoc™ XRS+ System (Bio-Rad Laboratories, Inc., CA, US) and analyzed by Image J.
Real-Time qPCR
Total RNA in cells and livers were extracted using Trizol reagent, and the first-strand complementary DNA (cDNA) was synthesized with PrimeScript RT Master Mix, followed by intercalator qPCR assay using TB green Premix Ex Taq II. The primers for real-time qPCR (RT-qPCR) were listed in Table 1. The expression of target genes was normalized to the expression of GAPDH and analyzed by the 2−△△ct method.
Immunofluorescence Assay
Cells were rinsed with PBS for 3 times and fixed with formaldehyde for 30 min, followed by permeabilized treatment with 0.3% Triton X-100 for 20 min. Five percent of BSA was used to block the cells for 1 h, and cells were incubated with primary antibody overnight. After washing with PBS for 3 times, the cells were incubated with FITC-conjugated secondary antibody for 2 h and then Hoechst for 30 min. Images were obtained by an inverted fluorescence microscope (Nikon, Tokyo, Japan).
3-(4,5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide Assay
After pretreatment of apigenin and administration of H2O2, 20 μL 5 mg/mL MTT was added and incubated with the cells for 4 h shielded from light. One hundred fifty microliters of dimethyl sulfoxide was added and the absorbance was detected at 490 nm.
RNA Interference
HepG2 cells were transfected with 50 nM NIK small interfering RNA (siRNA) and scrambled siRNA for negative control using lipofectamine 6000 transfection reagent (Beyotime Biotechnology, Shanghai, China) for 6 h according to the manufacturers’ directions. The knockdown efficiency was identified by RT-qPCR 48 h after the transfection. The concentration of apigenin used in the transfection experiment was 40 μM.
Statistical Analysis
Statistical analysis was performed on Software SPSS 19.0 (SPSS version 19.0, SPSS Inc., Chicago, US). All experimental data were presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) followed by least significant difference (LSD) test was used. p < 0.05 was considered statistically significant.
RESULT
Apigenin Alleviated CCl4-Induced Acute Liver Injury
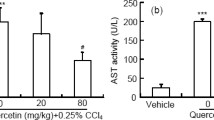
We first established CCl4-induced acute liver injury by intraperitoneal administration of CCl4 (2.5 mL/kg) for 12 h after pretreatment of apigenin with a different concentration for 7 days in mice. CCl4 was a classic and well-established model for acute liver injury [21], and raised ALT and AST activities in serum, as well as histopathological analysis, have been recognized as a marker for liver damage. Multifocal hepatic parenchymal necrosis was observed in CCl4-treated mice. On the contrary, apigenin and bifendate markedly ameliorated the pathological changes (Fig. 1a). Moreover, we found significantly increased serum ALT and AST levels in mice treated with CCl4, compared with the control group. However, apigenin and bifendate markedly reversed this change (Fig. 1b and c), which indicated that apigenin could inhibit liver injury induced by CCl4.

Apigenin attenuated liver injury induced by CCl4 in vivo. Mice were pre-treated with a different concentration of apigenin (50, 100, and 200 mg/kg) for 7 days, followed by 12-h dosing of CCl4 (n = 6). Histopathological examination and analysis of necrosis area were used to determine the liver injury (a), and ALT and AST in serum were measured (b). Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
Apigenin Reduced Oxidative Stress Induced by CCl4 in Mice
Although the mechanism through which CCl4 exerts its effect of liver injury is yet to determine, researchers have pointed out that liver damage may be induced by free radical metabolites [22], which was associated with oxidative stress. It has been reported that CCl4 treatment could lead to lipid peroxidation, thereby compromising the antioxidant system. MDA, the end product of lipid peroxidation, can be used as a marker to determine the severity of this process. On the other hand, the level of endogenous radical scavengers, such as SOD, CAT, and GSH-Px, the antioxidant enzymes, and GSH, non-enzyme molecules, indicates the damage degree of the antioxidant system. SOD, CAT, and GSH-Px activities as well as GSH level in the liver tissue of the model group were significantly decreased in comparison with the control group (Fig. 2a–d). In addition, the MDA level of the model group was dramatically upregulated (Fig. 2e). On the contrary, compared with the model group, increased level of SOD, CAT, GSH-Px, and GSH was observed in mice treated with a different concentration of apigenin and bifendate, as well as alleviated MDA level (Fig. 2a–e). Taken together, the results implied that apigenin could restore the antioxidant activity and suppress lipid peroxidation in mice, therefore inhibiting oxidative stress induced by CCl4.

Apigenin mitigated oxidative stress and inflammation induced by CCl4 in vivo. SOD (a), CAT (b), GSH-Px (c), GSH (d), and MDA (e) in liver tissue were measured for identifying the degree of oxidative stress. Pro-inflammatory cytokines TNF-α (f), IL-6 (g), and anti-inflammatory cytokine IL-10 (h) were detected using ELISA for inflammation. Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
Apigenin Ameliorated CCl4-Induced Inflammation in Mice
In addition to oxidative stress induced by CCl4, activated inflammatory response also causes liver injury in CCl4-induced hepatotoxin. Free radicals not only cause liver damage through oxidative stress but also initiate inflammation, via recruiting and activating resident macrophages (in the case of liver tissue, Kupffer cells) that produce and release pro-inflammatory mediators, such as TNF-α and IL-6 [4]. In our study, ELISA assay was used to detect the content of pro-inflammatory cytokines: TNF-α, IL-6, and anti-inflammatory cytokine IL-10 in the liver. The content of TNF-α, IL-6, and IL-10 in the model group was substantially upregulated compared with the control group (Fig. 2f–h). However, after pretreatment with a different concentration of apigenin and bifendate, a significant reduction of those pro-inflammatory cytokines and an increase of the anti-inflammatory cytokine were observed in comparison with the model group (Fig. 2f–h). The results indicated that apigenin alleviated inflammatory response induced by CCl4.
Apigenin Inhibited the Non-canonical NF-кB Pathway in CCl4-Induced Acute Liver Injury
The hepatoprotective activity of apigenin in vivo via anti-oxidation and anti-inflammation was confirmed, but the mechanism was yet to discover. It has been reported that hepatic NIK was identified as a novel trigger of the liver inflammation and destruction program under pathological conditions like drugs or other toxins exposure and oxidative stress [9]. Therefore, gene expression and protein levels were investigated to determine the effect of apigenin on the non-canonical NF-кB pathway.
In the resting state, an ubiquitin ligase complex containing TRAF2/3 and c-IAP1/2 induces the ubiquitination and subsequent degradation of a crucial factor, NIK. Upon stimulation, the complex undergoes signal-dependent self-degradation, leading to the accumulation of NIK and consequent activation of the non-canonical NF-κB pathway. In our study, we found a decline in the protein level of those negative regulators, including TRAF2/3 and c-IAP1, after CCl4 administration, as well as an increase in NIK, indicating the activation of the non-canonical pathway induced by CCl4. In contrast, compared with the model group, the TRAF2/3 and c-IAP1 levels in the groups treated with a different concentration of apigenin increased significantly, and the NIK level decreased dramatically (Fig. 3a). Unlike the canonical NF-кB pathway which relies on inducible degradation of IкBα and nuclear translocation of the p50/RelA dimer [23], the non-canonical pathway depends on phosphorylation-induced p100 processing and activation of p52/RelB complex [24]. Consistently, our results showed that CCl4 treatment markedly increased the transformation from p100 to p52 and the amount of RelB in comparison with the control group, indicating the activation of the non-canonical NF-кB pathway in CCl4-induced acute liver injury in mice. Nevertheless, apigenin pretreatment substantially restored the variations (Fig. 3a). In conformity with Western blot, real-time qPCR results showed an increased mRNA level of NIK and NF-κB2 in CCl4-induced mice. On the contrary, compared with the model group, the mRNA level of NIK and NF-κB2 significantly decreased after apigenin pretreatment (Fig. 3b and c). These observations suggested that apigenin might block the non-canonical NF-кB pathway to protect against CCl4-induced acute liver injury.

Apigein had an inhibitory effect on the non-canonical NF-κB pathway in CCl4-induced liver injury. The protein level of TRAF2, TRAF3, c-IAP1, NIK, RelB, p100 and p52 in liver tissue were detected (a). The mRNA level of NIK (b) and NF-κB2 (c) in liver tissue were measured. Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
Apigenin Reduced H2O2-Induced HepG2 Cell Injury
H2O2, a reactive non-radical, can permeate cytoplasmic membranes and be converted to more reactive species such as hydroxyl radical and singlet oxygen. H2O2 is the precursor for the generation of hydroxyl radical, which is a strong initiator of lipid peroxidation [25]. H2O2-induced oxidative stress in HepG2 cells has been used to verify the efficacy and mechanism of new agents. In our study, the in vitro model was designed by exposing HepG2 cells to H2O2 to validate the finding of the protective effect of apigenin in vivo. The cell viability was tested by MTT assay and the level of ALT and AST in medium supernatant was measured as indicators of cell function. Compared with the vehicle-treated cells, a significant decrease of cell viability and substantial ALT and AST level elevation in the medium of H2O2-treated cells was observed. On the contrary, upregulated cell viability and reduced level of ALT and AST were detected in the group treated with apigenin and bifendate (Fig. 4a and b), indicating the cytoprotective effect of apigenin pretreatment.

Apigenin alleviated H2O2-induced oxidative stress and inflammation in vitro. HepG2 cells were treated with a different concentration of apigenin (10, 20, and 40 μM) for 24 h, followed by 24-h exposure to H2O2 (n = 3). Cell viability after pretreatment of apigenin and exposure to H2O2 was detected by MTT assay (a). ALT and AST (b) in medium supernatant were measured. SOD (c), GSH (d), TNF-α (e) and IL-6 (f) in cells were detected. Fluorescent of intracellular ROS was identified (g). Scale bars represent 50 μM. Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
Apigenin Ameliorated Oxidative Stress Induced by H2O2in vitro
The quantities of SOD and GSH in HepG2 cell induced with H2O2 were tested, as well as the intracellular ROS level. A remarkable decrease of SOD and GSH in the cell lysate of the H2O2-treated cells was observed, compared with the vehicle-treated cells (Fig. 4c and d). And, the intracellular ROS level was significantly raised after H2O2 exposure (Fig. 4g). However, apigenin pretreatment of a different concentration, as well as bifendate, markedly enhanced the activity of SOD and GSH and alleviated the intracellular ROS level (Fig. 4c, d, and g). These findings suggested that apigenin had a cytoprotective effect against H2O2-induced oxidative stress in HepG2 cells.
Apigenin Alleviated H2O2-Induced Upregulated Expression of Pro-inflammatory Cytokines
The gene expression of pro-inflammatory cytokines was identified to determine the anti-inflammatory activity of apigenin in vitro. A remarkable increase in TNF-α and IL-6 mRNA levels was detected in cells treated with H2O2 compared with the vehicle-treated group. Contrariwise, the pretreatment of apigenin and bifendate significantly reduced the gene expression of TNF-α and IL-6 (Fig. 4e and f), revealing the anti-inflammatory effect of apigenin against H2O2-induced cell injury.
Apigenin Inhibited the Expression of NIK and NF-κB2 in H2O2-Induced Cell Injury
To determine the effect of apigenin on H2O2-induced oxidative stress, the protein level of NF-κB2 was detected by immunofluorescence assay, and the mRNA level of NIK and NF-κB2 was identified using real-time qPCR. Cells exposed to H2O2 demonstrated showed increased NF-κB2 (green) colocalizing with the nuclear stain (Hoechst, blue) compared with the control group (Fig. 5a), and H2O2-induced cell injury largely triggered the gene expression of NIK and NF-κB2 (Fig. 5b and c), which indicated that H2O2-induced oxidative stress activated the non-canonical NF-κB pathway. On the other hand, apigenin pretreatment of a different concentration for 24 h markedly reduced NF-κB2 nuclear colocalization (Fig. 5a) and suppressed gene expression of NIK and NF-κB2 (Fig. 5b and c). These data indicated that apigenin might carry out its cytoprotective effect against H2O2-induced oxidative stress via inhibiting the activity of NIK and NF-κB2.

Apigenin reduced NIK and NF-κB2 expression in H2O2-induced HepG2 cells. HepG2 cells were stained with a monoclonal antibody against NF-κB2 p100/p52 (green) and Hoechst (blue) nuclear stain. Scale bars represent 50 μM (a). The mRNA level of NIK (b) and NF-κB2 (c) in HepG2 cells were measured. Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
The Effect of Apigenin Was Abolished in the Absence of NIK
To further verify the involvement of the non-canonical NF-κB pathway in the protective effect of apigenin, HepG2 cells were transfected with siNIK to silence NIK mRNA and inhibit the pathway. The ROS level and mRNA level of pro-inflammatory cytokines were observed to determine whether the anti-oxidative and anti-inflammatory activity were abolished when NIK was absent. The result from RT-qPCR showed that the transfection was successful (Fig. 6a). Same as demonstrated before, the ROS level was significantly increased after the exposure to H2O2 and pretreatment of apigenin can reverse the situation (Fig. 6b). However, the knockdown of NIK can significantly decrease the level of ROS and cancel the anti-oxidative effect of apigenin (Fig. 6b). In addition, silencing NIK can remarkably downregulate the mRNA level of TNF-α and IL-6 and eliminate the anti-inflammatory activity of apigenin (Fig. 6c and d). These results showed that apigenin functioned dependently on the non-canonical NF-κB pathway.

The effect of apigenin was abolished by the transfection of siNIK. To explore the mechanism underlying the activity of apigenin, siNIK was used. The mRNA level of NIK after the transfection was detected (a). The level of ROS in HepG2 cells was identified (b). (a) Control, (b) model, (c) H2O2 + apigenin, (d) H2O2+ siNIK, (f) H2O2+ siNIK + apigenin. The mRNA level of TNF-α (c) and IL-6 (d) was determined. Scale bars represent 50 μM. Data are expressed in the form of means ± SD. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the model group.
DISCUSSION
In this study, we investigated the hepatoprotective effect of apigenin against CCl4-induced acute liver injury in mice and H2O2-induced oxidative stress in HepG2 cells. And, we further investigated its anti-oxidative and anti-inflammatory activity and explored the underlying possible mechanism. The results we got confirmed that apigenin exerted its hepatoprotective effect in vivo and in vitro through anti-oxidation and anti-inflammation via inhibiting the non-canonical NF-κB pathway.
Hepatocellular damage and the subsequent disruption of the plasma membrane induced by CCl4 cause the release of the content of hepatocytes including intracellular enzymes such as ALT and AST into the extracellular space [26]. The released enzymes ultimately enter into circulation and thereby increase the serum levels of ALT and AST [27]. The level of serum ALT and AST activity reflects damage to hepatocytes and is considered to be a highly sensitive and fairly specific preclinical and clinical biomarker of hepatotoxicity [27]. Our study showed pretreatment of apigenin restrained ALT and AST levels in serum in mice challenged by CCl4, which implies the repressed damage of liver cells and restoration of the cell membrane function induced by apigenin.
Inflammation underlies a wide variety of physiological and pathological processes. It has been widely appreciated that inflammation is a double-edged sword. On the one hand, it plays a vital role in the initial host defense against many infections. On the other hand, excessive activation of inflammatory cytokines promotes the pathogenesis of various liver diseases [28, 29]. In the liver, inflammation is particularly important because it underlies the pathogenesis of a number of diseases, such as alcoholic and nonalcoholic steatohepatitis, ischemia/reperfusion (I/R), liver injury, and cirrhosis [28]. A growing number of studies have implicated cytokines and cytokine-dependent pathways in the development of various liver diseases including liver failure and hepatic inflammation [30]. CCl4-induced acute liver injury is characterized by systemic and local inflammation with the recruitment of macrophages and neutrophils into the liver vasculature [31]. Elevation of pro-inflammatory cytokines like TNF-α and IL-6 is strongly associated with liver injury [32]. In the present study, we demonstrated that apigenin markedly restored the dysregulated TNF-α and IL-6 in mice challenged by CCl4, indicating the anti-inflammatory effect of apigenin in vivo.
The condition of oxidative stress results from the imbalance between antioxidant defense mechanisms and ROS [33]. Liver diseases are nearly always characterized by increased oxidative stress induced by excessive ROS. Oxidative stress is a major molecular event triggered by CCl4 [34]. After activation by CYP450, CCl3· is produced and converted into CCl3OO· in the presence of oxygen, which is more reactive than the former and far more likely to abstract a hydrogen from polyunsaturated fatty acids, thereby initiating the process of lipid peroxidation [6]. MDA accumulated in the liver, the final product of lipid peroxidation, can be detected to estimate the severity of CCl4-induced lipid peroxidation. CAT, SOD, and GSH-Px enzymes constitute the primary part of the enzymatic antioxidant defense system against oxidative stress [35]. The decrease in those antioxidant enzymes and an antioxidant molecule GSH after CCl4 administration can serve as the hallmark of oxidative injury in the liver. In our study, the pretreatment of apigenin significantly repressed the increase of MDA in the liver induced by CCl4 and markedly enhanced SOD, CAT, GSH, and GSH-Px activity. The results indicated that apigenin can restore the balance between accumulation of ROS and antioxidant defense mechanism, and the hepatoprotective effect of apigenin partly results from attenuating oxidative stress.
HepG2 retains many of the phenotypic and genotypic characteristics of liver cells; therefore, this cell line has been used in various studies concerning traditional medicines for their hepatoprotective effect [36]. In our study, we demonstrated the protective effect of apigenin against H2O2-induced oxidative stress in HepG2 cells. Substantial decrease of ALT and AST in medium supernatant after the pretreatment of apigenin was observed. The anti-inflammatory activity of apigenin was shown by the diminished mRNA level of TNF-α and IL-6 compared with the model group. And, the balance between excessive ROS and antioxidants was restored as the increased SOD and GSH level and attenuate ROS level were identified.
In order to explore the potential mechanism underlying the hepatoprotective effect of apigenin, the non-canonical NF-κB pathway was probed by Western blot and real-time qPCR. Recently, the non-canonical NF-κB pathway has been shown to play an important role in liver injury [9, 15]. Abnormal activation of hepatic NIK is involved in many liver diseases, such as nonalcoholic fatty liver disease (NAFLD), liver steatosis [37], liver injury, and fibrosis [9]. In resting cells, the level of NIK is extremely low due to ubiquitination and degradation completed by an ubiquitin E3 ligase complex containing TRAF2 and cIAP1/2 [38]. And, TRAF3 acts as an adaptor that binds to NIK and recruits NIK to the complex [39]. NIK can be activated by many stimuli, including oxidative stress, cytokines, and endotoxins [9]. Upon activation, stimulation causes TRAF3 degradation, thus uncoupling NIK from the ubiquitin E3 ligase complex and leading to the accumulation of NIK [40]. As the crucial component of non-canonical NF-κB pathway, NIK further induces p100 processing by phosphorylation and ubiquitylation via the activation of the IκB kinase α (IKKα) [41], leading to the generation of p52 and the nuclear translocation of p52 and RelB [38]. It has been reported that a high level of NIK mRNA was detected in CCl4-induced acute liver injury [9, 15]. And, inhibition of NIK by a small-molecule inhibitor B022 completely blocked CCl4-induced nuclear translocation of p52 and had a protective effect against liver injury [15]. It has been shown that the non-canonical NF-κB pathway was strongly associated with CCl4-induced acute liver injury. In this study, we observed reduction in the protein level of negative regulators, TRAF2/3 and cIAP1, accumulation of NIK, and increased transformation from p100 to p52 after CCl4-induced acute liver injury, which indicated that toxin like CCl4 may mediate injury via activating the non-canonical NF-κB pathway. In addition, increased colocalization of NF-κB2 and the nuclear stain and enhanced expression of NIK and NF-κB2 mRNA was identified in H2O2-induced HepG2 cells, implying that oxidative stress activated the accumulation of NIK and the nuclear translocation of p52, which was associated with the increased activity of the non-canonical NF-κB pathway. However, apigenin was shown to restore all the dysregulation, revealing its inhibitory effect on the non-canonical NF-κB pathway. Moreover, silencing NIK by siNIK transfection abolished the anti-inflammatory and anti-oxidative activity of apigenin against H2O2-induced oxidative stress in HepG2 cells, demonstrating that apigenin exerted its protective effect via inhibiting the non-canonical NF-κB pathway.
CONCLUSION
In summary, our study demonstrated the hepatoprotective activity of apigenin in CCl4-induced acute liver injury in mice and H2O2-induced oxidative stress in HepG2 cells. This effect can be attributed to its antioxidant and anti-inflammatory activities through inhibiting the non-canonical NF-κB pathway. All in all, our study provided evidence for the potential of apigenin to be a therapeutic agent for liver injury.
References
Bhakuni, Ganesh Singh, Onkar Bedi, Jitender Bariwal, Rahul Deshmukh, and Puneet Kumar. 2016. Animal models of hepatotoxicity. Inflammation Research 65. Springer International Publishing: 13–24. https://doi.org/10.1007/s00011-015-0883-0.
Protzer, Ulrike, Mala K. Maini, and Percy A. Knolle. 2012. Living in the liver: hepatic infections. Nature Reviews Immunology 12. Nature Publishing Group: 201–213. https://doi.org/10.1038/nri3169.
Sun, B., and M. Karin. 2008. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene 27: 6228–6244. https://doi.org/10.1038/onc.2008.300.
Torres, Lucillia R. de O., Fernanda C. de Santana, Francisco L. Torres-Leal, Illana L.P. de Melo, Luciana T. Yoshime, Emidio M. Matos-Neto, Marília C.L. Seelaender, Cintia M.M. Araújo, Bruno Cogliati, and Jorge Mancini-Filho. 2016. Pequi (Caryocar brasiliense Camb.) almond oil attenuates carbon tetrachloride-induced acute hepatic injury in rats: Antioxidant and anti-inflammatory effects. Food and Chemical Toxicology 97. Pergamon: 205–216. https://doi.org/10.1016/J.FCT.2016.09.009.
Huang, Cheng, Yang Yang, Wan-Xia Li, Xiao-Qin Wu, Xiao-Feng Li, Tao-Tao Ma, Lei Zhang, Xiao-Ming Meng, and Jun Li. 2015. Hyperin attenuates inflammation by activating PPAR-γ in mice with acute liver injury (ALI) and LPS-induced RAW264.7 cells. International Immunopharmacology 29: 440–447. https://doi.org/10.1016/j.intimp.2015.10.017.
Weber, Lutz W.D., Meinrad Boll, and Andreas Stampfl. 2003. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Critical Reviews in Toxicology 33.Taylor & Francis: 105–136. https://doi.org/10.1080/713611034.
Rashid, Kahkashan, Krishnendu Sinha, and Parames C. Sil. 2013. An update on oxidative stress-mediated organ pathophysiology. Food and Chemical Toxicology 62. Pergamon: 584–600. https://doi.org/10.1016/J.FCT.2013.09.026.
Abouzied, Mekky M., Heba M. Eltahir, Ashraf Taye, and Mahran S. Abdelrahman. 2016. Experimental evidence for the therapeutic potential of tempol in the treatment of acute liver injury. Molecular and Cellular Biochemistry 411. Springer US: 107–115. https://doi.org/10.1007/s11010-015-2572-2.
Shen, Hong, Sheng Liang, Chen Zheng, Jiang Lin, Su Haoran, Lei Yin, M. Bishr Omary, and Liangyou Rui. 2014. Mouse hepatocyte overexpression of NF-κB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology 60. John Wiley & Sons, Ltd: 2065–2076. https://doi.org/10.1002/hep.27348.
Ai, Xiao-Yu, Yuan Qin, Hui-Jua Liu, Zhan-Hong Cui, Meng Li, Jia-Huan Yang, Wei-Long Zhong, et al. 2017. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 8. Impact Journals, LLC: 100216–100226. https://doi.org/10.18632/oncotarget.22145.
Zarnegar, Brian J., Yaya Wang, Douglas J. Mahoney, Paul W. Dempsey, Herman H. Cheung, Jeannie He, Travis Shiba, et al. 2008. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nature Immunology 9: 1371–1378. https://doi.org/10.1038/ni.1676.
Dejardin, Emmanuel, Nathalie M. Droin, Mireille Delhase, Elvira Haas, Yixue Cao, Constantin Makris, Zhi-Wei Li, Michael Karin, Carl F. Ware, and Douglas R. Green. 2002. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 17: 525–535.
Claudio, Estefania, Keith Brown, Sun Park, Hongshan Wang, and Ulrich Siebenlist. 2002. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nature Immunology 3: 958–965. https://doi.org/10.1038/ni842.
Coope, H.J., P.G.P. Atkinson, B. Huhse, M. Belich, J. Janzen, M.J. Holman, G.G.B. Klaus, L.H. Johnston, and S.C. Ley. 2002. CD40 regulates the processing of NF-kappaB2 p100 to p52. The EMBO Journal 21: 5375–5385. https://doi.org/10.1093/emboj/cdf542.
Ren, Xiaomeng, Xinzhi Li, Linna Jia, Deheng Chen, Hai Hou, Liangyou Rui, Yujun Zhao, and Chen Zheng. 2017. A small-molecule inhibitor of NF-κB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. The FASEB Journal 31. Federation of American Societies for Experimental BiologyBethesda, MD, USA: 711–718. https://doi.org/10.1096/fj.201600840R.
Jiang, Bijie, Hong Shen, Chen Zheng, Lei Yin, Linsen Zan, and Liangyou Rui. 2015. Carboxyl terminus of HSC70-interacting protein (CHIP) down-regulates NF-κB-inducing kinase (NIK) and suppresses NIK-induced liver injury. The Journal of Biological Chemistry 290. American Society for Biochemistry and Molecular Biology: 11704–11714. https://doi.org/10.1074/jbc.M114.635086.
Lefort, Émilie C., and Jonathan Blay. 2013. Apigenin and its impact on gastrointestinal cancers. Molecular Nutrition & Food Research 57: 126–144. https://doi.org/10.1002/mnfr.201200424.
Zhou, Qian, Hui Xu, Wenzhe Yu, Edmund Li, and Mingfu Wang. 2019. Anti-inflammatory effect of an apigenin-Maillard reaction product in macrophages and macrophage-endothelial cocultures. Oxidative Medicine and Cellular Longevity 2019. Hindawi Limited: 9026456. https://doi.org/10.1155/2019/9026456.
Ali, Fahad, Falaq Naz Rahul, Smita Jyoti, and Yasir Hasan Siddique. 2014. Protective effect of apigenin against N-nitrosodiethylamine (NDEA)-induced hepatotoxicity in albino rats. Mutation Research, Genetic Toxicology and Environmental Mutagenesis 767: 13–20. https://doi.org/10.1016/j.mrgentox.2014.04.006.
Balez, Rachelle, Nicole Steiner, Martin Engel, Sonia Sanz Muñoz, Jeremy Stephen Lum, Yizhen Wu, Dadong Wang, et al. 2016. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Scientific Reports 6. Nature Publishing Group: 31450. https://doi.org/10.1038/srep31450.
Tan, Cheau Yih, Ruenn Chai Lai, Winnie Wong, Yock Young Dan, Sai-Kiang Lim, and Han Kiat Ho. 2014. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Research & Therapy 5. BioMed Central: 76. https://doi.org/10.1186/scrt465.
Sadek, K.M., and E.A. Saleh. 2014. Fasting ameliorates metabolism, immunity, and oxidative stress in carbon tetrachloride-intoxicated rats. Human & Experimental Toxicology 33: 1277–1283. https://doi.org/10.1177/0960327114527629.
Vallabhapurapu, Sivakumar, Atsushi Matsuzawa, Weizhou Zhang, Ping-Hui Tseng, Jonathan J. Keats, Haopeng Wang, Dario A.A. Vignali, P. Leif Bergsagel, and Michael Karin. 2008. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nature Immunology 9: 1364–1370. https://doi.org/10.1038/ni.1678.
Sun, Shao-Cong. 2011. Non-canonical NF-κB signaling pathway. Cell Research 21. Nature Publishing Group: 71–85. https://doi.org/10.1038/cr.2010.177.
Yarnpakdee, Suthasinee, Soottawat Benjakul, Hordur G. Kristinsson, and Hilma Eiðsdóttir Bakken. 2015. Preventive effect of Nile tilapia hydrolysate against oxidative damage of HepG2 cells and DNA mediated by H2O2 and AAPH. Journal of Food Science and Technology 52: 6194–6205. https://doi.org/10.1007/s13197-014-1672-4.
Amacher, David E. 1998. Serum transaminase elevations as indicators of hepatic injury following the administration of drugs. Regulatory Toxicology and Pharmacology 27: 119–130. https://doi.org/10.1006/rtph.1998.1201.
Ozer, Josef, Marcia Ratner, Martin Shaw, Wendy Bailey, and Shelli Schomaker. 2008. The current state of serum biomarkers of hepatotoxicity. Toxicology 245. Elsevier: 194–205. https://doi.org/10.1016/J.TOX.2007.11.021.
Kubes, Paul, and Wajahat Z. Mehal. 2012. Sterile inflammation in the liver. Gastroenterology 143. W.B. Saunders: 1158–1172. https://doi.org/10.1053/J.GASTRO.2012.09.008.
Luan, Jingyun, and Ju. Dianwen. 2018. Inflammasome: a double-edged sword in liver diseases. Frontiers in Immunology 9: 2201. https://doi.org/10.3389/fimmu.2018.02201.
Tacke, Frank, Tom Luedde, and Christian Trautwein. 2009. Inflammatory pathways in liver homeostasis and liver injury. Clinical Reviews in Allergy & Immunology 36: 4–12. https://doi.org/10.1007/s12016-008-8091-0.
Jaeschke, H., Gregory J. Gores, Arthur I. Cederbaum, Jack A. Hinson, Dominique Pessayre, and John J. Lemasters. 2002. Mechanisms of hepatotoxicity. Toxicological Sciences 65. Narnia: 166–176. https://doi.org/10.1093/toxsci/65.2.166.
Hong, Feng, Won-Ho Kim, Zhigang Tian, Barbara Jaruga, Edward Ishac, Xuening Shen, and Bin Gao. 2002. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-xL proteins. Oncogene 21. Nature publishing group: 32–43. https://doi.org/10.1038/sj.onc.1205016.
Chong, Wai Chin, Madhur D. Shastri, and Rajaraman Eri. 2017. Endoplasmic reticulum stress and oxidative stress: a vicious Nexus implicated in bowel disease pathophysiology. International Journal of Molecular Sciences 18. Multidisciplinary Digital Publishing Institute (MDPI). https://doi.org/10.3390/ijms18040771.
Niu, Xiaofeng, Fang Liu, Weifeng Li, Wenbing Zhi, Qing Yao, Jinmeng Zhao, Guoxiang Yang, Xiumei Wang, Qin Lin, and Zehong He. 2017. Hepatoprotective effect of fraxin against carbon tetrachloride-induced hepatotoxicity in vitro and in vivo through regulating hepatic antioxidant, inflammation response and the MAPK-NF-κB signaling pathway. Biomedicine & Pharmacotherapy 95. Elsevier Masson: 1091–1102. https://doi.org/10.1016/J.BIOPHA.2017.09.029.
Cigremis, Yilmaz, Huseyin Turel, Kevser Adiguzel, Muslum Akgoz, Asim Kart, Musa Karaman, and Hasan Ozen. 2009. The effects of acute acetaminophen toxicity on hepatic mRNA expression of SOD, CAT, GSH-Px, and levels of peroxynitrite, nitric oxide, reduced glutathione, and malondialdehyde in rabbit. Molecular and Cellular Biochemistry 323. Springer US: 31–38. https://doi.org/10.1007/s11010-008-9961-8.
Krithika, Rajesh, Ramasamy Mohankumar, Ramtej J. Verma, Pranav S. Shrivastav, Illiyas L. Mohamad, Palani Gunasekaran, and Srinivasan Narasimhan. 2009. Isolation, characterization and antioxidative effect of phyllanthin against CCl4-induced toxicity in HepG2 cell line. Chemico-Biological Interactions 181: 351–358. https://doi.org/10.1016/j.cbi.2009.06.014.
Liu, Yan, Sheng Liang, Yi Xiong, Hong Shen, Yong Liu, and Liangyou Rui. 2017. Liver NF-κB-inducing kinase promotes liver steatosis and glucose counterregulation in male mice with obesity. Endocrinology 158. The Endocrine Society: 1207–1216. https://doi.org/10.1210/en.2016-1582.
Sun, Shao-Cong. 2017. The non-canonical NF-κB pathway in immunity and inflammation. Nature Reviews Immunology 17: 545–558. https://doi.org/10.1038/nri.2017.52.
Gardam, Sandra, Frederic Sierro, Antony Basten, Fabienne Mackay, and Robert Brink. 2008. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28: 391–401. https://doi.org/10.1016/j.immuni.2008.01.009.
Liao, Gongxian, Minying Zhang, Edward W. Harhaj, and Shao-Cong Sun. 2004. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. Journal of Biological Chemistry 279: 26243–26250. https://doi.org/10.1074/jbc.M403286200.
Xiao, Gutian, Edward W. Harhaj, and Shao-Cong Sun. 2001. NF-κB-Inducing Kinase Regulates the Processing of NF-κB2 p100. Molecular Cell 7. Cell Press: 401–409. https://doi.org/10.1016/S1097-2765(01)00187-3.
Acknowledgments
This study was conceived by XW. XW and SWY designed the study. SWY performed all experiments in the current study. NX and HLL provided experimental assistance. SWY wrote the original draft. BSH, ZC, and XW reviewed the manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20180574, BK20171064), the Technology Incubator Scheme (Social Programs) of Jiangning District (2018Ca12), the Research Foundation of Jiangsu Provincial Medical Youth Talent, the Project of Inviogorating Health Care through Science, Technology and Education (QNRC2016858), the Nanjing Medical University Science Research and Development Foundation (2016NJMU013), the Natural Science Research Fund (General Program) for the Higher Education of Jiangsu Province of China (18KJB350005), and the Zhenjiang City 2017 Science and Technology Innovation Fund (SH2017043).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures performed in studies involving animals were in accordance with the ethical standards of the Institutional Animal Care and Use Committee of China Pharmaceutical University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yue, S., Xue, N., Li, H. et al. Hepatoprotective Effect of Apigenin Against Liver Injury via the Non-canonical NF-κB Pathway In Vivo and In Vitro. Inflammation 43, 1634–1648 (2020). https://doi.org/10.1007/s10753-020-01238-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-020-01238-5