
Abstract
Gypenoside IX (GP IX) is a pure compound isolated from Panax notoginseng. Gypenosides have been implicated to benefit the recovery of enormous neurological disorders. By suppressing the activation of astrocytes, gypenosides can improve the cognitive impairment. However, so far, little is known about whether GP IX could restrain the inflammatory responses in astrocytes or reactive astrogliosis. In present study, the anti-inflammatory effects of GP IX were investigated in reactive astrocytes induced by proinflammatory mediators both in vitro and in vivo. GP IX significantly reduced the production of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) at either protein or mRNA level in glial cell line C6 cells stimulated by lipopolysaccharide (LPS)/TNF-α combination. It also alleviated the astrogliosis and decreased the production of inflammatory mediators in brain cortex of LPS-treated mice. Further study disclosed that GP IX inhibited nuclear translocation of nuclear factor kappa B (NFκB) and reduced its transcriptional activity. Meanwhile, GP IX significantly attenuated the phosphorylation of NFκB, inhibitor of kappa B (IκB), Akt, and p38 mitogen-activated protein kinase (MAPK) under inflammatory conditions both in vitro and in vivo. These findings indicated that GP IX might suppress reactive astrogliosis by suppressing Akt/p38 MAPK/NFκB signaling pathways. And GP IX might be a promising drug candidate or prodrug for the therapy of neuroinflammatory disorders characterized with reactive astrogliosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Neuroinflammation is a kind of inflammatory response that occurs in the progression of neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and amylotrophic lateral sclerosis [21, 25]. As a type of glial cell in the central nervous system (CNS), astrocytes actively participate in the regulation of neuroinflammatory responses in neurological diseases [22]. Once activated, astrocytes display prominent morphological and molecular changes, including hypertrophy of astrocytic processes and upregulation of glial fibrillary acidic protein (GFAP), the key constituent of astrocyte intermediate filaments [22]. Reactive astrocytes secrete and respond to a number of important cytokines such as interleukin-6 (IL-6) [16], tumor necrosis factor-α (TNF-α) [10], interleukin-1β (IL-1β) [12, 13], thereby, affect the cellular state of surrounding microglia and neurons as well as that of themselves.
Many signaling pathway molecules are involved in the inflammatory responses of astrocytes. For instance, nuclear factor kappa B (NFκB), a transcription factor, has been revealed to participate in astrocyte-mediated neuroinflammation in the context of spinal cord injury and experimental autoimmune encephalitis [3, 4]. PI3K/Akt pathway has been proposed for the active regulation of LPS-primed reactive astrocytes [35], which has a cross talk with NFκB pathway [11]. Additionally, astrocyte activation is strongly linked to p38 mitogen-activated protein kinase (MAPK) signaling [18]. Deactivation of these inflammation-related signaling pathways may contribute to the alleviation of inflammatory responses in astrocytes.
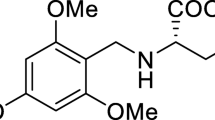
Panax notoginseng (Burk.) F.H. Chen has a long history in treating cerebral vascular disease in China as it promotes fibrinolysis, decreases fibrinogen, and prevents platelet aggregation [19]. Gypenosides from P. notoginseng have been shown to benefit the recovery of many neurological disorders, such as cerebral ischemia [33, 36], Alzheimer’s disease [5], Parkinson’s disease [32], and anxiety [26, 37]. And it was reported that gypenosides can improve the cognitive impairment by suppressing the activation of astrocytes [36]. However, so far, little is known whether gypenoside IX (GP IX, Fig. 1), one of the molecules within gypenosides, can suppress reactive astrogliosis. Here, the effects of GP IX on the prevention of reactive astrogliosis both in vitro and in vivo were investigated for the first time and the possible underlying mechanisms were discussed. Our results showed that GP IX alleviated the inflammatory responses in C6 astrocyte cells stimulated by LPS/TNF-α combination and in brain cortex of mice induced by LPS. Furthermore, GP IX suppressed the activation of p38 MAPK/Akt/NFκB signaling pathways both in vitro and in vivo. These findings indicated that GP IX might be a promising drug candidate or prodrug for the therapy of neuroinflammatory disorders characterized with astrocyte activation.

The chemical structure of GP IX.
MATERIALS AND METHODS
Reagents
GP IX (purity > 98%) was provided by the Shanghai Research Center for Standardization of Chinese Medicines (Shanghai, China). Lipopolysaccharide from Escherichia coli 055:B5 and NG-Methyl-L-arginine acetate salt (L-NMMA) were obtained from Sigma-Aldrich Company (St. Louis, MO, USA). Recombinant rat TNF-α was purchased from Peprotech (Rocky Hill, NJ, USA). Primary antibodies against phospho-NFκB (cat# 3033S), NFκB (cat# 6956), phospho-IκB (cat# 2859S), IκB (cat# 4812S), phospho-p38 MAPK (cat# 9211), p38 MAPK (cat# 9212), phospho-Akt (cat# 9271), Akt (cat# 9272), β-actin (cat# 4967L), and GAPDH (cat# 5174) were provided by Cell Signaling Technology (Danvers, MA, USA). Antibody against histone (ab181973) was obtained from Abcam (Cambridge, London, UK). Secondary antibodies conjugated with horseradish peroxidase were purchased from Life Technologies (Grand Island, NY, USA).
Cell Culture and Treatment
Rat C6 glial cell line was obtained from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibico Co., NY, USA) containing 10% fetal bovine serum and antibiotics (100 IU/ml of penicillin and 100 μg/ml of streptomycin) at 37 °C in a humidified atmosphere with 5% CO2. Prior to drug treatment, C6 cells were seeded in 96-well plates at a density of 1.5 × 106 cells/ml and cultured overnight. For cell viability analysis, the cells were treated with GP IX (0, 3.125, 6.25, 12.5, 25, and 50 μM) or L-NMMA (4 μM) for 24 h. Cell viability after treatment was measured using CCK-8 kit according to the manual of the manufacturer (Dojindo, Kumamoto, Japan).
Proinflammatory Factor Measurement
The cells were seeded in 96-well plates at a density of 1.5 × 106 cells/ml and cultured overnight. Then, they were stimulated with LPS (1 μg/ml) and TNF-α (10 ng/ml) for 24 h after pre-treated with GP IX (0, 3.125, 6.25, 12.5, 25, and 50 μM) or L-NMMA (4 μM) for 2 h. The released NO in the medium was determined by measuring nitrite concentration using Griess assay as described previously [15]. In brief, the culture medium was collected and mixed with equal volume of Griess reagent (Sigma-Aldrich). Optical absorbance of the mixture was measured immediately at 540 nm. The concentration of nitrate in the medium was quantified by a standard curve of sodium nitrate. The data was expressed as the fold change over the control. Concentrations of TNF-α and IL-6 secreted in the medium by the cells were determined, respectively, using ELISA kits according to the manufacturer’s instructions (eBioscience, San Diego, CA, USA).
NFκB-Luciferase Assay
C6 cells (7 × 105 cells/ml) were seeded in 24-well plates and cultured overnight before transfection. After grown to 90% confluence, the cells were co-transfected with NFκB reporter plasmid (pNFκB-luc, Beyotime, Nantong, Jiangsu, China) and Renilla-luciferase plasmid using lipofectamine 2000 according to the manufacturer’s instruction (Life Technologies). Six hours later, the cells were serum-starved and treated with GP IX (10, 50, 100 μM) for 2 h followed by LPS (1 μg/ml) and TNF-α (10 ng/ml) stimulation for 24 h. The cells were then lysed and subjected to NFκB-luciferase assay with Dual-Luciferase Reporter Gene Assay kit (Promega, WI, USA). The final NFκB-luciferase activity was normalized to the Renilla-luciferase activity within the same sample.
Animals
All animal experiments were carried out according to a protocol approved by the University Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine (SHUTCM). Five-week-old male C57BL/6 mice were obtained from Experimental Animal Center of SHUTCM and acclimatized for 1 week with free access to food and water. Thereafter, the mice were randomly divided into six groups, namely control group, LPS group, dexamethasone (Dex) group, and GP IX-low, -middle, -high groups (n = 9/group). The control group mice were intraperitoneally (i.p.) injected with PBS daily. The GP IX group mice were administrated i.p. with GP IX at doses of 3 (GP IX-L), 10 (GP IX-M), and 30 (GP IX-H) mg/kg/day, respectively, while the Dex group mice were given Dex i.p. at the dose of 3 mg/kg/day. On the day 7, all mice except that in the control group were i.p. injected with LPS (3 mg/kg). On the day 8, after given respective drug treatment as before for 2 h, the mice were anesthetized with excessive urethane and the brain cortices were dissected for further analysis.
Immunohistochemistry
For the histological analysis, mice were anesthetized with 2% pentobarbital sodium and perfused transcardially with 0.1 M PBS, followed by 4% paraformaldehyde. Coronal sections of brain at 20 μm-thick were obtained on a Leica 1950 cryostat. Immunohistochemistry (IHC) procedure was performed according to the method described previously (He et al., 2013). In brief, the sections were permeabilized and blocked with 10% donkey serum in PBS containing 0.3% Triton X-100 for half an hour. Consequently, they were incubated with primary antibody against GFAP (Santa Cruz Biotechnology, cat#3670) at 4 °C overnight followed by thoroughly washing with PBS for three times. After incubation with secondary antibody conjugated with Alexa 488 (Invitrogen, cat#A21208), the sections were mounted on slides. Fluorescence was visualized using an inverted fluorescent microscope (Olympus IX 81, Japan).
Quantitative RT-PCR
Total RNAs were isolated from the tissues or cells using Trizol reagent and transcripted into cDNA with RevertAid First Strand cDNA Synthesis kit (Life Technologies). Real-time PCR was performed using Taqman SYBR kit (Life Technologies). The expression of respective genes was normalized to that of β-actin or GAPDH within the same sample using delta delta CT method. The sequences of primers used were listed in Tables 1 and 2.
Sample Preparation for Western Blotting Analysis
C6 cells cultured in 6-well plates were pre-treated with or without GP IX (25 μM) for 6 h followed by stimulation of LPS and TNF-α for 1 h. For the translocation of NFκB analysis, cytoplasmic and nuclear protein fractions were extracted with Ne-Per Nuclear and Cytoplasmic extraction kit (Thermo Fisher Scientific, Waitham, MA, USA) and dissolved in lysis buffer (CelLytic™ MT mammalian tissue lysis reagent with protease and phosphatase inhibitor cocktails). For the other purposes, the cells were directly lysed in lysis buffer. For the brain cortices, they were homogenized in lysis buffer with a homogenizer (Polytron, PT-MR 1600 E, Switzerland). All protein samples were centrifuged at 12000 rpm for 10 min at 4 °C. Afterwards, they were subjected to western blotting analysis as described elsewhere [15, 34].
Immunocytochemistry
C6 cells were pre-treated with or without GP IX (25 μM) for 6 h before the stimulation of LPS and TNF-α. One hour later, the cells were subjected to immunocytochemistry (ICC) procedure as described previously [17].
Statistical Analysis
All data were expressed as mean ± S.D. All the comparisons among groups were conducted by one-way ANOVA with Dunnett’s post hoc test. Differences were regarded as statistically significant as P value < 0.05.
RESULTS
GP IX Prevented the Production of Inflammatory Mediators in C6 Cells Stimulated by LPS and TNF-α
To determine whether GP IX had direct cytotoxicity on C6 cells, the cell viability was measured using CCK-8 kit. As revealed in Fig. 2a, GP IX treatment from 3.125 to 50 μM for 24 h did not influence the cell viability. Compared with the un-stimulated cells, C6 cells stimulated with LPS and TNF-α combination released much higher level of NO as shown by increased nitrite concentration in the culture medium (Fig. 2b, P < 0.01 or P < 0.001). GP IX pre-treatment dose-dependently reduced the release of NO (P < 0.01 or P < 0.001) with an IC50 around 25 μM. Further western blotting assay exposed that GP IX (25 μM) could markedly reduce the elevation of iNOS induced by LPS and TNF-α (Fig. 2c, P < 0.001), and mitigate the secretion of IL-6 and TNF-α into the medium (Fig. 2d, e, P < 0.001 and P < 0.01). L-NMMA, the iNOS inhibitor used as a positive drug, pre-treatment inhibited the release of NO significantly (P < 0.001). However, it induced the expression of iNOS (P < 0.05). Meanwhile, L-NMMA pre-treatment prevented the secretion of TNF-α significantly (P < 0.01).

GP IX prevented the production of inflammatory mediators in C6 cells induced by LPS and TNFα. a GP IX treatment at different doses (0, 3.125, 6.25, 12.5, 25, 50 μM) for 24 h did not change the cell viability of C6 cells. b GP IX pre-treatment at different doses (3.125, 6.25, 12.5, 25, 50 μM) and L-NMMA pre-treatment (4 μM) for 2 h followed by LPS and TNF-α stimulation for 24 h inhibited the NO production in C6 cells. c GP IX pre-treatment (25 μM) for 2 h suppressed the protein expression of iNOS in C6 cells upon LPS and TNF-α stimulation for 24 h. d, e GP IX pre-treatment (25 μM) prevented the elevation of IL-6 and TNF-α concentration in C6 cell culture medium. Model group, treated with LPS (1 μg/ml) and TNF-α (10 ng/ml). *P < 0.05; **P < 0.01; ***P < 0.001 vs model group, mean ± S.D. The data shown were one of the representative results from at least three independent experiments.
Upon stimulation by LPS and TNF-α combination, mRNA expression of inflammatory mediators including iNOS, TNF-α, IL-6, IL-1β, and COX-2 was increased significantly in C6 cells (Fig. 3, P < 0.01 or P < 0.001). Pre-treatment with L-NMMA prevented the upregulation of iNOS, TNF-α, and COX-2 (P < 0.05 or P < 0.001) at mRNA level but did not change that of IL-6. Moreover, L-NMMA even induced much higher mRNA expression of IL-1β (P < 0.001). In contrast, GP IX pre-treatment counteracted the upregulated mRNA expression of the inflammatory mediators induced by LPS and TNF-α combination. These results indicated that GP IX could attenuate the inflammatory responses in C6 cells stimulated with LPS and TNF-α.

GP IX prevented the mRNA expressions of inflammatory mediators in C6 cells induced by LPS and TNFα. a–e GP IX pre-treatment (25 μM) for 2 h suppressed the mRNA expressions of iNOS, TNFα, IL-6, IL1β, and COX-2 in C6 cells stimulated with LPS (1 μg/ml) and TNF-α (10 ng/ml) for 24 h. The concentration of L-NMMA was used at 4 μM. Model group, treated with LPS and TNF-α combination. *P < 0.05; **P < 0.01; ***P < 0.001 vs model group, mean ± S.D. The data shown were one of the representative results from at least three independent experiments.
GP IX Deactivated p38MAPK/Akt/NFκB Signaling in C6 Cells Induced by LPS and TNF-α
LPS and TNF-α stimulation enhanced NFκB activation and translocation in astrocytes. To determine whether GP IX pre-treatment could inhibit the process, C6 cells were transfected with NFκB reporter plasmid. Our un-published data showed that the transfection efficiency of NFκB reporter plasmid was over 60% in C6 cells. As exposed in Fig. 4a, GP IX pre-treatment dose-dependently reduced NFκB-luciferase activity in C6 cells induced by LPS and TNF-α (P < 0.01 or P < 0.001). On cultured primary astrocytes, LPS and TNF-α stimulation induced the nuclear translocation of NFκB, which was weakened by GP IX pre-treatment (Fig. 4b). Consistently, the phosphorylation of NFκB in nucleus and IκB in cytoplasm of C6 cells was reduced by GP IX (Fig. 4c, d P < 0.01 and P < 0.05). Meanwhile, the phosphorylation of p38MAPK and Akt was also ameliorated significantly by GP IX (Fig. 4e, f, P < 0.01 and P < 0.001). These results implicated that the attenuated inflammatory responses in astrocytes by GP IX was closely associated with p38MAPK/Akt/NFκB signaling pathways.

GP IX deactivated p38MAPK/Akt/NFκB signaling in C6 cells induced by LPS and TNFα. a GP IX pre-treatment for 2 h dose-dependently alleviated NFκB-luciferase activity in C6 cells induced by LPS (1 μg/ml) and TNF-α (10 ng/ml) for 24 h. b GP IX pre-treatment (25 μM) for 6 h weakened NFκB nuclear translocation in C6 cells induced by LPS and TNF-α stimulation for 1 h. Scale bar, 25 μm. c, d GP IX pre-treatment (25 μM) for 6 h reduced the phosphorylation of NFκB in nucleus and IκB in cytoplasm of C6 cells induced by LPS and TNF-α stimulation for 1 h. e, f GP IX pre-treatment (25 μM) for 6 h significantly ameliorated the phosphorylation of p38 MAPK and Akt of C6 cells induced by LPS and TNF-α stimulation for 1 h. The data shown were one of the representative results from at least three independent experiments. Model group, treated with LPS and TNF-α combination. *P < 0.05; **P < 0.01; ***P < 0.001 vs model group, N = 4/group, mean ± S.D.
GP IX Ameliorated Inflammatory Responses in Brain Cortex of LPS-Induced Mice
In brain cortex of LPS-treated mice, the mRNA expression of inflammatory genes, such as iNOS, TNF-α, IL-6, and COX-2, was upregulated significantly (Fig. 5, P < 0.05, P < 0.01 or P < 0.001). Dex, the positive drug, inhibited the elevation of these inflammatory genes induced by LPS (P < 0.01 or P < 0.001). GP IX, especially used at 30 mg/kg/day, prevented the increase of the inflammatory genes (P < 0.05, P < 0.01, or P < 0.001). Therefore, the highest dose was chosen for the consequent studies. Consistent with its effect on the mRNA expression levels of inflammatory mediators, GP IX treatment reduced the production of TNF-α, IL-6, and iNOS at protein levels in brain cortex (Fig. 6, P < 0.05 or P < 0.01).

GP IX ameliorated inflammatory responses in brain cortex of LPS-induced mice. a–d GP IX pre-treatment prevented the increase of mRNA expression of the inflammatory genes, such as iNOS, TNFα, IL-6, and COX-2. LPS, 3 mg/kg; Dex, 3 mg/kg/day; IX-L, 3 mg/kg/day; IX-M, 10 mg/kg/day; IX-H, 30 mg/kg/day. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group, N = 4/group, mean ± S.D.

GP IX treatment reduced the production of TNFα, IL-6, and iNOS in brain cortex. a, b GP IX inhibited the protein expressions of TNF-α and IL-6. c GP IX suppressed protein expression of iNOS. LPS, 3 mg/kg; Dex, 3 mg/kg/day; GP IX, 30 mg/kg/day. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group, N = 5/group, mean ± S.D. The data shown were one of the representative results from at least three independent experiments.
Furthermore, GP IX administration could alleviate astrogliosis in LPS-treated mice. As shown in Fig. 7a, LPS induced prominent astrogliosis in mouse brain cortex. The cell bodies of the activated astrocytes became enlarged with retracted end feet. After GP IX and Dex treatment, the cell morphology resumed normal and was similar to that in the control mice. Meanwhile, both mRNA and protein analysis exposed that GP IX and Dex could impede the elevation of GFAP in brain cortex of LPS-treated mice (Fig. 7b, c, P < 0.05, P < 0.01, or P < 0.001).

GP IX administration deactivated astrocyte activation. a Immunostaining results showed that GP IX treatment decreased GFAP positive astrocytes in cortex of LPS-induced mice. Scale bar, 50 μm. b GP IX inhibited mRNA expression of GFAP. N = 4/group. c GP IX suppressed protein expression of GFAP. LPS, 3 mg/kg; Dex, 3 mg/kg/day; GP IX, 30 mg/kg/day. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group, N = 4/group, mean ± S.D. The data shown were one of the representative results from at least three independent experiments.
GP IX Attenuated the Phosphorylation of p38 MAPK/Akt/NFκB Signaling Molecules in Brain Cortex of LPS-Induced Mice
In brain cortex of LPS-treated mice, the phosphorylation of NFκB, IκB, p38 MAPK, and Akt was enhanced significantly (Fig. 8, P < 0.05 or P < 0.01). Dex administration attenuated the phosphorylation of NFκB and IκB but did not alter that of p38 MAPK and Akt. In contrast, GP IX administration alleviated the phosphorylation of NFκB, IκB, p38 MAPK, and Akt, suggesting the participation of the signaling pathways in the preventive effect of GP IX against astrogliosis.

GP IX attenuated the phosphorylation of p38MAPK/AKT/NFκB signaling molecules in brain cortex of LPS-induced mice. a–d GP IX administration alleviated the phosphorylation of NFκB, IκB, p38 MAPK and AKT in brain cortex of LPS-induced mice. LPS, 3 mg/kg; Dex, 3 mg/kg/day; GP IX, 30 mg/kg/day. *P < 0.05; **P < 0.01 vs LPS group, N = 4/group, mean ± S.D. The data shown were one of the representative results from at least three independent experiments.
DISCUSSION
Reactive astrogliosis is a ubiquitous hallmark of neuroinflammation in CNS diseases. In present study, GP IX was found to inhibit astrocyte activation, suppress phosphorylation of p38 MAPK, Akt, and NFκB, and prevent the production of inflammatory mediators, such as iNOS, TNF-α, IL-6, IL-1β, and COX-2 under inflammatory conditions both in vitro and in vivo. However, our un-published data showed that GP IX did not change the profile of inflammatory mediators or suppress p38 MAPK/Akt/NFκB signaling in normal C6 cells (Supplementary Figs. 1 and 2). The findings might facilitate its potential use in the alleviation of reactive astrogliosis of CNS diseases.
Astrocytes express receptors that recognize pathogenic molecules, such as toll-like receptor-4, and can directly respond to proinflammatory stimuli [2, 30]. Astrocytes can also express iNOS, which, by generating NO, is a potent and pleiotropic mediator of several biological functions. Although the physiological roles of NO are numerous, it has been clearly demonstrated that it has bidirectional actions on neuron survival [8]. L-NMMA is an iNOS activity inhibitor [29] in our experiments, which displayed significant inhibition on the release of NO from C6 cells stimulated by LPS and TNF-α combination. However, L-NMMA slightly induced the protein expression of iNOS in cells stimulated with LPS and TNF-α, which might reflect the compensatory mechanism of the cells in response to the inhibition of iNOS activity. Meanwhile, it also effectively prevented the release of TNF-α but did not change that of IL-6, which partially indicated its un-specific inhibition on iNOS activity. In present study, GP IX could markedly reduce the elevation of iNOS induced by LPS and TNF-α at both mRNA and protein levels, suggesting that GP IX modulated iNOS activity in a different way from L-NMMA.
COX-2 is an enzyme responsible for the production of proinflammatory prostaglandins in various models of inflammation [27]. Prostaglandins sustain homeostatic functions and mediate inflammatory response [24]. Cytokines have been known to act as major regulators of inflammatory responses associated with brain infection and injury. During inflammation, TNF-α plays a key role in host defense by triggering and regulating the cytokine signaling cascade in neuronal cells, whereas IL-6, a multifunctional cytokine, exerts major regulatory effects on the inflammatory response [7, 23]. IL-1β is thought to damage neurons in epilepsy directly [31]. The current study demonstrated that GP IX markedly suppressed the production of COX-2, TNF-α, IL-6, and IL-1β, further substantiated the neuroprotective potential of GP IX by mitigating inflammation.
NFκB has been considered as a major target in the treatment of diseases associated with inflammation. In the resting stage, NFκB exists as an inactive complex bound to the inhibitory protein, IκB, in the cytoplasm [28]. Once phosphorylated, NFκB will be translocated to nucleus and trigger the transcription of downstream proinflammatory molecules. PI3K/Akt pathway is known to be involved in the expression of inflammatory mediators in microglia through the activation of NFκB following IκB degradation [14]. Additionally, the MAPK pathway also plays a critical role in hippocampal astrocytes activation [9]; some MAPKs such as p38 MAPK have become important targets for anti-inflammatory molecules [20]. In present study, GP IX was found to inhibit NFκB nuclear translocation and transcription activity. Furthermore, it deactivated both Akt and p38 MAPK signaling pathways, suggesting that GP IX inhibited the activation of astrocytes through these pathways.
In CNS, besides astrocytes, microglia and oligodendrocytes can produce inflammatory responses upon stimulation by proinflammatory mediators [1, 6]. In present study, GP IX was found to reduce the production of inflammatory mediators in the cortex of LPS-induced mice. However, the anti-inflammatory effect of GP IX in mice might be a collective response of CNS glial cells. Moreover, although astrogliosis in mouse cortex was suppressed by GP IX, one could not exclude that it might reflect its indirect inhibitory effect on microglia or oligodendrocytes. Further investigation is needed to explore if GP IX could reduce the inflammatory responses in other CNS cells in addition to astrocytes.
CONCLUSIONS
To our knowledge, this is the first study showing that GP IX suppressed reactive astrogliosis induced by proinflammatory mediators. The novel findings might pave the road for the clinical application of GP IX in the relief of astrocyte-mediated neuroinflammation.
References
Ballerini, P., F. Diomede, N. Petragnani, S. Cicchitti, I. Merciaro, Mfxb Cavalcanti, and O. Trubiani. 2017. Conditioned medium from relapsing-remitting multiple sclerosis patients reduces the expression and release of inflammatory cytokines induced by LPS-gingivalis in THP-1 and MO3.13 cell lines. Cytokine 96: 261–272. doi:10.1016/j.cyto.2017.04.022.
Berger, J.V., A.O. Dumont, M.C. Focant, M. Vergouts, A. Sternotte, A.G. Calas, S. Goursaud, and E. Hermans. 2012. Opposite regulation of metabotropic glutamate receptor 3 and metabotropic glutamate receptor 5 by inflammatory stimuli in cultured microglia and astrocytes. Neuroscience 205: 29–38. doi:10.1016/j.neuroscience.2011.12.044.
Bozic, I., D. Savic, D. Laketa, I. Bjelobaba, I. Milenkovic, S. Pekovic, N. Nedeljkovic, and I. Lavrnja. 2015. Benfotiamine attenuates inflammatory response in LPS stimulated BV-2 microglia. PloS One 10 (2): e0118372. doi:10.1371/journal.pone.0118372.
Brambilla, R., T. Persaud, X. Hu, S. Karmally, V.I. Shestopalov, G. Dvoriantchikova, D. Ivanov, L. Nathanson, S.R. Barnum, and J.R. Bethea. 2009. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. Journal of Immunology 182 (5): 2628–2640. doi:10.4049/jimmunol.0802954.
Cai, H., Q. Liang, and G. Ge. 2016. Gypenoside attenuates beta amyloid-induced inflammation in N9 microglial cells via SOCS1 signaling. Neural Plasticity 2016: 6362707. doi:10.1155/2016/6362707.
Chung, D.W., K.Y. Yoo, I.K. Hwang, D.W. Kim, J.Y. Chung, C.H. Lee, J.H. Choi, et al. 2010. Systemic administration of lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in endothelium and increases microglia in the mouse hippocampus. Cellular and Molecular Neurobiology 30 (4): 531–541. doi:10.1007/s10571-009-9477-0.
Dheen, S.T., C. Kaur, and E.A. Ling. 2007. Microglial activation and its implications in the brain diseases. Current Medicinal Chemistry 14 (11): 1189–1197.
Estevez, A.G., N. Spear, S.M. Manuel, L. Barbeito, R. Radi, and J.S. Beckman. 1998. Role of endogenous nitric oxide and peroxynitrite formation in the survival and death of motor neurons in culture. Progress in Brain Research 118: 269–280.
Gorina, R., M. Font-Nieves, L. Marquez-Kisinousky, T. Santalucia, and A.M. Planas. 2011. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59 (2): 242–255. doi:10.1002/glia.21094.
Hamby, M.E., A.R. Gragnolati, S.J. Hewett, and J.A. Hewett. 2008. TGF beta 1 and TNF alpha potentiate nitric oxide production in astrocyte cultures by recruiting distinct subpopulations of cells to express NOS-2. Neurochemistry International 52 (6): 962–971. doi:10.1016/j.neuint.2007.10.010.
Hussain, A.R., S.O. Ahmed, M. Ahmed, O.S. Khan, S. Al Abdulmohsen, L.C. Platanias, K.S. Al-Kuraya, and S. Uddin. 2012. Cross-talk between NFkB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PloS One 7 (6): e39945. doi:10.1371/journal.pone.0039945.
Johnson, K.M., R. Milner, and S.J. Crocker. 2015. Extracellular matrix composition determines astrocyte responses to mechanical and inflammatory stimuli. Neuroscience Letters 600: 104–109. doi:10.1016/j.neulet.2015.06.013.
Keene, S.D., T.M. Greco, I. Parastatidis, S.H. Lee, E.G. Hughes, R.J. Balice-Gordon, D.W. Speicher, and H. Ischiropoulos. 2009. Mass spectrometric and computational analysis of cytokine-induced alterations in the astrocyte secretome. Proteomics 9 (3): 768–782. doi:10.1002/pmic.200800385.
Lee, J.Y., B.S. Jhun, Y.T. Oh, J.H. Lee, W. Choe, H.H. Baik, J. Ha, K.S. Yoon, S.S. Kim, and I. Kang. 2006. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neuroscience Letters 396 (1): 1–6. doi:10.1016/j.neulet.2005.11.004.
Li, H.L., H. Wu, B.B. Zhang, H.L. Shi, and X.J. Wu. 2016. MAPK pathways are involved in the inhibitory effect of berberine hydrochloride on gastric cancer MGC 803 cell proliferation and IL-8 secretion in vitro and in vivo. Molecular Medicine Reports 14 (2): 1430–1438. doi:10.3892/mmr.2016.5361.
Li, Y., L. Zhao, H. Fu, Y. Wu, and T. Wang. 2015. Ulinastatin suppresses lipopolysaccharide induced neuro-inflammation through the downregulation of nuclear factor-kappaB in SD rat hippocampal astrocyte. Biochemical and Biophysical Research Communications 458 (4): 763–770. doi:10.1016/j.bbrc.2015.01.155.
Liu, H.S., H.L. Shi, F. Huang, K.E. Peterson, H. Wu, Y.Y. Lan, B.B. Zhang, et al. 2016. Astragaloside IV inhibits microglia activation via glucocorticoid receptor mediated signaling pathway. Scientific Reports 6: 19137. doi:10.1038/srep19137.
Nahirnyj, A., I. Livne-Bar, X. Guo, and J.M. Sivak. 2013. ROS detoxification and proinflammatory cytokines are linked by p38 MAPK signaling in a model of mature astrocyte activation. PloS One 8 (12): e83049. doi:10.1371/journal.pone.0083049.
Ng, T.B. 2006. Pharmacological activity of sanchi ginseng (Panax notoginseng). The Journal of Pharmacy and Pharmacology 58 (8): 1007–1019. doi:10.1211/jpp.58.8.0001.
Oh, Y.T., J.Y. Lee, J. Lee, J.H. Lee, J.E. Kim, J. Ha, and I. Kang. 2010. Oleamide suppresses lipopolysaccharide-induced expression of iNOS and COX-2 through inhibition of NF-kappaB activation in BV2 murine microglial cells. Neuroscience Letters 474 (3): 148–153. doi:10.1016/j.neulet.2010.03.026.
Orre, M., W. Kamphuis, L.M. Osborn, A.H. Jansen, L. Kooijman, K. Bossers, and E.M. Hol. 2014. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiology of Aging 35 (12): 2746–2760. doi:10.1016/j.neurobiolaging.2014.06.004.
Pekny, M., M. Pekna, A. Messing, C. Steinhauser, J.M. Lee, V. Parpura, E.M. Hol, M.V. Sofroniew, and A. Verkhratsky. 2016. Astrocytes: a central element in neurological diseases. Acta Neuropathologica 131 (3): 323–345. doi:10.1007/s00401-015-1513-1.
Raivich, G., M. Bohatschek, C.U. Kloss, A. Werner, L.L. Jones, and G.W. Kreutzberg. 1999. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Research. Brain Research Reviews 30 (1): 77–105.
Ricciotti, E., and G.A. FitzGerald. 2011. Prostaglandins and inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology 31 (5): 986–1000. doi:10.1161/ATVBAHA.110.207449.
Rizzo, F., G. Riboldi, S. Salani, M. Nizzardo, C. Simone, S. Corti, and E. Hedlund. 2014. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cellular and Molecular Life Sciences 71 (6): 999–1015. doi:10.1007/s00018-013-1480-4.
Shin, K.S., T.T. Zhao, H.S. Choi, B.Y. Hwang, C.K. Lee, and M.K. Lee. 2014. Effects of gypenosides on anxiety disorders in MPTP-lesioned mouse model of Parkinson’s disease. Brain Research 1567: 57–65. doi:10.1016/j.brainres.2014.04.015.
Subbaramaiah, K., and A.J. Dannenberg. 2003. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends in Pharmacological Sciences 24 (2): 96–102. doi:10.1016/S0165-6147(02)00043-3.
Sun, S.C., and S.C. Ley. 2008. New insights into NF-kappaB regulation and function. Trends in Immunology 29 (10): 469–478. doi:10.1016/j.it.2008.07.003.
Szabo, C., G.J. Southan, C. Thiemermann, and J.R. Vane. 1994. The mechanism of the inhibitory effect of polyamines on the induction of nitric oxide synthase: role of aldehyde metabolites. British Journal of Pharmacology 113 (3): 757–766.
van Neerven, S., A. Nemes, P. Imholz, T. Regen, B. Denecke, S. Johann, C. Beyer, U.K. Hanisch, and J. Mey. 2010. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. Journal of Neuroimmunology 229 (1–2): 169–179. doi:10.1016/j.jneuroim.2010.08.005.
Viviani, B., S. Bartesaghi, F. Gardoni, A. Vezzani, M.M. Behrens, T. Bartfai, M. Binaglia, et al. 2003. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. The Journal of Neuroscience 23 (25): 8692–8700.
Wang, P., L. Niu, L. Gao, W.X. Li, D. Jia, X.L. Wang, and G.D. Gao. 2010. Neuroprotective effect of gypenosides against oxidative injury in the substantia nigra of a mouse model of Parkinson’s disease. The Journal of International Medical Research 38 (3): 1084–1092.
Wang, X.J., T. Sun, L. Kong, Z.H. Shang, K.Q. Yang, Q.Y. Zhang, F.M. Jing, et al. 2014. Gypenosides pre-treatment protects the brain against cerebral ischemia and increases neural stem cells/progenitors in the subventricular zone. International Journal of Developmental Neuroscience 33: 49–56. doi:10.1016/j.ijdevneu.2013.12.001.
Wu, H., Y. Gao, H.L. Shi, L.Y. Qin, F. Huang, Y.Y. Lan, B.B. Zhang, Z.B. Hu, and X.J. Wu. 2016. Astragaloside IV improves lipid metabolism in obese mice by alleviation of leptin resistance and regulation of thermogenic network. Scientific Reports 6: 30190. doi:10.1038/srep30190.
Xie, Z.F., G. Xin, Y.X. Xu, Y. Su, and K.S. Li. 2016. LPS-primed release of HMGB-1 from cortical astrocytes is modulated through PI3K/AKT pathway. Cellular and Molecular Neurobiology 36 (1): 93–102. doi:10.1007/s10571-015-0223-5.
Zhang, G.L., J.P. Deng, B.H. Wang, Z.W. Zhao, J. Li, L. Gao, B.L. Liu, et al. 2011. Gypenosides improve cognitive impairment induced by chronic cerebral hypoperfusion in rats by suppressing oxidative stress and astrocytic activation. Behavioural Pharmacology 22 (7): 633–644. doi:10.1097/FBP.0b013e32834afef9.
Zhao, T.T., K.S. Shin, H.S. Choi, and M.K. Lee. 2015. Ameliorating effects of gypenosides on chronic stress-induced anxiety disorders in mice. BMC Complementary and Alternative Medicine 15: 323. doi:10.1186/s12906-015-0856-4.
Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (81530096, 81673626, 81603354), Shanghai Sailing Program (17YF1417700), Natural Science Foundation of Shanghai (17ZR1430200), Educational Commission of Shanghai in China (2015YSN15), and Shanghai E-Research Institute of Bioactive Constituent in TCM plan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Xiaoshuang Wang and Liu Yang contribute equally to this work.
Electronic Supplementary Material
Supplementary Fig. 1
(DOCX 164 kb)
Supplementary Fig. 2
(DOCX 182 kb)
Rights and permissions
About this article

Cite this article
Wang, X., Yang, L., Yang, L. et al. Gypenoside IX Suppresses p38 MAPK/Akt/NFκB Signaling Pathway Activation and Inflammatory Responses in Astrocytes Stimulated by Proinflammatory Mediators. Inflammation 40, 2137–2150 (2017). https://doi.org/10.1007/s10753-017-0654-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0654-x