
Abstract
Cyanobacteria characterized by exceptional tolerance to environmental stresses often become pioneer settlers in habitats with harsh conditions. There, they can constitute the core of microbial communities. The taxonomic composition of the cyanobacterial component of algal–bacterial consortia dwelling in a habitat with particularly harsh conditions (rock baths at the coast of Kandalaksha Bay of the White Sea) has been elucidated for the first time. Two workflows of the taxonomic analysis of the cyanobacteria were tested including the combinations of two programs and two databases (QIIME+Greengenes and Usearch+NCBI GenBank). Our results obviated the need of the using of a complex approach combining morphological and metagenomic analyses for revealing the taxonomic structure of cyanobacteria in natural habitats. Our results show that the cyanobacterial component of the consortia from the habitats with harsh and highly volatile environmental conditions is enriched with non-diazotrophic and diazotrophic non-branched filamentous cyanobacteria.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phototrophic bacteria of the B10 phylum (Cyanobacteria) are typical for terrestrial and aquatic bacterial communities. In extreme environments cyanobacteria can exist within biofilms, multicomponent stable structures typically formed on air–water-solid substrate interface (Nikolaev & Plakunov, 2007). Adverse environmental conditions play a key role in biofilm formation since the biofilms stabilize environmental conditions and mitigate stresses (Paerl & Pinckney, 1996; Nikolaev & Plakunov, 2007).
In ecosystems of the White Sea cyanobacteria often exist in mucous layers on the surface of macrophytes, hydroid polyps, and stones (Gorelova et al., 2013). The supralittoral zone of the White Sea is characterized by sharp daily and seasonal changes in temperature, salinity, illumination, and humidity. Under the volatile conditions, cyanobacteria, heterotrophic bacteria, and eukaryotic microalgae exist within the biofilms (Mueller et al., 2005; Vincent, 2007). In many cases, cyanobacteria of subsections III and IV predominate over other components of the biofilms (Komárek & Anagnostidis, 2005; Mueller et al., 2005; Comte et al., 2007). Nostoc, Calothrix, Scytonema, and Oscillatoria are all capable of forming visually obvious crusts, biofilms, and microbial mats on the rock and ice surfaces (Vincent, 2000). Trichomes incorporated into the mats become desiccated to aid survival under the harsh conditions of polar night; the photosynthetic activity is restored during a short period in spring (Elster & Šabacká, 2006).
Microbial consortia of the White Sea region have attracted little study. Pesciaroli et al. (2012, 2015a, b) carried out a metagenomic study based on 16S ribosomal RNA gene (16SrRNA) of the marine bacteria. Belevich et al. (2015, 2017) described the diversity of White Sea eukaryotic picoalgae, while Krasnova et al. (2014, 2015) characterized microbial communities of meromictic lakes gradually separating from the sea. Several authors (Chekanov et al., 2014; Gorelova et al., 2009, 2012, 2015; Ismagulova et al., 2018) have described isolation and characterization of microalgal strains from the associations with invertebrates as well as microalgae dwelling in the rock baths of the Kandalaksha Bay. Cyanobacterial communities of this region have so far escaped the attention of researchers.
However, there have been metagenomic studies aimed at characterizing cyanobacteria conducted in regions with similar conditions (Iceland, the Wadden Sea, the Baltic Sea, Arctica, Antarctica) at the genus/species level (Varin et al., 2012; Maccario et al., 2014; Ininbergs et al., 2015; Palinska et al., 2017; Vogt et al., 2018).
In the water column of these aquatic systems unicellular cyanobacteria constitute the main group of primary producers. Palinska et al. (2017) investigated the Blue Lagoon (Iceland) establishing that Cyanobacterium aponinum I. Moro, N. Rascio, N. LaRocca, M. DiBella & C. Andreoli was the dominant cyanobacterium in this lake, while two genera of filamentous cyanobacteria (Halomicronema Abed, Garcia-Pichel et Hernández-Mariné, and Phormidium Kützing ex Gomont) were minor components. Ininbergs et al. (2015) showed that Synechococcus Nägeli and Cyanobium Rippka et Cohen-Bazire are the dominant picocyanobacteria in the Baltic Sea.
Filamentous and in some cases unicellular cyanobacteria are the dominant microbial group in the extreme littoral and supralittoral zones of the seas, ice, and snow. Maccario et al. (2014) reported on finding Nostocales and Chroococcales in snow communities; Varin et al. (2012) showed that cyanobacteria are dominants of the microbial mats on the Arctic and Antarctic ice shelves. Vogt et al. (2018) investigated the cyanobacterial populations. In the intertidal flats in the Wadden Sea (part of the North Sea), Coleofasciculus Siegesmund, J.R. Johansen et Friedl, Hydrocoleum Kützing ex Gomont, and Lyngbya C. Agardh ex Gomont (Oscillatoriales, III subsection) were the dominants.
Despite the essential role of cyanobacteria in ecosystems their systematics is far from being established (Komárek, 2018) and this complicates the analysis of their diversity. Recently, Komárek et al. (2014) proposed a commonly accepted system based on 16SrRNA gene and a complex of ultrastructural, ecological and physiological data. It is employed in widely used databases such as AlgaeBase or NCBI GenBank.
Use of a polyphasic approach (Komárek, 2018) presumes the involvement of highest possible number of the classification criteria. Accordingly, based on data from morphology, ecology and 16SrRNA sequencing is needed for correctly identify the cyanobacteria.
The aim of the present study was to characterize cyanobacterial diversity in supralittoral rock baths applying combined morphological and NGS sequencing based approaches.
Materials and methods
Environmental data
Samples with cyanobacteria were collected at the Rugozerskaya Gulf of Kandalaksha Bay in the White Sea (Louhi Region, Republic of Karelia, Russia) (Table 1). The samples were collected in the afternoons of the 19 and 20 July 2017. Air and water temperature were 17°C and 15°C, respectively; it was partly cloudy. The sample stations differed in height above the sea level (at high tide) and light regime (Table 1).
Sampling
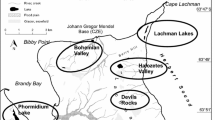
There were three sampling stations (Fig. 1a): (I) black rocks on the seacoast of Probkina Gubka Bay on the Kindo Peninsula (‘Maria’s Stones’, 66°32′24′′N; 33°11′2′′E); (II) stony slope on the coast of Probkina Gubka Bay; (III) the littoral pool on Pokormezhny Island (66°28′51′′N; 33°24′19′′E). The samples were taken from three locations on I (1, 2, 3), one on II (4), and one on III (5). The Black Maria’s Stones were located at the lower border of the littoral zone, and almost completely covered with water during high tide. Because of their black color and absence of shading, they were strongly heated by direct sunlight. The rock pools on the Maria’s Stones were filled with water from rains or splashes, though sometimes the water evaporated completely. The rock pool on the stony slope shaded by trees was approximately 3.5 m above the sea level at high tide. It was filled by freshwater inflow. The north-western coast of Pokormezhny Island, where the pool was located, is almost flat and 600 m long. It is totally covered by water during high tide, and almost dry and covered with crystals of salt during low tide.

Sampling stations and sampling locations in the Rugozerskaya Gulf of the Kandalaksha Bay of the White Sea (a); overview of the sampling locations (1–5) and corresponding sampling stations (I, II and III): the rock baths on the Pokormezhny Island (b) and on the Maria’s Stones (c), and their location towards the sea (d and e, respectively). SS sampling station
The samples (5–10 ml water) were collected from the rock baths in the supralittoral zone and then transferred to 15-ml sterile plastic tubes, frozen at − 24°C and stored at this temperature until metagenomic analysis. Sample names and origin are summarized in Table 1. Water salinity was measured with a Kelilong RHS-10ATC refractometer (Kelilong Electron Co. Ltd, China) calibrated with a series of standard NaCl solutions.
Microscopy
Samples were studied using a Leica DM 2500 photomicroscope equipped with DFC 7000 T camera (both Leica Microsystems, Germany) with both bright-field and fluorescent modes. Fluorescence was excited by a UV-lamp HXP 120 (Leica Microsystems, Germany). To detect the presence of cyanobacterial phycobilin pigments, cell fluorescence was performed using a Y3 band-pass filter (565–610 nm).
Morphological diversity of cyanobacteria in the samples was studied using the systematics from Bergey’s manual (Castenholz & Waterbury, 1989) which classifies the following subsections: I (simple unicellular), II (baeocyte-forming unicellular), III (heterocyst-free filamentous), IV (heterocyst-forming filamentous, without true branching), and V (heterocyst-forming filamentous, with true branching). We also used morphological descriptions from CyanoDB (Komárek & Hauer, 2013) and AlgaeBase (Guiry & Guiry, 2018) databases.
DNA isolation, amplification and sequencing
Genomic DNA was isolated from the samples by PowerSoil DNA Isolation Kit (MO BIO Laboratories, Inc., USA) in accord with the manufacturer’s protocol. The sequence corresponding to the variable loop V4 of the 16SrRNA gene was amplified by polymerase chain reaction (PCR) using the primers pair F515: 5′-gtgccagcmgccgcggtaa-3′ and R806: 5′-ggactacvsgggtatctaat-3′ reported by Bates et al. (2011), fused with Illumina adapters, a pad and a linker of two bases, along with barcodes. Amplification was performed by Encyclo DNA polymerases mixture (Evrogen, Russia). 25 μl of PCR mixture contained 10–15 ng genomic DNA, 0.05 μM of each deoxynucleotide triphosphates, 0.2 μM of each primer, one activity unit of the polymerase and the polymerase buffer system. DNA was amplified on a Mastercycler Gradient DNA amplifier (Eppendorf, Germany) under the following amplification profile: 98°C–15 s, 62°C–15 s, 72°C–15 s, 30 cycles, initial denaturation 98°C–60 s, final elongation 72°C–10 min. PCR products were purified by a MinElute Gel Extraction Kit (Qiagen, Germany).
Sequencing libraries were prepared according to Illumina MiSeq Reagent Kit Preparation Guide. PCR products were denatured in 0.1 M NaOH and diluted to DNA concentration of 15 pM. Obtained solution (510 µl) was mixed with 14 pM Phi phage library (Illumina, USA) (90 µl) as a control and sequenced on a MiSeq benchtop sequencer (Illumina, USA) using a Miseq 500 cycles kit (Illumina, USA) for 2 × 250 bp paired-ends sequencing.
Sequence data analysis
Direct and reverse sequences were combined. The dataset was cleaned from adaptor sequences, chimeric sequences and sequences of unsatisfactory quality. Sequences less than 200 nt long and higher than 1000 nt long were removed. The values were grouped into operational taxonomic units, OTU, by UCLUST (Edgar, 2010) and UPARSE (Edgar, 2013) clustering algorithms. Sequences were assigned to the same OTU if their homology was ≥ 97%. To make the metagenomic analysis more robust we applied two workflows of NGS data mining and homolog search. The first workflow included analysis in QIIME v 1.9.1 (Caporaso et al., 2010) in combination with Greengenes 16S rRNA-oriented (DeSantis et al., 2006) database (QIIME+GG workflow). The second workflow included the analysis in USEARCH v 10.0.240 (Edgar, 2010) software in combination with the OTU homolog search in the NCBI GenBank database (Benson et al., 2008) by BLAST (Altschul et al., 1997) (Usearch+GB workflow). In the latter case, taxonomic names corresponding to OTUs were manually checked vs. AlgaBase and CyanoDB databases. The program QIIME v 1.9.1 (algorithm UCLUST) was chosen due to its common use and multifunctionality (Kim et al., 2013). The program USEARCH v 10.0.240 (algorithm UPARSE) is also widely spread (Kim et al., 2013), and it has an advantage over QIIME in OTU clustering (Edgar, 2013). The database Greengenes is well-crafted, rapid and chimera-checked (DeSantis et al., 2006) and is often used in tandem with QIIME, hence that combination was chosen for the first workflow. The second workflow (Usearch+GB) was chosen for several reasons. First, the algorithm UPARSE is characterized by the ability to generate OTUs perfectly representing almost all detectable biological species (Edgar, 2013). Second, it provides the option of manual taxonomic identification and further checking it in independently compiled databases using recent taxonomy (AlgaeBase and CyanoDB). Nevertheless, the data processing through NCBI GenBank is slower and the data are not checked thoroughly. We adopted system proposed by Komárek et al. (2014) as the most authoritative for OTU classification and phylogeny reconstruction.
To confirm the taxonomic assignment and reconstruct evolutionary relationships of the cyanobacteria corresponding to OTUs, their sequences were aligned with fragments of bacterial 16SrRNA from the phylogenetic study (Komárek, 2015) using Muscle (Edgar, 2004) in MEGA 6.06 (Tamura et al., 2013). The phylogenetic tree was constructed with the neighbor-joining (NJ) method (Saitou & Nei, 1987). Accuracy of the tree topology was tested with a bootstrap (Felsenstein, 1985) and interior branch length (Rzhetsky & Nei 1992; Dopazo, 1994) methods (1,000 replicates).
Results
Description of the samples
Samples MS-1 and MS-2 (Table 1) consisted of water and fragments of cyanobacteria-containing reddish-brown biofilm. Samples MS-3, PG and PI were water with soil particles and macroscopic flocs of cyanobacteria. All the samples had green unicellular algae, motile dinoflagellates, heterotrophic bacteria and cyanobacteria. Haematococcus was the predominant chlorophyte in all samples studied. Fragments of other material, including rotifers and parts of other invertebrates, fungal spores and pollen were also observed. Sample PG also contained unicellular Chlorophyceae microalgae:most likely, Brachiomonas (Fig. S1a) and Chlamydomonas, Klebsormidium (Fig. S1b), pennate diatoms (Fig. S1c), green coccoid algae (Fig. S1d, e, f), and non-photosynthetic eukaryotic microorganisms. Biofilm-containing samples (MS-1, MS-2) were characterized by microorganisms with a well-developed extracellular matrix.
The salinity of Maria’s Stones samples (MS-1, MS-2,MS-3) was in the range of 1–6‰ which is less than that of sea water. Water in PG was fresh (Table 1). Sample PI had salinity of 130‰ (Table 1) and salt crystals present.
Microscopy
All five subsections of cyanobacteria were represented in the samples. However, only samples PG and PI had subsections V (branched filamentous heterocyst-forming cyanobacteria, probably Stigonema Agardh ex Bornet et Flahault) and I (non-baeocyte forming unicellular cyanobacteria) (Fig. 2a, b). Cyanobacteria of III subsection were predominant (MS-2, PG) or constituted one of the dominants (equally IV subsections in MS-1 and MS-3) in all samples other than PI. In the latter, subsection I was the dominant (Fig. 2c, d, e, f, g). MS-2 was represented exclusively by III subsection cyanobacteria. The non-heterocystous filaments in MS-2 formed bundles up to 300 µm in diameter with parallel-oriented trichomes that fitted Microcoleus Desmazières ex Gomont. Irregular fascicles (shortened trichomes) forming brownish-green non-branched filaments without heterocysts were observed in all the samples which fitted the description of Wilmottia O.Strunecky, J.Elster & J.Komárek. Samples MS-1 and MS-3 were characterized by many filaments of subsection IV (Fig. 2c, d, e, g). In MS-3 these included isopolar brown trichomes with occasional pseudo-branches (Fig. 2c). These filaments were cylindrical; average cells width and length were of 13 and of 3 µm, respectively. Another representative of subsection IV was found in the sample MS-1 and this fitted Rivularia Agardh ex Bornet et Flahault (Fig. 2d, g). Heteropolar brown trichomes with hemispherical basal and cylindrical intercalary heterocysts and yellow-brownish funnel-like sheaths widened at the ends in MS-3 (Fig. 2e) fitted Calothrix Agardh ex Bornet et Flahault.

Representative cyanobacteria of different morphological types encountered in the samples: a branched filamentous heterocyst-forming cyanobacteria, supposedly the genus Stigonema, in the sample PG (arrow); b Merismopedia-like unicellular cyanobacteria (UC), sample MS-1; c isopolar brown trichomes of the filamentous heterocyst-forming cyanobacteria from the IV subsection (1) and the filamentous heterocyst-free cyanobacteria from the III subsection (2), sample MS-3; d, e heteropolar brown filaments with hemispherical basal and cylindrical intercalary heterocysts without funnel-like sheaths widened at the ends (d), and with them (arrows) (e) in the samples MS-1 and MS-3, respectively; f fluorescence microphotograph of the filamentous heterocyst-free cyanobacteria from the III subsection, sample MS-3, e the representatives of the sample MS-1: cyanobacteria from the III (2) and IV (1) subsections, and the cells of carotenogenetic green algae Haematococcus spp. (Hs). UC unicellular cyanobacteria, Hc heterocyst, Hs cells of the green algae Haematococcus spp.
Metagenomic study
Based on QIIME+GG and Usearch+GB analysis, cyanobacteria were the predominant prokaryotes organisms in all samples. There was no significant difference in percentage of cyanobacterial reads obtained with the two workflows (Table 2).
The composition of the samples is shown in Table 3. Based on QIIME+GG there were six genera (Phormidium, Leptolyngbya Anagnostidis et Komárek, Rivularia, Nostoc Vaucher ex Bornet et Flahault, Chroococcidiopsis Geitler, Toxopsis Lamprinou, Skaraki, Kotoulas, Economou-Amili et Pantazidou) observed in the samples. Seven OTUs were identified only at family level (two belonging to Xenococcaceae, four to Nostocaceae, and one to Chamaesiphonaceae). Phormidium, Leptolyngbya, Chroococcidiopsis, and Nostocaceae were dominant. Based on Usearch+GB, five species were found: Wilmottia murrayi (West & G.S.West) Strunecký, Elster & Komárek, Plectolyngbya hodgsonii Taton, Wilmotte, Marda, Elster & Komárek, Microcoleus vaginatus Gomont ex Gomont, Microcoleus autumnalis (Gomont) Strunecky, Komárek & J.R.Johansen, and Stigonema elegans N.L.Gardner. In addition two OTUs, identified to genus (Nostoc, Chroococcidiopsis), six OTUs, identified to family (one Leptolyngbyaceae, two Rivulariaceae, three Godleyaceae) and two OTUs, identified to order level (Pleurocapsales, Chroococcales) presented. Species W. murrayi, M. vaginatus, P. hodgsonii, members of the family Godleyaceae and the order Chroococcales were dominant.
Thus, three subclasses were observed in all samples: Synechococcophycidae, Oscillatoriophycidae, Nostochopycidae. In MS-1, MS-2, and PI, Oscillatoriophycidae were predominant, while Nostochopycidae and Synechococcophycidae were predominant in MS-3 and PG.
The second workflow (Usearch+GB), which is based on recent taxonomic study, proved to be more convenient for our study.
Taxonomic assignment
For a better taxonomy assignment of OTUs obtained with the second workflow, a phylogenetic tree was constructed. On the NJ-based tree (Fig. 3), containing OTUs of predominant cyanobacteria, clusters corresponding to the five of the eight orders were distinguished. The OTUs identified as P. hodgsonii from MS-1, MS-3, PG, and PI and also Leptolyngbya sp. from PI clustered with Synechococcales. The single representative of Pleurocapsales from MS-1 nested in this cluster, as did the OTU corresponding to Chroococcales from PI. OTUs corresponding to Nostocales separated into Godleyaceae and Rivulariaceae; while Stigonema elegans from sample PG clustered with data from similar species. OTUs related to Oscillatoriales fitted with clusters corresponding to Microcoleus (MS-1, MS-2, PG) and Wilmottia (MS-1, MS-2, MS-3, PI).

Phylogenetic tree of cyanobacteria based on the 46 sequences of 16SrRNA gene. The taxa corresponding to the OTUs obtained in the present study and their sample IDs are underlined. The tree was calculated by the NJ algorithm. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test/the confidence probability that the interior branch length is greater than zero are shown near the corresponding branches. Only bootstrap values more 50% are shown. The taxa corresponding to the cyanobacterial OTUs from present work are highlighted. Scale bar: number of base substitutions per site
Annotation of plastid 16S rRNA sequences
The cyanobacterial 16SrRNA sequences were characterized by a high homology with plastid 16SrRNA. Based on BLAST search, they corresponded to plastid 16SrRNA of eukaryotic microalgae. Based on chloroplast ribosomal DNA sequences, Table 4 shows the eukaryotic algae in the samples. There were Chlorophyceae (Haematococcus, Chlamydomonas, Brachiomonas), Klebsormidiophyceae (Klebsormidium), Bacillariophyceae (Bacillaria and other diatoms), Trebouxiophyceae, Ulvophyceae, Chlorokybophyceae, and Eustigmatophyceae. These data were in good accord with observations through microscopy.
Discussion
The supralittoral baths on the White Sea coastal rocks contained algal–bacterial communities with cyanobacteria as the predominant prokaryotes. The sites included small freshwater pools and pools with moderately high or very high salinity. In case of eukaryotic algae, there were obvious differences, with the greatest diversity in freshwater sample PG: with diatoms, green, and other algae. Both microscopy and metagenomic data indicated a fewer algal genera in the saline samples. Haematococcus was the most abundant microalga. The genus, which is known to show resistance to the high solar irradiation and salinity typical of sea coastal rock pools (Chekanov et al., 2014).
In microbial communities, cyanobacteria play a key role in the regulation of interaction between prokaryotic and eukaryotic microorganisms. Their ability to form massive surface structures, mucous sheath, and capsules is well known (Baulina, 2012). They take part in the biofilm and microbial mat formation (Mueller et al., 2005; Vincent, 2007; Burow et al., 2012, 2013; Nozhevnikova et al., 2015) and this ability manifests itself under stressful conditions since it helps to mitigate the deleterious effects of stressors. Vincent (2007) described Cyanobacterial mechanisms for protection against long-term UV-irradiation and low temperatures.
The metagenomic study revealed the presence of Oscillatoriales in all samples. After processing according to the QIIME+GG workflow, these OTUs were related to Phormidium. This differed from the result obtained using the Usearch+GB workflow which split the OTUs into Microcoleus autumnalis and Wilmottia Murrayi except for MS-3 and PI, with only the latter. These data appeared to correlated with microscopy. Usearch+GB revealed the presence of Plectolyngbya hodgsonii in MS-1, MS-3, PG, and PI as well as Leptolyngbya sp. in PI, thus potentially corresponding to subsection III of Komárek & Hauer (2013). Both microorganisms were related to Synechococcales. However, according to the QIIME+GG workflow, Leptolyngbya previously belonged to Pseudoanabaenales, which is not present in the recent cyanobacterial system (Komárek et al., 2014). The difference between the results obtained by the two workflows in this case was due to the different taxonomy accepted in the corresponding databases. The older system was adopted by the Greengenes database, whereas a recently established taxonomy was in the GenBank database. Some species of Phormidium are currently synonymous with species of Microcoleus (P. autumnale Gomont = M. autumnalis) and Wilmottia (P. murrayi (West & G.S.West) Anagnostidis & Komárek = W. murrayi). Plectolyngbya hodgsonii, which is the only species of this genus from the Antarctic, is phylogenetically close to Leptolyngbya (Taton et al., 2011; Komárek, 2015). Microcoleus spp. are known to be the dominant cyanobacteria of littoral microbial mats according to some current taxonomy (Burow et al., 2012, 2013; Nozhevnikova et al., 2015), though they had previously been identified as Phormidium (Elster et al., 2002; Mueller et al., 2005). Microcoleus and Phormidium had been considered very close to each other taxonomically, so it was difficult to distinguish these two genera before the revision (Hašler et al., 2012).
All samples except MS-2 contained the OTUs corresponding to Nostocales. Notably, the QIIME+GG workflow assigned an OTU from MS-1 to Rivularia sp. (also Nostocales, where it was transferred after the recent revision system). According to recent classification, heterocyst-forming cyanobacteria form monophyletic group (order Nostocales) in phylum B10. Thus, most of the Nostocales OTUs in the samples corresponded to subsection IV. The Usearch+GB workflow assigned an OTU in the sample PG to the true branching Stigonema sp. and this fitted with microscopy. However, this taxon was not present in this sample in QIIME+GG results, perhaps due to a difference in the calculation algorithm.
Based on metagenomic analysis, the highest diversity of heterocystous cyanobacteria was in PG and PI. Most of their OTUs according to Usearch+GG calculation were assigned to the family Godlyeaceae (Hauer et al., 2014). Based on microscopy, a large number of heterocyst-containing isopolar filaments were present in these samples. Although the identification of Godleyaceae solely by morphology is problematic (Hauer et al., 2014), we suggested these organisms belong here, relying on metagenomic analysis evidence. Morphological observations on MS-3 showed two other cyanobacteria, Calothrix and Rivularia, although they were not revealed by the metagenomic study. Probably this was due to insufficient resolution using the method based on the 16SrRNA V4 fragment and hence incapability to resolve fully microbial systematics at lower taxonomic levels. Obviously, a more detailed analysis is needed in this case.
Microscopy indicated that unicellular cyanobacteria were present in MS-1, PG, and PI, whereas metagenomic analysis of OTUs, potentially corresponding to this morphology type, did not provide consistent results. Based on the QIIME+GG workflow, they were assigned to Xenococcaceae (Chroococcales) in MS-1 and PG, and to Chroococcidiopsis sp. (Chroococcales) in PI. Based on the Usearch+GB workflow, this OTU was assigned to non-filamentous cyanobacteria related to Pleurocapsales in MS-1. This was in line with our observations of baeocyte-forming microorganisms typical of this order. Moreover, Xenococcaceae are now related to Pleurocapsales (Komárek et al., 2014). In another two samples, the OTUs potentially corresponding to subsection I also were detected and assigned to Chroococcales or Chroococcidiopsidales.
In the samples characterized by a weak salinity (MS-1, MS-2, MS-3), non-heterocystous filamentous cyanobacteria predominated. In the freshwater sample (PG), heterocyst-forming and non-heterocystous representatives were quite similar in amount, whereas in PI (the hypersaline sample) unicellular cyanobacteria were predominant. Thus, the cyanobacterial composition of these samples was strongly influenced by the salinity.
Taxonomic assignment of the predominant cyanobacteria
Despite a high practical and ecological significance of cyanobacteria, their systematics is far from being fully established. The clusters resolved on the NJ-based phylogenetic tree corresponded to five out of eight orders of the currently accepted system; most belonged to the cluster in the Synechococcales. They were characterized by a high homology with 16SrRNA fragment sequences of P. hodgsonii (Leptolyngbyaceae) from the Antarctic region (Komárek, 2015). Most likely, filamentous cyanobacteria of this group are typical owing to the adverse conditions of polar and sub-polar zones. Conclusions from the genetic data were thus supported in this case by ecological similarity. Despite the possibility of paraphyletic origin of the Synechococcales (Komárek, 2016), the deduced plylogeny of Leptolyngbyaceae is reliably supported by the sequencing data, so it can be regarded as a well-distinguished monophyletic group. The Oscillatoriales is also represented by unicellular and filamentous heterocyst-free organisms (Komárek, 2016). Wilmottia and Microcoleus observed in the samples studied and they were assigned to the relatively well-described families Coelofasciculaceae and Microcoleaceae, respectively. These filamentous cyanobacteria are well-studied taxa with a clear phylogeny (Komárek, 2016). Despite the scarce molecular data on Microcoleus (Komárek, 2016), we regarded these genera as monophyletic groups. Wilmottia murrayi is also an endemic species from the polar region (Komárek, 2015). Some OTUs from the samples were considered as representatives of the monophyletic Nostocales (Komárek, 2016). The sequences from the PG, PI, and MS-3 samples were assigned to Godleyaceae and certain sequences from PG to the Stigonemataceae. The phylogeny of these Nostocales families is weakly elucidated and their members are represented by only a small amount of molecular data (Komárek, 2016). However, these clusters were supported by relatively high bootstrap values. Due to a small length of the 16SrRNA gene fragment used in this analysis, it was impossible to resolve the phylogeny of Pleurocapsales and Chroococcales and assign the related OTU below the level of orders. ThePleurocapsales have not been described fully in the recent phylogenetic studies (Komárek et al., 2014; Komárek, 2015, 2016; Wanigatunge et al., 2014). Ishida et al. (2001), who focused on this taxon, suggested their polyphyletic nature. Nevertheless, a predominant OTU (from MS-1) was characterized by close homology to several Pleurocapsales sequences and was assigned as such. Likewise, we assigned OTUs with a high homology to Pseudocapsa and Gloeocapsa (Chroococcaceae) 16SrRNA to Chroococcales. However, most of the genera in this order have scarcely been studied genetically (Komárek, 2016), so their monophyletic origin remains questionable.
Difficulties in studying cyanobacterial diversity
Our study based on comparison of metagenomic data and microscopy made the diversity of cyanobacteria in the supralittoral communities of the White Sea region clear. Our investigations were based on comparing the metagenomic data and microscopic observations. The diversity of cyanobacteria from this region has been described and novel cyanobacterial gene sequences were obtained for future analyses.
However, the study also demonstrated a difficulty for studies of the kind as there is the lack of an established taxonomic system (see Introduction). Information about some cyanobacterial groups is still lacking, especially molecular data on strains (Komárek, 2016). The situation is complicated by frequent taxonomic revisions and reclassifications (Komárek, 2016, 2018). This leads to a high number of synonyms, new taxa and conflicting systems established by different authors. There are also contradictions between the classification based on studies of in situ populations and those based on isolated strains in vitro (Komárek, 2018). The description of genera and species are based on the type of strains cultivated under laboratory conditions (Rippka et al., 1979). However, cyanobacteria are characterized by a high degree of phenotypic plasticity (Baulina, 2012). Therefore, their characteristics may differ between those shown in nature and cultivated under a range of laboratory conditions (Baulina, 2012). This leads to frequent mistakes in their identification, hence wrong names are frequently encountered in public databases such as NCBI GenBank. Moreover, some databases use different cyanobacterial systems, for example, the widely used 16SrRNA-based Greengenes and GenBank. Our work demonstrates how these discrepancies can lead to contradictory results in qualitative and quantitative assessment of cyanobacterial biodiversity by formal automatic analysis of metagenomic data. Thus, manual checking of the data is utterly important for precise taxonomic analysis. Additionally, the present research uses short fragments of 16SrRNA shown to be valueless for identification at the genus or even family level.
Conclusion
In conclusion, the commonly accepted species concept is largely inapplicable for prokaryotic organisms. Their classification does not rely on traditional taxa but on strains, designated by strain and genus name or binary species name, although this does not make them Linnaean species. The species concept is characterized by a complex of cytological, ecological, and molecular data. Therefore, a polyphasic approach based on the most complete set of all characters is needed to describe the prokaryotic organisms in laboratory culture and in their natural habitat with the necessary level of accuracy and precision.
Abbreviations
- 16SrRNA :
-
Gene of 16S ribosomal RNA
- NJ:
-
Neighbor-joining algorithm
- PCR:
-
Polymerase chain reaction
- OTU:
-
Operation taxonomic unit
- QIIME+GG:
-
Workflow of sequence data analysis in QIIME software and homolog search in the Greengenes database
- Usearch+GB:
-
Workflow of sequence data analysis in Usearch software and homolog search in the GenBank NCBI database
References
Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller & D. J. Lipman, 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25: 3389–3402.
Bates, S. T., D. Berg-Lyons, J. G. Caporaso, W. A. Walters, R. Knight & N. Fierer, 2011. Examining the global distribution of dominant archaeal populations in soil. The ISME Journal 5: 908–917.
Baulina, O. I., 2012. Ultrastructural Plasticity of Cyanobacteria. Springer, New York: 202.
Belevich, T. A., L. V. Ilyash, I. A. Milyutina, M. D. Logacheva, D. V. Goryunov & A. V. Troitsky, 2015. Metagenomic analyses of White Sea picoalgae: first data. Biochemistry 80: 1514–1521.
Belevich, T. A., L. V. Ilyash, I. A. Milyutina, M. D. Logacheva & A. V. Troitsky, 2017. Phototrophic picoeukaryotes of Onega Bay, the White Sea: abundance and species composition. Moscow University Biological Sciences Bulletin 72: 109–114.
Benson, D. A., I. Karsch-Mizrachi, D. J. Lipman, J. Ostell & D. L. Wheeler, 2008. GenBank. Nucleic Acids Research 36: D25–D30.
Burow, L. C., D. Woebken, B. M. Bebout, P. J. McMurdie, S. W. Singer, J. Pett-Ridge, et al., 2012. Hydrogen production in photosynthetic microbial mats in the Elkhorn Slough estuary, Monterey Bay. The ISME Journal l 6: 863.
Burow, L. C., D. Woebken, I. P. Marshall, E. A. Lindquist, B. M. Bebout, L. Prufert-Bebout, et al., 2013. Anoxic carbon flux in photosynthetic microbial mats as revealed by metatranscriptomics. The ISME Journal 74: 817.
Caporaso, J. G., J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, Peña A. Gonzalez, J. K. Goodrich, J. I. Gordon & G. A. Huttley, 2010. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7: 335–336.
Castenholz, R. W. & J. B. Waterbury, 1989. Taxa of the cyanobacteria. In Staley, J. T., M. P. Bryant, N. Pfenning & J. G. Holt (eds), Bergey’s Manual of Systematic Bacteriology. Williams and Wilkins, Baltimore: 1727–1728.
Chekanov, K., E. Lobakova, I. Selyakh, L. Semenova, R. Sidorov & A. Solovchenko, 2014. Accumulation of astaxanthin by a new Haematococcus pluvialis strain BM1 from the White Sea coastal rocks (Russia). Marine Drugs 12: 4504–4520.
Comte, K., M. Šabacká, A. Carre-Mlouka, J. Elster & J. Komárek, 2007. Relationships between the Arctic and the Antarctic cyanobacteria: three Phormidium-like strains evaluated by a polyphasic approach. FEMS Microbiol Ecology 59: 366–376.
DeSantis, T. Z., P. Hugenholtz, N. Larsen, M. Rojas, E. L. Brodie, K. Keller, T. Huber, D. Dalevi, P. Hu & G. L. Andersen, 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72: 5069–5072.
Dopazo, J., 1994. Estimating errors and confidence intervals for branch lengths in phylogenetic trees by a bootstrap approach. Journal of Molecular Evolution 38: 300–304.
Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797.
Edgar, R. C., 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edgar, R. C., 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods 10: 996–998.
Elster, J. & M. Šabacká, 2006. Response of cyanobacteria and algae from Antarctic wetland habitats to freezing and desiccation stress. Polar Biology 30: 31–37.
Elster, J., J. Svoboda, S. Ohtani & H. Kanda, 2002. Feasibility studies on future phycological research in polar regions. Polar Bioscience 15: 114–122.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Gorelova, O. A., I. A. Kosevich, O. I. Baulina, T. A. Fedorenko, A. Z. Torshkhoeva & E. S. Lobakova, 2009. Associations between the White Sea invertebrates and oxygen-evolving phototrophic microorganisms. Moscow University Biological Sciences Bulletin 64: 16–22.
Gorelova, O. A., O. I. Baulina, A. E. Solovchenko, T. A. Fedorenko, T. R. Kravtsova, O. B. Chivkunova, O. A. Koksharova & E. S. Lobakova, 2012. Green microalgae isolated from associations with white sea invertebrates. Microbiology 81: 505–507.
Gorelova, O. A., O. I. Baulina, I. A. Kosevich & E. S. Lobakova, 2013. Associations between the White Sea colonial hydroid Dynamena pumila and microorganisms. Journal of the Marine Biological Association of the United Kingdom 93: 69–80.
Gorelova, O. A., O. I. Baulina, A. E. Solovchenko, K. A. Chekanov, O. B. Chivkunova, T. A. Fedorenko & E. S. Lobakova, 2015. Similarity and diversity of the Desmodesmus spp. microalgae isolated from associations with White Sea invertebrates. Protoplasma 252: 489–503.
Guiry, M. D., & Guiry, G. M., 2018. AlgaeBase [Internet]. Galway: World-Wide Electronic Publication, National University of Ireland; [available on internet at http://www.algaebase.org].
Hašler, P., P. Dvořák, J. R. Johansen, M. Kitner, V. Ondřej & A. Poulíčková, 2012. Morphological and molecular study of epipelic filamentous genera Phormidium, Microcoleus and Geitlerinema (Oscillatoriales, Cyanophyta/Cyanobacteria). Fottea 12: 341–356.
Hauer, T., M. Bohunická, J. R. Johansen, J. Mareš & E. Berrendero-Gomez, 2014. Reassessment of the cyanobacterial family Microchaetaceae and establishment of new families Tolypothrichaceae and Godleyaceae. Journal of Phycology 50: 1089–1100.
Ininbergs, K. B., J. Larsson & M. Ekman, 2015. Microbial metagenomics in the Baltic Sea: recent advancements and prospects for environmental monitoring. Ambio 44: 439–450.
Ishida, T., M. M. Watanabe, J. Sugiyama & A. Yokota, 2001. Evidence for polyphyletic origin of the members of the orders of Oscillatoriales and Pleurocapsales as determined by 16S rDNA analysis. FEMS Microbiology Letters 201: 79–82.
Ismagulova, T., K. Chekanov, O. Gorelova, O. Baulina, L. Semenova, I. Selyakh, O. Chivkunova, E. Lobakova, O. Karpova & A. Solovchenko, 2018. A new subarctic strain of Tetradesmus obliquus – part I: identification and fatty acid profiling. Journal of Applied Phycology 30(5): 2737–2750.
Kim, M., K. H. Lee, S. W. Yoon, B. S. Kim, J. Chun & H. Yi, 2013. Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics & informatics 11: 102–113.
Komárek, J., 2015. About endemism of cyanobacteria in freshwater habitats of maritime Antarctica. Algological Studies 148: 15–32.
Komárek, J., 2016. A polyphasic approach for the taxonomy of cyanobacteria: principles and applications. European Journal of Phycology 51: 346–353.
Komárek, J., 2018. Several problems of the polyphasic approach in the modern cyanobacterial system. Hydrobiologia 811: 7–17.
Komárek, J. & K. Anagnostidis, 2005. Cyanoprokaryota. 2. Oscillatoriales. In Büdel, B., L. Krienitz, G. Gärtner & M. Schagerl (eds), Süsswasserflora von Mitteleuropa 19. Elsevier/Spektrum, Heidelberg: 759.
Komárek, J. & Hauer, T., 2013. CyanoDB. cz. On-line database of cyanobacterial genera. Word-wide electronic publication, University of South Bohemia and Institute of Botany AS CR [available on internet at http://www.cyanodb.cz].
Komárek, J., J. Kaštovský, J. Mareš & J. R. Johansen, 2014. Taxonomic classification of cyanoprokaryotes (cyanobacterial genera), using a polyphasic approach. Preslia 86: 295–335.
Krasnova, E. D., A. N. Pantyulin, D. N. Matorin, D. A. Todorenko, T. A. Belevich, I. A. Milyutina & D. A. Voronov, 2014. Cryptomonad alga Rhodomonas sp. (Cryptophyta, Pyrenomonadaceae) bloom in the redox zone of the basins separating from the White Sea. Microbiology 83: 270–277.
Krasnova, E. D., A. V. Kharcheva, I. A. Milyutina, D. A. Voronov & S. V. Patsaeva, 2015. Study of microbial communities in redox zone of meromictic lakes isolated from the White Sea using spectral and molecular methods. Journal of the Marine Biological Association of the United Kingdom 95: 1579–1590.
Maccario, L., T. M. Vogel & C. Larose, 2014. Potential drivers of microbial community structure and function in Arctic spring snow. Frontiers in Microbiology 5: 413.
Mueller, D. R., W. F. Vincent, S. Bonilla & I. Laurion, 2005. Extremophiles, extremotrophs and broadband pigmentation strategies in a high arctic ice shelf ecosystem. FEMS Microbiology Ecology 53: 73–87.
Nikolaev, Yu A & V. K. Plakunov, 2007. Biofilm – “City of microbes” or an analogue of multicellular organisms? Microbiology 76: 125–138.
Nozhevnikova, A. N., E. A. Botchkova & V. K. Plakunov, 2015. Multi-species biofilms in ecology, medicine, and biotechnology. Microbiology 84: 731–750.
Paerl, H. W. & J. L. Pinckney, 1996. A mini-review of microbial consortia: their roles in aquatic production and biogeochemical cycling. Microbial Ecology 31: 225–247.
Palinska, K. A., J. C. Vogt & W. Surosz, 2017. Biodiversity analysis of the unique geothermal microbial ecosystem of the Blue Lagoon (Iceland) using next-generation sequencing (NGS). Hydrobiologia 811: 93–102.
Pesciaroli, C., F. Cupini, L. Selbmann, P. Barghini & M. Fenice, 2012. Temperature preferences of bacteria isolated from seawater collected in Kandalaksha Bay, White Sea, Russia. Polar Biology 35: 435–445.
Pesciaroli, C., B. Rodelas, B. Juarez-Jiménez, P. Barghini & M. Fenice, 2015a. Bacterial community structure of a coastal area in Kandalaksha Bay, White Sea, Russia: possible relation to tidal hydrodynamics. Annals of Microbiology 65: 443–453.
Pesciaroli, C., P. Barghini, F. Cerfolli, B. Bellisario & M. Fenice, 2015b. Relationship between phylogenetic and nutritional diversity in Arctic (Kandalaksha Bay) seawater planktonic bacteria. Annals of Microbiology 65: 2405–2414.
Rippka, R., J. Deruelles, J. B. Waterbury, M. Herdman & R. Y. Stanier, 1979. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology 111: 1–61.
Rzhetsky, A. & M. Nei, 1992. A simple method for estimating and testing minimum-evolution trees. Molecular Biology and Evolution 9: 945–967.
Saitou, N. & M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406–425.
Tamura, K., G. Stecher, D. Peterson, A. Filipski & S. Kumar, 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30: 2725–2729.
Taton, A., A. Wilmotte, J. Šmarda, J. Elster & J. Komárek, 2011. Plectolyngbya hodgsonii: a novel filamentous cyanobacterium from Antarctic lakes. Polar Biology 34: 181–191.
Varin, T., C. Lovejoy, A. D. Jungblut, W. F. Vincent & J. Corbeil, 2012. Metagenomic analysis of stress genes in microbial mat communities from Antarctica and the High Arctic. Applied and Environmental Microbiology 78: 549–559.
Vincent, W. F., 2000. Cyanobacterial dominance in the dominance in the polar regions. In Whitton, B. A. & M. Potts (eds), Ecology of the Cyanobacteria: Their Diversity in Time and Space. Kluwer Academic Publishers, Dordrecht: 321–340.
Vincent, W. F., 2007. Cold tolerance in cyanobacteria and life in the cryosphere. In Seckbach, J. (ed.), Algae and Cyanobacteria in Extreme Environments. Springer, Heidelberg: 287–301.
Vogt, J. C., D. C. Albach & K. A. Palinska, 2018. Cyanobacteria of the Wadden Sea: seasonality and sediment influence on community composition. Hydrobiologia 811: 103–117.
Wanigatunge, R. P., D. N. Magana-Arachchi, N. V. Chandrasekharan & S. A. Kulasooriya, 2014. Genetic diversity and molecular phylogeny of cyanobacteria from Sri Lanka based on 16S rRNA gene. Environmental Engineering Research 19: 317–329.
Acknowledgements
Microscopic studies were carried out using equipment at the Center of Microscopy of White Sea Biological Station of Moscow State University. This work was supported partially by the Ministry of Science and Education of the Russian Federation (Grant Number 02.a03.21.0008 of 24 June 2016) and partially by Council for Grants of the President of the Russian Federation. The dedicated technical assistance of Dr. Yuriy Khlopko is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Stefano Amalfitano
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kublanovskaya, A., Chekanov, K., Solovchenko, A. et al. Cyanobacterial diversity in the algal–bacterial consortia from Subarctic regions: new insights from the rock baths at White Sea Coast. Hydrobiologia 830, 17–31 (2019). https://doi.org/10.1007/s10750-018-3844-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-018-3844-0