
Abstract
Umbelliferone (UMB) is a natural coumarin that has diverse biological activities. However, its potential to protect against liver fibrosis has not been reported yet. This study aimed to investigate the protective effect of UMB against carbon tetrachloride (CCl4)-induced liver fibrosis in rats. Rats received CCl4 and UMB for 8 weeks and samples were collected for analyses. CCl4 induced a significant increase in serum levels of liver function markers and pro-inflammatory cytokines. Treatment with UMB significantly ameliorated liver function markers and pro-inflammatory cytokines and prevented CCl4-induced histological alterations. CCl4 promoted significant upregulation of α-smooth muscle actin (SMA), collagen I, collagen III, NF-κB p65, TGF-β1, and p-Smad3. Masson’s trichrome staining revealed a significant fibrogenesis in CCl4-induced rats. Treatment with UMB suppressed TGF-β1/Smad3 signaling and downregulated α-SMA, collagen I, collagen III, and NF-κB p65. In addition, UMB diminished malondialdehyde and nitric oxide levels, boosted reduced glutathione and antioxidant enzymes, and upregulated the expression of PPARγ. In conclusion, our results demonstrated that UMB prevented CCl4-induced liver fibrosis by attenuating oxidative stress, inflammation, and TGF-β1/Smad3 signaling, and upregulating PPARγ. Therefore, UMB may be a promising candidate for preventing hepatic fibrogenesis, given that further research is needed to delineate the exact molecular mechanisms underlying its antifibrotic efficacy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Chronic liver disease is a progressive destruction and regeneration of the liver tissue leading to hepatic fibrosis and cirrhosis. It represents a major health problem and a cause of morbidity and mortality [1]. Liver fibrosis is a ubiquitous wound-healing response to chronic tissue injury caused by hepatitis, biliary obstruction, nonalcoholic fatty liver disease, metabolic liver disease, and others [2]. It is a complex and multifactorial process associated with increased deposition of collagen-rich extracellular matrix (ECM). If left untreated, fibrosis can develop into cirrhosis, and consequently hepatocellular carcinoma (HCC), liver failure, and death [3]. Hepatic stellate cells (HSCs) play the central role in fibrogenesis of the liver. These cells are located between hepatocytes and endothelial cells of the sinusoids and being activated during liver injury [4]. Activated HSCs acquire myofibroblast-like phenotypes and become the source of activated myofibroblasts and portal fibroblasts. Consequently, the production and deposition of fibrous ECM proteins increase, leading to liver fibrosis [5]. The fibrotic activity of HSCs could be promoted by different factors, including inflammatory cytokines and reactive oxygen species (ROS) [6, 7]. In addition, the transforming growth factor beta 1 (TGF-β1)/Smad3 signaling was evidenced to induce liver fibrosis. Activated TGF-β1/Smad3 signaling induces the synthesis and production of ECM rich in collagen I and III [8, 9]. Therefore, attenuation of TGF-β1/Smad3 signaling and suppression of oxidative stress and inflammation represent an efficient strategy to inhibit fibrogenesis.
Peroxisome proliferator-activated receptor gamma (PPARγ) is a ligand-activated nuclear receptor that participates in many biological processes, mainly adipogenesis and metabolism [10]. In addition to its metabolic functions, it possesses a potent anti-inflammatory efficacy [11], and can induce the expression of antioxidant enzymes [12] and suppress TGF-β1/Smad3 signaling [13]. Previously, we have reported that upregulation of PPARγ can protect against hepatocarcinogenesis [14], and liver injury induced by cyclophosphamide [15,16,17,18], methotrexate [19, 20], and azoxymethane [21]. In addition, multiple studies have revealed the role of PPARγ in ameliorating hepatic fibrosis [13, 22, 23].
Umbelliferone (UMB) is a natural product found in plants of the Rutaceae and Umbelliferae families. It is also known as 7-hydroxycoumarin and commonly used as a sunscreen agent [24]. UMB possesses several pharmacological activities, including antioxidant, anti-inflammatory, antidiabetic, and antitumor effects [24, 25]. UMB prevented cyclophosphamide hepatotoxicity by upregulating PPARγ as we have recently reported [17]. In a rat model of hyperammonemia, UMB protected the brain and liver against the deleterious effects of excess ammonia [26]. In addition, UMB has been recently demonstrated to prevent liver injury in db/db mice [27]. In these studies, the effects of UMB were mediated mainly via suppression of oxidative stress and inflammation. These beneficial effects make UMB a good candidate for the protection against liver fibrosis. Therefore, we investigated the efficacy of UMB to ameliorate fibrosis induced by carbon tetrachloride (CCl4) in rats, pointing to its ability to modulate TGF-β1/Smad3 signaling and PPARγ expression. CCl4 is frequently used to induce liver injury [28,29,30,31] and fibrogenesis in rodents [6, 32].
MATERIALS AND METHODS
Reagents and Chemicals
UMB, CCl4, malondialdehyde (MDA), reduced glutathione (GSH), thiobarbituric acid, pyrogallol, Griess reagent, trichloroacetic acid, and 5,5′-dithio-bis-[2-nitrobenzoic acid] were purchased from Sigma (USA). Assay kits for aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), gamma-glutamyltransferase (γGT), bilirubin, and albumin were supplied by Spinreact (Girona, Spain), and the Bradford protein assay kit was purchased from BioBasic (Markham, ON, Canada). Primary antibodies for TGF-β1, p-Smad3, Smad3, PPARγ, nuclear factor-kappaB (NF-κB) p65, and β-actin, and secondary antibodies were supplied by Novus Biologicals (Centennial, CO, USA). Assay kits for tumor necrosis factor alpha (TNF-α) and interleukin (IL)-6 were supplied by R&D Systems (USA), and TRIzol was purchased from Invitrogen (USA). All other chemicals were obtained from standard commercial supplies.
Experimental Animals and Treatments
Male Wistar rats, weighing about 160–180 g, obtained from the National Institute of Ophthalmology (Egypt) were included in this study. The animals were housed at normal temperature (23 ± 2 °C) and 12-h light/dark cycle. A standard diet and water were provided ad libitum, and the experimental protocol and all animal procedures were approved by the Institutional Animal Ethics Committee of Beni-Suef University (Egypt).
Thirty rats were divided into five groups (n = 6) as follows (Fig. 1):
-
Group I
(Control): received intraperitoneal (i.p.) injection of 1 ml/kg olive oil twice/week and 1% carboxymethyl cellulose (CMC) by daily oral gavage for 8 weeks
-
Group II
(CCl4): received i.p. injection of 1 ml CCl4/olive oil (1:1 v/v) twice/week [33] and 1% CMC by daily oral gavage for 8 weeks
-
Group III
(CCl4 + 25 mg UMB): received i.p. injection of CCl4 as group II and 25 mg/kg UMB dissolved in 1% CMC daily by oral gavage for 8 weeks
-
Group IV
(CCl4 + 50 mg UMB): received i.p. injection of CCl4 as group II and 50 mg/kg UMB dissolved in 1% CMC daily by oral gavage for 8 weeks
-
Group V
(CCl4 + 100 mg UMB): received i.p. injection of CCl4 as group II and 100 mg/kg UMB dissolved in 1% CMC daily by oral gavage for 8 weeks

A schematic diagram showing the experimental design.
The doses of UMB used in this study were selected based on our previous studies reporting its antioxidant and hepatoprotective effects in vivo [17, 26], and was regularly adjusted based on the body weight changes. All rats were sacrificed under anesthesia at the end of 8 weeks. Blood samples were collected for serum separation, and liver was excised and washed in cold phosphate-buffered saline (PBS). Pieces from the liver were fixed in 10% neutral buffered formalin, and other samples were homogenized in cold PBS (10% w/v), while others were kept frozen at − 80 °C for gene and protein expression analysis.
Assay of Liver Function Markers and Pro-inflammatory Cytokines
Serum levels of ALT, AST, ALP, γGT, bilirubin, and albumin were measured using commercially available kits (Spinreact, Girona, Spain). TNF-α and IL-6 were assayed in serum of control and experimental rats using specific ELISA kits (R&D Systems, USA).
Assay of MDA, Nitric Oxide, and Antioxidants
MDA [34] and nitric oxide (NO) [35] levels were measured in the liver homogenate (10% w/v in PBS). GSH, superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) were determined according to the methods of Beutler et al. [36], Marklund and Marklund [37], Aebi [38], and Matkovics et al. [39], respectively.
Histological Examination
The liver samples were fixed in 10% neutral buffered formalin for 48 h and then processed for the preparation of paraffin blocks. Five-micrometer sections were cut using a rotatory microtome and then stained with hematoxylin and eosin (H&E) and Masson’s trichrome (MT) and examined using a light microscope. The MT-stained areas in the liver sections were quantified using ImageJ (version 1.32j, NIH, USA).
Gene Expression Analysis
The effect of UMB on collagen I (COL1A1) and collagen III (COL3A1) mRNA expression levels in the liver of CCl4-intoxicated rats was evaluated using quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR) as previously described [19]. Total RNA was isolated from the liver samples using TRIzol. The isolated RNA was quantified, and samples with A260/A280 of 1.8 or more were used for reverse transcription. Amplification of the cDNA was carried out by SYBR Green and the following primers: COL1A1 F: 5′-GTACATCAGCCCAAACCCCA-3′ and R: 5′-CAGGATCGGAACCTTCGCTT-3′, COL3A1 F: 5′-AGGGCAGGGAACAACTGATG-3′ and R: 5′-GGTCCCACATTGCACAAAGC-3′, and β-actin F: 5′- AGGAGTACGATGAGTCCGGC-3′ and R: 5′-CGCAGCTCAGTAACAGTCCG-3′. The obtained data were analyzed using the 2−ΔΔCt method [40] and normalized to β-actin.
Western Blotting
The liver samples were homogenized in RIPA buffer with proteinase inhibitors and centrifuged at 10,000 rpm, and the clear supernatant was collected. The Bradford protein assay kit was used to determine protein concentration, and 40 μg proteins was subjected to 10% SDS-PAGE followed by transfer to nitrocellulose membranes. The membrane was blocked for 1 h at room temperature and incubated with antibodies for TGF-β1, p-Smad3, Smad3, PPARγ, NF-κB p65, and β-actin overnight at 4 °C. The membranes were incubated with the secondary antibodies, developed using the enhanced chemiluminescence kit (BIO-RAD, USA), and the obtained bands were quantified using ImageJ (version 1.32j, NIH), and the results were presented as percent of control.
Statistical Analysis
The results were analyzed using GraphPad Prism5 (GraphPad software, San Diego, CA, USA) and expressed as mean ± standard error of the mean (SEM). Different parameters among the experimental groups were compared by one-way ANOVA, followed by Tukey’s post hoc test. The difference was considered significant at P < 0.05.
RESULTS
UMB Inhibits CCl4-Induced Functional and Histological Alterations in the Liver of Rats
The circulating levels of liver function markers were measured to determine the protective effect of UMB on CCl4-induced liver injury. CCl4-administered rats exhibited a significant increase in serum ALT (Fig. 2a; P < 0.001), AST (Fig. 2b; P < 0.001), ALP (Fig. 2c; P < 0.001), and γGT (Fig. 2d; P < 0.001). Concurrent treatment with UMB for 8 weeks notably ameliorated serum levels of the liver function markers in CCl4-induced rats. The high dose of UMB (100 mg/kg) produced a significant (P < 0.001) reduction in serum AST levels when compared with the 25 mg/kg dose (Fig. 2b). Serum bilirubin was increased significantly (P < 0.001) in CCl4-induced rats and remarkably decreased in 25, 50, and 100 mg/kg UMB-treated groups (Fig. 2e). In contrast, serum albumin was decreased (P < 0.001) in CCl4-induced rats as represented in Fig. 2f. UMB significantly improved serum albumin levels when administered at 25 (P < 0.05), 50 (P < 0.001), and 100 (P < 0.001) mg/kg into CCl4-induced rats.

Umbelliferone inhibits CCl4-induced liver injury in rats. Treatment with UMB ameliorated serum levels of a ALT, b AST, c ALP, d γGT, e bilirubin, and f albumin in CCl4-induced rats. Data are expressed as mean ± SEM, n = 6. *P < 0.05 and ***P < 0.001.
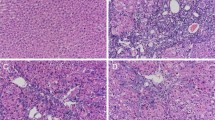
The hepatoprotective effect of UMB was further confirmed by the histopathologic examination (Fig. 3). H&E-stained sections of control rats showed normal histological structure of the liver (Fig. 3a). CCl4-induced rats exhibited multiple alterations, including focal necrosis, steatosis of hepatocytes, cytoplasmic vacuolization of hepatocytes, Kupffer cell activation, and fibroblast proliferation (Fig. 3b, c). On the other hand, CCl4-induced rats that received 25 (Fig. 3d), 50 (Fig. 3e), and 100 mg/kg UMB (Fig. 3f) showed a notable improvement in the histological appearance of the liver with slight cytoplasmic vacuolations (Fig. 3d, f). The histopathological lesions in the liver of all experimental groups are summarized in Table 1.

Umbelliferone prevents CCl4-induced histological alterations in the liver of rats. H&E-stained liver sections in a control rats showing normal histological structure of hepatic lobule; b and c CCl4-induced rats showing steatosis of hepatocytes [b; arrow], cytoplasmic vacuolization of hepatocytes [c; short arrow], and fibroblast proliferation in the portal triad [c; long arrow]; and CCl4-induced rats treated with 25 mg/kg (d), 50 mg/kg (e), and 100 mg/kg (f) showing slight cytoplasmic vacuolization (arrow). (×400).
UMB Prevents CCl4-Induced Liver Fibrosis in Rats
Fibrosis in the liver of CCl4-intoxicated rats was evaluated using MT staining and assessment of the expression levels of α-SMA and collagen. Image analysis of the MT-stained liver sections of CCl4-induced rats revealed an increase in ECM deposition (P < 0.001) when compared with the control rats (Fig. 4a and 5b). Oral supplementation of all UMB doses for 8 weeks reduced ECM deposition significantly (P < 0.001).

Umbelliferone attenuates CCl4-induced liver fibrosis in rats. a Masson’s trichrome-stained liver sections in (a) control rats showing normal histochemical reaction for collagen fibers, (b and c) CCl4-induced rats showing strong positive histochemical reaction for collagen fibers (arrow), and CCl4-induced rats treated with 25 (d), 50 (e), and 100 mg/kg umbelliferone (f) showing slight positive histochemical reaction for collagen fibers (arrow). (×400). b Quantification of Masson’s trichrome-positive area showing a significant increase in collagen deposition and the remarkable ameliorative effect of umbelliferone. c–e Umbelliferone downregulated the protein expression levels of α-SMA (c), and mRNA abundance of collagen I (d) and collagen III (e) in the liver of CCl4-induced rats. Data are expressed as mean ± SEM, n = 6, ***P < 0.001.

Umbelliferone downregulates TGF-β1/Smad3 signaling in the liver of CCl4-induced rats. a Representative blots of TGF-β1, p-Smad3, Smad3, and β-actin. Treatment with umbelliferone significantly decreased TGF-β1 protein expression (b) and Smad3 phosphorylation (c) in the liver of CCl4-induced rats. Data are expressed as mean ± SEM, n = 6, ***P < 0.001.
Liver α-SMA was remarkably (P < 0.001) upregulated in CCl4-intoxicated rats when compared with the control group as represented in Fig. 4c. The mRNA abundance of COL1A1 (Fig. 4d) and COL3A1 (Fig. 4e), determined by qRT-PCR, showed a notable (P < 0.001) upregulation in CCl4-induced rats. In contrast, CCl4-intoxicated rats that received different doses of UBM exhibited a significant decrease in the expression levels of α-SMA, COL1A1, and COL3A1.
UMB Downregulates Hepatic TGF-β1/Smad3 Signaling in CCl4-Intoxicated Rats
TGF-β1 protein expression was significantly upregulated in the liver of CCl4-intoxicated rats as represented in Fig. 5a, b. Similarly, Smad3 phosphorylation was remarkably (P < 0.001) increased in CCl4-intoxicated rats (Fig. 5a, c). Treatment with 25, 50, and 100 mg/kg UMB suppressed TGF-β1 expression and Smad3 phosphorylation.
UMB Attenuates CCl4-Induced Oxidative Stress in the Liver of Rats
MDA, a marker of lipid peroxidation, was increased significantly (P < 0.001) in the liver of rats that received CCl4 as represented in Fig. 6a. Similarly, NO was significantly (P < 0.001) elevated in the liver of rats received CCl4 (Fig. 6b). Treatment with 25, 50, and 100 mg/kg UMB produced a significant decrease in hepatic MDA (P < 0.01; P < 0.001; P < 0.001) and NO levels (P < 0.001; P < 0.001; P < 0.001).

Umbelliferone attenuates CCl4-induced oxidative stress in the liver of rats. Umbelliferone decreased MDA (a) and NO levels (b), and increased GSH (c), SOD (d), CAT (e), and GPx (f) in the liver of CCl4-induced rats. Data are expressed as mean ± SEM, n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001.
On the other hand, the GSH content was reduced significantly (P < 0.001) in CCl4-induced rats (Fig. 6c). UMB supplementation significantly increased the GSH content when administered at 25 (P < 0.05), 50 (P < 0.05), and 100 mg/kg body weight (P < 0.01). Activities of SOD (Fig. 6d), CAT (Fig. 6e), and GPx (Fig. 6f) were significantly (P < 0.001) declined in CCl4-induced rats. The 25 mg/kg UMB did not induce a significant improvement in SOD activity, whereas it enhanced the activity of both CAT (P < 0.05; Fig. 6e) and GPx (P < 0.001; Fig. 6f). The higher UMB doses (50 and 100 mg/kg) enhanced the activity of SOD, CAT, and GPx significantly in the liver of CCl4-induced rats.
UMB Prevents CCl4-Induced Inflammation in Rats
To investigate the potential of UMB to attenuate CCl4-induced inflammation in rats, we determined the expression levels of the NF-κB p65 subunit in the liver and circulating levels of TNF-α and IL-6.
CCl4-induced rats showed a notable (P < 0.001) increase in the expression of the NF-κB p65 subunit in the liver. Oral supplementation of 25, 50, or 100 mg/kg UMB for 8 weeks downregulated NF-κB p65 in the liver of CCl4-induced rats (P < 0.001) as depicted in Fig. 7a.

Umbelliferone prevents CCl4-induced inflammation in rats. a Umbelliferone downregulated NF-κB p65 protein expression levels in the liver of CCl4-iduced rats. b and c Umbelliferone decreased serum levels of TNF-α (b) and IL-6 (c) in CCl4-induced rats. Data are expressed as mean ± SEM, n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001.
TNF-α and IL-6 in serum of CCl4-induced rats showed a significant (P < 0.001) increase when compared with those of the control group as represented in Fig. 7b, c, respectively. Treatment of the CCl4-induced rats with 25, 50, or 100 mg/kg UMB significantly ameliorated serum TNF-α (Fig. 7b) and IL-6 (Fig. 7c).
UMB Upregulates PPARγ Expression in the Liver of CCl4-Induced Rats
Several studies have demonstrated the role of PPARγ in the inhibition of hepatic fibrosis [13, 22, 23]. Therefore, we determined whether the antifibrosis effect of UMB was associated with PPARγ activation. CCl4-induced rats showed a significant (P < 0.001) downregulation of hepatic PPARγ protein expression when compared with the control rats (Fig. 8). In contrast, treatment of the CCl4-induced rats with 25, 50 or 100 mg/kg UMB resulted in a significant increase in the protein expression levels of PPARγ.

Umbelliferone upregulates PPARγ expression in the liver of CCl4-induced rats. Data are expressed as mean ± SEM, n = 6, ***P < 0.001.
DISCUSSION
UMB has displayed several pharmacological and biological effects, including antioxidant, anti-inflammatory, and antidiabetic [24, 25]. However, its potential to protect against liver fibrosis has not been reported yet. Herein, we investigated the ameliorative effect of UMB on liver fibrosis induced by CCl4 in rats. Our findings introduced novel information on the hepatoprotective mechanism of UMB and pointed to the involvement of TGF-β1/Smad3 signaling and PPARγ modulation in mediating its effect. In the present study, the repeated administration of CCl4 (twice per week for 8 weeks) induced hepatocellular damage and fibrosis evidenced by the increased serum ALT, AST, ALP, γGT, and bilirubin coupled with decreased serum albumin levels. Liver injury due to CCl4 administration was confirmed by the histopathological manifestations, including focal hepatic necrosis, steatosis of hepatocytes, cytoplasmic vacuolization of hepatocytes, Kupffer cell activation, and fibroblast proliferation. Staining with MT demonstrated a remarkable increase in fibrous tissue and liver fibrosis in CCl4-intoxicated rats. Liver fibrosis was further confirmed by the significant increase in the expression of α-SMA, collagen I, and collagen III. Interestingly, all alterations induced by the chronic administration of CCl4 were significantly attenuated by UMB. Treatment with different doses of UMB remarkably ameliorated the liver function markers and prevented histological alterations induced by CCl4. In addition, UMB inhibited fibroblast proliferation and suppressed the production and deposition of fibrous ECM as evidenced by downregulation of α-SMA and collagen expression. Collagen I and collagen III, the main components of ECM, are synthesized by fibroblasts, and their excessive secretion and accumulation represent the characteristic pathological feature in liver fibrosis [5]. Excessive ECM deposition along with limited remodeling occur during chronic liver disease and lead to scar deposition and fibrosis marked by excessive deposition of ECM extensively rich in both collagen I and collagen III [41]. Recent studies have demonstrated increased hepatic collagen and α-SMA expression in CCl4-intoxicated rodents [6, 32]. Therefore, UMB reversed the progression of fibrosis by suppressing the excessive deposition of ECM via downregulating collagen I, collagen III, and α-SMA.
To explore the mechanism underlying the ameliorative efficacy of UMB on CCl4-induced liver injury and fibrosis, we determined its modulatory effect on TGF-β1/Smad3 signaling. Previous studies have demonstrated the central role of TGF-β1 in liver fibrosis by promoting myofibroblast-like cell formation and inducing the excessive accumulation of ECM components. TGF-β1 binds to its transmembrane receptor and activates Smad signaling, leading to increased expression of the ECM proteins [8]. Herein, CCl4-intoxicated rats showed a significant increase in liver TGF-β1 expression and Smad3 phosphorylation levels. Accordingly, previous studies have reported increased TGF-β1 and Smad3 expression in CCl4-induced rats [6, 32]. In a rat model of CCl4/diethylnitrosamine (DEN)-induced hepatocarcinogenesis, we have recently shown that increased deposition of ECM and liver fibrosis were associated with TGF-β1/Smad3 signaling activation [14]. Here, treatment with UMB decreased the protein expression and phosphorylation levels of TGF-β1 and Smad3, respectively, in the liver of CCl4-intoxicated rats. Therefore, suppressed TGF-β1/Smad3 signaling is the main mechanism by which UMB reversed the progression of fibrosis in CCl4-intoxicated rats. This notion has been supported by the study of Lang et al. who investigated the role of TGF-β1 silencing in the attenuation of fibrosis [42]. siRNA-mediated downregulation of TGF-β1 inhibited the proliferation of HSCs and suppressed the production of collagen I and collagen III, leading to prevention of liver fibrosis in rats [42].
Oxidative stress plays a critical role in liver fibrogenesis as revealed by several studies [6, 14, 32]. Increased ROS production has been associated with the activation of HSCs and production of collagen-rich ECM [6, 7]. In addition, oxidative stress-mediated fibrosis occurs due to excessive ROS production and its subsequent hepatocyte injury via membrane lipid peroxidation [43]. CCl4-derived free radicals cause hepatocyte membrane damage and leakage of the enzymes into the circulation [44], as confirmed by the elevated serum levels of ALT, AST, ALP, and γGT. Within the liver, CCl4 is metabolized into the highly reactive trichloromethyl and peroxyl radicals, via the action of cytochrome P450. These radicals initiate lipid peroxidation and can bind covalently to the cellular macromolecules [44]. In addition, CCl4 has been recently reported to induce nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)-mediated ROS production in the liver of rats [6]. In our study, oxidative stress was monitored by measuring MDA, NO, GSH, and antioxidant enzymes. MDA, a marker of lipid peroxidation, and NO levels were significantly increased in CCl4-intoxicated rats. Lipid peroxidation is mediated via CCl4-derived radicals and increased ROS generation, whereas increased NO could be attributed to increased expression of inducible nitric oxide synthase (iNOS). NF-κB is a transcription factor very well known to be activated by ROS. Activated NF-κB promotes the expression of several inflammatory mediators, including TNF-α, IL-6, and iNOS [16]. Here, CCl4 increased the expression of liver NF-κB p65 and serum levels of TNF-α and IL-6. Chronic inflammation has been reported to precede fibrosis and to be associated with the development of cirrhosis [45]. Apoptotic bodies derived from damaged hepatocytes due to inflammation can activate Kupffer cells and HSCs, promoting inflammation and fibrogenesis in the liver [5]. Histological examination of the liver of CCl4-induced rats revealed activated Kupffer cells associated with fibroblast proliferation. Activated Kupffer cells can stimulate HSCs through the release of cytokines and chemokines [5, 46]. In addition, ROS and NO produced by the active Kupffer cells can stimulate DNA damage, apoptosis, and expression of pro-inflammatory genes, leading to fibrogenesis [47]. In conjunction with increased MDA and NO, CCl4-induced rats exhibited a decrease in liver GSH content and the activity of SOD, CAT, and GPx. GSH is an essential antioxidant that scavenges free radicals and maintains protein sulfydryl groups, and SOD, CAT, and GPx are antioxidant enzymes that protect the cells against the deleterious effects of free radicals and oxidants. Therefore, depletion of these antioxidant defenses can lead to cell injury and death. In accordance with our findings, recent studies have demonstrated increased lipid peroxidation and diminished GSH and SOD in the liver of CCl4-intoxicated rats [6, 32]. Treatment of the CCl4-intoxicated rats with UMB significantly decreased MDA and NO and boosted the antioxidant defenses. In addition, UMB suppressed inflammation in CCl4-intoxicated rats as revealed by the downregulated NF-κB and decreased TNF-α and IL-6. We have previously reported the efficacy of UMB to prevent cyclophosphamide hepatotoxicity via suppression of lipid peroxidation, NF-κB and iNOS, and enhancement of the antioxidant defense system [17]. Therefore, attenuation of oxidative stress and inflammation plays a central role in the ameliorative effect of UMB against liver fibrogenesis.
The crucial role of oxidative stress and inflammation in chronic liver disease and the deleterious effects of ROS and pro-inflammatory cytokines on hepatocytes support the use of UMB as an antifibrotic agent. The antioxidant, anti-inflammatory, and antifibrotic potential of UMB could be explained by its ability to activate the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and PPARγ signaling as we previously reported [17]. In support of our findings, Yin et al. have recently shown the ability of UMB to upregulate Nrf2 in the liver of diabetic mice [27]. When activated, the redox-sensitive Nrf2 can attenuate oxidative stress and suppress NF-κB, iNOS, and pro-inflammatory cytokines [16, 19, 48]. The ligand-activated nuclear receptor PPARγ possesses potent anti-inflammatory properties mediated via negative interference and/or transcriptional repression of NF-κB [11], and can directly induce expression of the antioxidant enzymes [12]. The role of PPARγ in attenuating the progression of liver fibrosis has been reported in different studies. Yang et al. have reported enhanced fibrogenesis in PPARγ-deficient mice after liver injury, whereas HSC activation and fibrosis were suppressed following in vivo PPARγ overexpression [22]. In CCl4-induced fibrosis in mice, PPARγ suppressed the differentiation of bone marrow-derived mesenchymal stem cells into myofibroblasts [49]. PPARγ can also interact with the fibrosis-related signaling pathway TGF-β1/Smad3. PPARγ has been reported to bind Smad3 and prevent the nuclear accumulation of p-Smad3, leading to suppression of TGF-β1 signaling [13]. We have previously reported a negative correlation between PPARγ upregulation and TGF-β1/Smad3 pathway in the liver of CCl4/DEN-induced rats [14]. The PPARγ agonist rosiglitazone exhibited antifibrotic function mediated via suppressing the early growth response protein 1 (Egr-1) which acts as a mediator of non-Smad TGF-β1 signaling [50]. The antifibrotic potential of UMB is therefore associated with its ability to upregulate PPARγ and subsequently attenuate inflammation and fibrosis. However, the role of PPARγ in mediating the antifibrotic mechanism of UMB needs to be further explored.
In conclusion, our results demonstrated that UMB prevented CCl4-induced liver fibrosis in rats. UMB attenuated oxidative stress, inflammation, expression of collagen and α-SMA, and deposition of ECM in the liver of rats. In addition, UMB suppressed TGF-β1/Smad3 signaling and upregulated the expression of PPARγ. Therefore, UMB may be a promising candidate for preventing the progression of fibrogenesis. However, further research is needed to delineate the exact molecular mechanisms underlying the antifibrotic efficacy of UMB.
References
Kim, W.R., R.S. Brown Jr., N.A. Terrault, and H. El-Serag. 2002. Burden of liver disease in the United States: summary of a workshop. Hepatology (Baltimore, Md.) 36: 227–242.
Zhang, C.Y., W.G. Yuan, P. He, J.H. Lei, and C.X. Wang. 2016. Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World Journal of Gastroenterology 22: 10512–10522.
Tacke, F., and C. Trautwein. 2015. Mechanisms of liver fibrosis resolution. Journal of Hepatology 63: 1038–1039.
Josan, S., K. Billingsley, J. Orduna, J.M. Park, R. Luong, L. Yu, R. Hurd, A. Pfefferbaum, D. Spielman, and D. Mayer. 2015. Assessing inflammatory liver injury in an acute CCl4 model using dynamic 3D metabolic imaging of hyperpolarized [1-(13)C] pyruvate. NMR in Biomedicine 28: 1671–1677.
Friedman, S.L. 2008. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiological Reviews 88: 125–172.
Cheng, Q., C. Li, C.-f. Yang, Y.-j. Zhong, D. Wu, L. Shi, L. Chen, Y.-w. Li, and L. Li. 2019. Methyl ferulic acid attenuates liver fibrosis and hepatic stellate cell activation through the TGF-β1/Smad and NOX4/ROS pathways. Chemico-Biological Interactions 299: 131–139.
Gandhi, C.R. 2012. Oxidative stress and hepatic stellate cells: a paradoxical relationship. Trends in Cell & Molecular Biology 7: 1–10.
Duan, W.J., X. Yu, X.R. Huang, J.W. Yu, and H.Y. Lan. 2014. Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. American Journal of Pathology 184: 2275–2284.
Gressner, A.M., R. Weiskirchen, K. Breitkopf, and S. Dooley. 2002. Roles of TGF-beta in hepatic fibrosis. Frontiers in Bioscience : a Journal and Virtual Library 7: d793–d807.
Rosen, E.D., and B.M. Spiegelman. 2001. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. The Journal of Biological Chemistry 276: 37731–37734.
Ricote, M., J.T. Huang, J.S. Welch, and C.K. Glass. 1999. The peroxisome proliferator-activated receptor (PPARgamma) as a regulator of monocyte/macrophage function. Journal of Leukocyte Biology 66: 733–739.
Yu, Y., Y. Wu, G. Wen, and W. Yang. 2014. Effect of pioglitazone on the expression of TLR4 in renal tissue of diabetic rats. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 30 (8): 793–797.
Lin, L.C., S.L. Hsu, C.L. Wu, W.C. Liu, and C.M. Hsueh. 2011. Peroxisome proliferator-activated receptor gamma (PPARgamma) plays a critical role in the development of TGFbeta resistance of H460 cell. Cellular Signalling 23: 1640–1650.
Mahmoud, A.M., H.M. Mohammed, S.M. Khadrawy, and S.R. Galaly. 2017. Hesperidin protects against chemically induced hepatocarcinogenesis via modulation of Nrf2/ARE/HO-1, PPARgamma and TGF-beta1/Smad3 signaling, and amelioration of oxidative stress and inflammation. Chemico-Biological Interactions 277: 146–158.
Mahmoud, A.M. 2014. Hesperidin protects against cyclophosphamide-induced hepatotoxicity by upregulation of PPARγ and abrogation of oxidative stress and inflammation. Canadian Journal of Physiology and Pharmacology 92: 717–724.
Mahmoud, A.M., and H.S. Al Dera. 2015. 18β-Glycyrrhetinic acid exerts protective effects against cyclophosphamide-induced hepatotoxicity: potential role of PPARγ and Nrf2 upregulation. Genes & Nutrition 10: 1–13.
Mahmoud, A.M., M.O. Germoush, M.F. Alotaibi, and O.E. Hussein. 2017. Possible involvement of Nrf2 and PPARgamma up-regulation in the protective effect of umbelliferone against cyclophosphamide-induced hepatotoxicity. Biomedicine & Pharmacotherapy 86: 297–306.
Alqahtani, S., and A.M. Mahmoud. 2016. Gamma-glutamylcysteine ethyl ester protects against cyclophosphamide-induced liver injury and hematologic alterations via upregulation of PPARgamma and attenuation of oxidative stress, inflammation, and apoptosis. Oxidative Medicine and Cellular Longevity 2016: 4016209.
Mahmoud, A.M., O.E. Hussein, W.G. Hozayen, and S.M. Abd El-Twab. 2017. Methotrexate hepatotoxicity is associated with oxidative stress, and down-regulation of PPARgamma and Nrf2: protective effect of 18beta-glycyrrhetinic acid. Chemico-Biological Interactions 270: 59–72.
Mahmoud, A.M., W.G. Hozayen, and S.M. Ramadan. 2017. Berberine ameliorates methotrexate-induced liver injury by activating Nrf2/HO-1 pathway and PPARgamma, and suppressing oxidative stress and apoptosis in rats. Biomedicine & Pharmacotherapy 94: 280–291.
Abdella, E., A. Mahmoud, and A. El-Derby. 2016. Brown seaweeds protect against azoxymethane-induced hepatic repercussions through up-regulation of peroxisome proliferator activated receptor gamma and attenuation of oxidative stress. Pharmaceutical Biology 54: 2496–2504.
Yang, L., C.-C. Chan, O.-S. Kwon, S. Liu, J. McGhee, S.A. Stimpson, L.Z. Chen, W.W. Harrington, W.T. Symonds, and D.C. Rockey. 2006. Regulation of peroxisome proliferator-activated receptor-γ in liver fibrosis. American Journal of Physiology - Gastrointestinal and Liver Physiology 291: G902–G911.
Yang, L., S.A. Stimpson, L. Chen, W.W. Harrington, and D.C. Rockey. 2010. Effectiveness of the PPARγ agonist, GW570, in liver fibrosis. Inflammation Research 59: 1061–1071.
Mazimba, O. 2017. Umbelliferone: sources, chemistry and bioactivities review. Bulletin of Faculty of Pharmacy, Cairo University 55: 223–232.
Ramesh, B., and K.V. Pugalendi. 2005. Antihyperlipidemic and antidiabetic effects of umbelliferone in streptozotocin diabetic rats. The Yale Journal of Biology and Medicine 78: 189–196.
Germoush, M.O., S.I. Othman, M.A. Al-Qaraawi, H.M. Al-Harbi, O.E. Hussein, G. Al-Basher, M.F. Alotaibi, H.A. Elgebaly, M.A. Sandhu, A.A. Allam, and A.M. Mahmoud. 2018. Umbelliferone prevents oxidative stress, inflammation and hematological alterations, and modulates glutamate-nitric oxide-cGMP signaling in hyperammonemic rats. Biomedicine & Pharmacotherapy 102: 392–402.
Yin, J., H. Wang, and G. Lu. 2018. Umbelliferone alleviates hepatic injury in diabetic db/db mice via inhibiting inflammatory response and activating Nrf2-mediated antioxidant. Bioscience Reports 38: BSR20180444.
Al-Sayed, E., O. Martiskainen, S.H. Seif el-Din, A.-N.A. Sabra, O.A. Hammam, N.M. El-Lakkany, and M.M. Abdel-Daim. 2014. Hepatoprotective and antioxidant effect of Bauhinia Hookeri extract against carbon tetrachloride-induced hepatotoxicity in mice and characterization of its bioactive compounds by HPLC-PDA-ESI-MS/MS. BioMed Research International 2014: 9.
Al-Rasheed, N., L. Faddah, N. Al-Rasheed, Y.A. Bassiouni, I.H. Hasan, A.M. Mahmoud, R.A. Mohamad, and H.I. Yacoub. 2016. Protective effects of silymarin, alone or in combination with chlorogenic acid and/or melatonin, against carbon tetrachloride-induced hepatotoxicity. Pharmacognosy Magazine 12: S337–S345.
Al-Sayed, E., and M.M. Abdel-Daim. 2014. Protective role of cupressuflavone from Cupressus macrocarpa against carbon tetrachloride-induced hepato- and nephrotoxicity in mice. Planta Medica 80: 1665–1671.
Fahmy, N.M., E. Al-Sayed, M.M. Abdel-Daim, M. Karonen, and A.N. Singab. 2016. Protective effect of Terminalia muelleri against carbon tetrachloride-induced hepato and nephro-toxicity in mice and characterization of its bioactive constituents. Pharmaceutical Biology 54: 303–313.
Li, X., L. Wang, and C. Chen. 2017. Effects of exogenous thymosin β4 on carbon tetrachloride-induced liver injury and fibrosis. Scientific Reports 7: 5872.
Hardjo, M., M. Miyazaki, M. Sakaguchi, T. Masaka, S. Ibrahim, K. Kataoka, and N.H. Huh. 2009. Suppression of carbon tetrachloride-induced liver fibrosis by transplantation of a clonal mesenchymal stem cell line derived from rat bone marrow. Cell Transplantation 18: 89–99.
Ohkawa, H., N. Ohishi, and K. Yagi. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Analytical Biochemistry 95: 351–358.
Green, L.C., D.A. Wagner, J. Glogowski, P.L. Skipper, J.S. Wishnok, and S.R. Tannenbaum. 1982. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Analytical Biochemistry 126: 131–138.
Beutler, E., O. Duron, and B.M. Kelly. 1963. Improved method for the determination of blood glutathione. The Journal of Laboratory and Clinical Medicine 61: 882–888.
Marklund, S., and G. Marklund. 1974. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase, FEBS. European Journal of Biochemistry 47: 469–474.
Aebi, H. 1984. [13] Catalase in vitro. Methods in Enzymology 105: 121–126.
Matkovics, B., L. Szabo, and I.S. Varga. 1998. Determination of enzyme activities in lipid peroxidation and glutathione pathways (in Hungarian). Laboratoriumi Diagnosztika 15: 248–249.
Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta c(t)) method. Methods 25 (4): 402–408.
Schuppan, D. 2015. Liver fibrosis: common mechanisms and antifibrotic therapies. Clinics and Research in Hepatology and Gastroenterology 39 (Suppl 1): S51–S59.
Lang, Q., Q. Liu, N. Xu, K.L. Qian, J.H. Qi, Y.C. Sun, L. Xiao, and X.F. Shi. 2011. The antifibrotic effects of TGF-β1 siRNA on hepatic fibrosis in rats. Biochemical and Biophysical Research Communications 409: 448–453.
Sun, J., Y. Wu, C. Long, P. He, J. Gu, L. Yang, Y. Liang, and Y. Wang. 2018. Anthocyanins isolated from blueberry ameliorates CCl4 induced liver fibrosis by modulation of oxidative stress, inflammation and stellate cell activation in mice. Food and Chemical Toxicology 120: 491–499.
Weber, L.W., M. Boll, and A. Stampfl. 2003. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Critical Reviews in Toxicology 33: 105–136.
Czaja, A.J., and H.A. Carpenter. 2004. Progressive fibrosis during corticosteroid therapy of autoimmune hepatitis. Hepatology (Baltimore, Md.) 39: 1631–1638.
Cohen-Naftaly, M., and S.L. Friedman. 2011. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therapeutic Advances in Gastroenterology 4: 391–417.
Wheeler, M.D., H. Kono, M. Yin, M. Nakagami, T. Uesugi, G.E. Arteel, E. Gabele, I. Rusyn, S. Yamashina, M. Froh, Y. Adachi, Y. Iimuro, B.U. Bradford, O.M. Smutney, H.D. Connor, R.P. Mason, S.M. Goyert, J.M. Peters, F.J. Gonzalez, R.J. Samulski, and R.G. Thurman. 2001. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radical Biology & Medicine 31: 1544–1549.
Satta, S., A.M. Mahmoud, F.L. Wilkinson, M. Yvonne Alexander, and S.J. White. 2017. The role of Nrf2 in cardiovascular function and disease. Oxidative Medicine and Cellular Longevity 2017: 18.
Jia, S., X. Liu, W. Li, J. Xie, L. Yang, and L. Li. 2015. Peroxisome proliferator-activated receptor gamma negatively regulates the differentiation of bone marrow-derived mesenchymal stem cells toward myofibroblasts in liver fibrogenesis. Cellular Physiology and Biochemistry 37: 2085–2100.
Wu, M., D.S. Melichian, E. Chang, M. Warner-Blankenship, A.K. Ghosh, and J. Varga. 2009. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. The American Journal of Pathology 174: 519–533.
Author information
Authors and Affiliations
Contributions
AMM, WGH, and IHH conceived the study and designed the experiments. AMM and ES performed the experiments. AMM analyzed the data, prepared the figures, and wrote the manuscript. All the authors participated in the assays, revised the manuscript, and approved the submission.
Corresponding author
Ethics declarations
The experimental protocol and all animal procedures were approved by the Institutional Animal Ethics Committee of Beni-Suef University (Egypt).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mahmoud, A.M., Hozayen, W.G., Hasan, I.H. et al. Umbelliferone Ameliorates CCl4-Induced Liver Fibrosis in Rats by Upregulating PPARγ and Attenuating Oxidative Stress, Inflammation, and TGF-β1/Smad3 Signaling. Inflammation 42, 1103–1116 (2019). https://doi.org/10.1007/s10753-019-00973-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-00973-8