
Abstract
Talitrids are semiterrestrial crustacean amphipods inhabiting sandy and rocky beaches; they generally show limited active dispersal over long distances. In this study we assessed levels of population genetic structure and variability in the talitrid amphipod Orchestia montagui, a species strictly associated to stranded decaying heaps of the seagrass Posidonia oceanica. The study is based on six populations (153 individuals) and covers five basins of the Mediterranean Sea (Tyrrhenian, Ionian, Adriatic, Western and Eastern basins). Samples were screened for polymorphisms at a fragment of the mitochondrial DNA (mtDNA) coding for the cytochrome oxidase subunit I gene (COI; 571 base pairs) and at eight microsatellite loci. MtDNA revealed a relatively homogeneous haplogroup, which clustered together the populations from the Western, Tyrrhenian and Eastern basins, but not the populations from the Adriatic and Ionian ones; microsatellites detected two clusters, one including the Adriatic and Ionian populations, the second grouping all the others. We found a weak geographic pattern in the genetic structuring of the species, with a lack of isolation by distance at either class of markers. Results are discussed in terms of probability of passive dispersal over long distances through heaps of seagrass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The geographic distribution of species and the capability to spread over different areas largely derive from their dispersal ability (Slatkin 1987). This is, in turn, not only dependent on the intrinsic biological features of each species, but also on the characteristics of the colonized environment. Many marine animals that are sedentary or scarcely mobile as adults rely on meroplanktonic larvae for their dispersal. For those marine species that lack such a type of larval stage, the only avenue for long-distance dispersal is through assisted dispersal. Whatever the mechanism(s) involved in achieving dispersal, gene flow is of central importance in shaping patterns of population genetic structuring. Levels of gene flow, the movement and exchange of individuals or entire populations, may either constrain evolution by preventing adaptation to local conditions or promote evolution by spreading new combinations and/or variants of genes throughout a species’ range (Slatkin 1987).
Talitrids are crustacean amphipods that live in the supralittoral zone. Their physiological adaptations to survive in such a harsh coastal zone confine them to areas with high moisture and humidity levels. No planktonic larval stage is present in talitrids; females carry the embryos in their brood pouch and active dispersal of adults extends only over a few tens of meters on the beaches. To disperse over long distances, talitrids rely on passive transport. Different means of transportation have been hypothesized (i.e. birds or humans; Wildish 1988); nonetheless, the most presumable way of dispersing is still believed to be on floating wrack or wood (De Matthaeis et al. 1995, 2000a; Pavesi et al. 2011), a phenomenon known as rafting.
Previous population genetic studies on talitrids have shown how species that are genetically highly structured are generally confined to sandy beaches where they burrow into the sand (e.g. Talitrus saltator, Deshayesorchestia deshayesii) or dig into rotting logs (Macarorchestia remyi) (De Matthaeis et al. 2000b; Pavesi et al. 2011). On the other hand, species associated with heaps of decaying wrack on rocky shores seem to be genetically homogeneous, even over long distances (De Matthaeis et al. 2000a). De Matthaeis et al. (2000a, b) interpreted these contrasting patterns in the homogeneity of talitrid gene pools in terms of probabilities for the animals to be dragged away by sea currents and thus be dispersed passively. These probabilities would consequently be low for species bound to sand beaches, but would be high for those species colonizing ephemeral habitats (e.g., accumulations of beached seagrass wrack) closer to the mean sea level.
Orchestia montagui Audouin 1,826 is a talitrid species that commonly occurs on decaying wrack accumulations of the seagrass P. oceanica (the so-called “banquette” formations) stranded on sandy and rocky Mediterranean coasts. O. montagui’s inherent dispersal extends over short distances only, but the banquette may act as a “motile platform” which increases the chances of passive dispersal for the species. If that were the case, one would expect populations to be genetically connected to each other over a certain geographic area. The intrinsic temporal instability of this environment (e.g., due to the removal of wrack through beach grooming, tourism, wave action) could also play a role, leading to frequent population crashes and genetic bottlenecks. The persistence in time of the species in a given area would then rely on cyclical phenomena of extinction and re-colonization.
A number of studies focused on the population genetic structure of talitrid amphipods and how this relates to the characteristics of the colonized environment. The vast majority of them is based on allozymes (Angelini et al. 1996; Cervelli et al. 2006; De Matthaeis et al. 1995, 2000a, b). For example, De Matthaeis et al. (2000a) applied a set of allozymic loci to analyse patterns of gene flow in 23 populations of O. montagui from the Tyrrhenian (mostly from islets surrounding Sardinia) and the Aegean Sea and found the species to be poorly structured. Only recently, some authors have used mitochondrial DNA in talitrids (Henzler and Ingólfsson 2007 on Orchestia gammarellus; Pavesi et al. 2011 on Macarorchestia remyi). Here we present the first study that applies simultaneously mitochondrial and nuclear markers in a talitrids species in order to describe its population structure. Our study acts as a preliminary and exploratory research, seeking to address the interplay between population genetic structure and abiotic forces in the talitrid species O. montagui, by using markers with different evolutionary properties. Thus, we used a fragment of the mitochondrial DNA (mtDNA) coding the Cytochrome Oxidase subunit I gene (COI), and a set of eight polymorphic nuclear microsatellite loci. These were identified and developed de novo for the species by our research group (Bonizzoni et al. 2011). Mitochondrial DNA depicts historical processes and/or detects sex-bias phenomena because of its maternal inheritance; microsatellites are rapidly evolving, non-coding nuclear markers able to unravel recent population processes. The study is based on six populations from five different Mediterranean basins (Tyrrhenian, Ionian, Adriatic, Western and Eastern). Populations from the Ionian and Adriatic basins are here screened molecularly for the first time. We aimed to obtain an estimation of the degree of spatial genetic structuring and variability on a macro-geographical scale. In particular, we wanted to test whether the theoretical high probability of passive transport through floating accumulations of P. oceanica wrack might shape the population structure of the species. We postulated the following two alternative scenarios: under high levels of gene flow, we would expect shallow population structuring among basins and a weak correlation between genetic and geographic distances. On the contrary, limited gene flow among basins would translate into a more clear phylogeographic pattern.
Materials and methods
Sampling localities
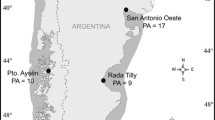
A total of 153 individuals from six populations of O. montagui were obtained from sample sites that cover five different Mediterranean basins (Tyrrhenian, Ionian and Adriatic Seas, Western and Eastern basins). We collected talitrids using an aspirator or by hand within accumulations of seagrass wrack stranded on different kinds of shore (i.e., sandy, rocky or pebble). Details on sampling localities and their geographic position are given in Table 1 and Fig. 1.

a Sampling locations and the four putative barriers identified by mtDNA and microsatellites. Barriers are shown as grey lines; numbers are bootstrap support values (above: mtDNA; below: microsatellites). The year-round gyres are located where the putative barriers have been found and the Atlantic-Ionian streams with its main derivates are also shown. b Results from Structure analysis for 147 individuals (8 microsatellite loci). Each vertical line represents one individual for K = 2 (two coloured clusters). Length of each line is proportional to the probability of each individual membership to either cluster. Cluster 1 in grey, cluster 2 in black. Codes for populations as in Table 1
Genetic analyses
mtDNA
Total genomic DNA was extracted from the posterior half of each amphipod (N = 77) using the Qiagen DNeasy Blood and Tissue Kit (Hilden, Germany) following the manufacturer’s protocol. A 571 base pair (bp) long fragment of the mtDNA, coding for the COI gene, was amplified using Folmer et al. (1994) universal primers via Polymerase Chain Reaction (PCR), for six to 15 individuals per populations (see Table 1 for details). Double-stranded amplifications, purification of PCR products and direct sequencing were performed as described in Ketmaier et al. (2008) and Pavesi et al. (2011). Sequences were edited and aligned using Sequencher 4.1 (Gene Code Corporation, Ann Arbor, MI, USA).
The number of variable and parsimony-informative sites and the absolute number of substitutions were calculated with Paup* v.4.0b10 (Swofford 2002). Arlequin v.3.11 (Excoffier et al. 2005) was used to calculate genetic (H) and nucleotide diversity (π n ), the mean number of pairwise differences between all pairs of haplotypes (π) for each population, and to perform a hierarchical analysis of molecular variance (AMOVA; Excoffier et al. 1992), which estimates the variance components between populations at different levels of hierarchical subdivision. For the AMOVA, populations were combined first into three groups (Sardinia; Apulia; Gozo island + Latium, on the basis of their geographic position and on the outcome of the phylogeographic analysis, which suggested a close relationship between the Gozo island and Latium populations) and later in two groups (Apulia; Sardinia + Gozo island, this analysis was meant to make the comparison of results with microsatellite data easier). Arlequin v.3.11 was also used to calculate F ST values between pairs of populations as a measure of their differentiation. Statistical significance of these values was assessed by 1,000 permutations after sequential Bonferroni correction for multiple tests (Rice 1989). Arlequin v.3.5 was used to calculate F ST’s confidence intervals (CI). A Mantel test (Mantel 1967) was performed with Arlequin v.3.11 to evaluate the possible relationship between geographic and genetic distances (Isolation by Distance; IBD). Geographic distances were measured as the shortest pairwise marine distances between populations, according to the circulation of surface currents (Blondel et al. 2010; Robinson et al. 2001). Genetic distances were based upon a population pairwise F ST values: the correlation between the two matrices, based on geographic and genetic data, was computed to perform the IBD test. To identify where possible barriers to gene flow between the different populations could exist, we used Barrier v.2.2 (Manni et al. 2004), which implements the Monmonier’s maximum difference algorithm. The robustness of the identified barriers was tested by running 100 bootstrapped F ST matrices (calculated with a function in R v.2.9.0; UCLA 2011). A statistical parsimony network to estimate gene genealogies among all haplotypes was constructed using Tcs v.1.13 (Clement et al. 2000), with a 95% connection limit, following Templeton et al. (1992).
Microsatellites
A total of 147 samples were genotyped (29 or 30 per population, see Table 1) at eight O. montagui specific polymorphic microsatellite loci: JZV, LMX, 309, QVF, VXW, O7 W, OLX and IMN (Bonizzoni et al. 2011). LFM was not analysed for microsatellite variation because the sample size was too small (N = 6). PCR conditions for these loci are described in Bonizzoni et al. (2011). Fragment sizes were determined on an ABI 3,100 automatic sequencer using the GeneMapper v.3.5 software and an internal size standard (GS500LIZ from Applied Biosystems). We used Microchecker v.2.2.3 (van Oosterhout et al. 2004) to test for the presence of null alleles and scoring errors in all microsatellites detected. As null alleles may affect the estimates of genetic differentiation, we calculated global F ST values using the ENA correction (Excluding Null Alleles) employing 10,000 bootstrap iterations in the FreeNa software as described by Chapuis and Estoup (2007).
Linkage disequilibrium among loci and departures from Hardy–Weinberg equilibrium were tested using Arlequin v.3.11 (Excoffier et al. 2005). P values were corrected using the sequential Bonferroni correction for multiple comparisons (Rice 1989). For each population, expected (H E) and observed (H O) heterozygosities (Arlequin v.3.1) and allelic richness (A R) (Fstat v.2.9.3, Goudet 1995, 2001) were calculated. GenAlex v.6.3 (Peakall et al. 2006) was used to determine the allelic frequencies per locus and population, the number of private alleles and their relative frequencies in each population.
AMOVA, pairwise F ST values, IBD test and Barrier analyses were performed on microsatellites with the same analytical schemes employed for the mtDNA. F ST’s confidence intervals (CI) were calculated with Fstat v.2.9.3. Structure v.2.3.2 (Pritchard et al. 2000) was run in order to infer population structure first by identifying the most probable number of clusters (K), and then by assigning individuals to the detected clusters. The software uses a Monte Carlo Markov Chain (MCMC) Bayesian clustering model that assumes the different loci are at Hardy–Weinberg and linkage equilibrium with one another within the population. One run for each value of K (K = 1–6) was performed, followed by nine independent replicates, assuming an admixture model with 100,000 replicates of the MCMC after a burn-in of 10,000 iterations. Visual inspection of values of summary statistics showed approximate stationarity after such a burn-in. The number of clusters was elaborated from the ΔK estimates using Evanno et al. (2005) method. Finally, the assignment of individuals to the inferred clusters was estimated according to the highest Q values (probability of membership) from the original run.
Results
Mitochondrial DNA
A total of 25 haplotypes from the 77 individuals of O. montagui sequenced for a 571 bp segment of COI (GeneBank Accession nos: JQ390313-JQ390337) were obtained. As expected for an mtDNA coding region sequences were generally A + T rich and anti-G biased; no stop codons and/or gaps were observed in the alignment. The absolute number of substitutions varied from 1 (within GOZ, SAR, SNG and between GOZ-SNG, GOZ-LFM, PMP-PSF) to 18 (LFM-PMP and PMP-PSF). As shown in Table 1, genetic diversity (H) varied from 0.362 (SNG) to 0.933 (LFM); the mean number of pairwise differences between all pairs of haplotypes (π) ranged from 0.381 (SNG) to 9.571 (PSF), whereas nucleotide diversity (π n) was the lowest in SNG and SAR (0.001) and peaked in PSF (0.017).
Table 2 summarizes the AMOVA results for the two geographical groups tested. In both analyses, the majority of variation was apportioned within populations (55.2 and 50.2%, respectively); 29% (3 groups) and 28.2% (2 groups) of the revealed variation was due to differences among populations within groups, and only 15.7 and 21.6% (3 and 2 groups, respectively) of the variation was due to difference among groups. Pairwise F ST values (Table 3) ranged between 0.22 (LFM-SAR) and 0.52 (SNG-GOZ); the comparison between the SNG and SAR populations (south and north of Sardinia, respectively) was statistically not significant. Furthermore, LFM (Tyrrhenian Sea) was significantly differentiated from the Sardinian populations (Western basin) but not from any other from the remaining basins included in the study. A Mantel test performed on all populations showed no statistical relationship between genetic and geographic distances (R = −0.000038; P = 0.807).
Barrier analysis (Fig. 1a) identified four putative barriers to gene flow. The first two barriers (99 and 98% of bootstrap support) were found between LFM and Sardinian populations (in agreement with the F ST results), and between north and south Sardinia (at odds with the F ST results). A third and fourth barrier (both 68% of bootstrap support) were detected between GOZ (Eastern basin) and SNG (Western basin) on one side, and GOZ and Apulian populations on the other side.
The statistical parsimony network is shown in Fig. 2. Most of the Sardinian, Latium and Maltese haplotypes cluster together and differ by single mutational steps, with the only exception of three haplotypes (two exclusive to GOZ, one exclusive to LFM). All Adriatic and all but one Ionian haplotypes are scattered in the network and separated by intervening missing haplotypes. Haplotypes differed by a minimum of 1 to a maximum of 29 mutations and the total number of haplotypes per population ranged from a minimum of three in SNG to a maximum of seven in PSF. Fourteen out of 25 haplotypes were carried by one individual only; three of the 11 remaining haplotypes were shared by individuals from different populations (GOZ + SAR + SNG; GOZ + LFM; GOZ + LFM + PMP). The maximum number of individuals sharing one haplotype was 12 in SNG.

Haplotype network of gene genealogies for all haplotypes. Circle size is proportional to haplotypes frequency; numbers indicate how many individuals carry that particular haplotype (if more than one); black dots indicates missing haplotypes
Microsatellites
All loci were polymorphic in the five populations analysed, with the number of alleles per locus ranging from six (LMX) to 13 (IMN). A few loci showed significant deviations from Hardy–Weinberg equilibrium after Bonferroni correction (probably due to the presence of null alleles). Average global F ST values not using and using ENA method were 0.13 and 0.12, respectively, and the corrected global F ST values per locus varied from 0.05 to 0.24, also being always very similar to the uncorrected ones (results not shown), suggesting in our case that even when present, null alleles have very little impact on the F ST estimates and should not lead to misleading interpretations (Chapuis et al. 2005; Chapuis and Estoup 2007; Meglecz et al. 2004; Villacorta et al. 2009). No scoring errors and no evidence of linkage disequilibrium were detected between any pairs of loci after Bonferroni correction. HE ranged from 0.536 (PSF) to 0.681 (GOZ), and H O ranged from 0.212 (PSF) to 0.635 (SNG) (Table 1).
AMOVA (Table 2) was initially performed by grouping populations according to the Structure outcome (2 groups: Apulia; Sardinia + GOZ) and then into three groups, to facilitate a direct comparison with mtDNA data. Both analyses identified most of the variation within populations (2 groups: 83.7%, P = 0.001; 3 groups: 85.3%, P = 0.001) and also indicated a similar partition of the variation among groups (2 groups: 8.1%, P = 0.081; 3 groups: 6.8%, P = 0.069) and among populations within groups (2 groups: 8.2%, P = 0.001; 3 groups: 7.9%, P = 0.001). F ST values (Table 3) were statistically significant for all comparisons between each pair of populations; values ranged from 0.043 (PMP-PSF) to 0.209 (SNG-PSF). The Mantel test detected no evidence of statistical relationship between genetic and geographic distances (R = −0.186; P = 0.633). Results from the analysis of population structure using Barrier (Fig. 1a) for the five populations and all the loci analysed showed evidence of three putative barriers, all with a bootstrap support greater than 60%. A first barrier was found between Apulian populations and GOZ (Eastern basin), a second barrier identified between SAR and SNG (north and south Sardinia) and the last barrier was found between SNG and GOZ.
Structure revealed the most likely number of clusters to be two (mean ΔK = 182, LnP(K) = −3250.35 ± 0.79) (Fig. 1b). Estimates of LnP(K) were consistent across independent runs and were neither bi- nor multimodal indicating appropriate mixing of the MCMC runs. The analysis identified one cluster including Apulian populations (PMP + PSF) and a second cluster grouping Sardinian (SNG) and GOZ populations. Q-values showed a very high membership of individuals either to the first (94%) or to the second (95%) cluster. SAR was in between the two clusters, individuals showed a mean average Q value of 51% to either clusters, and low membership of individuals with Q value >80% (14% to the first and 38% to the second cluster). Results from Barrier are consistent with those from Structure in terms of the existence of a boundary between the Apulians and all other populations; Barrier found a genetic discontinuity between SNG and GOZ while Structure assigned them to the same cluster (but one individual in GOZ with a very weak membership). The two populations are separated by a significant F ST value (0.091).
Discussion
Population genetic structure
To our knowledge, this is the first study employing markers of different origin (mitochondrial and nuclear) and with different evolutionary properties to elucidate the population genetic structure of a talitrid species. Both classes of markers identified the Apulian populations as divergent from the others. A pattern of Isolation by Distance (IBD) was detected neither at the mtDNA nor at the microsatellite level. COI-based pairwise F ST values indicated no significant divergence within the Western Mediterranean basin as well as between Tyrrhenian versus. Eastern, Ionian and Adriatic Seas. All pairwise F ST values calculated on microsatellites were statistically significant. For both classes of markers, Barrier detected 3–4 barriers to gene flow while multiple AMOVAs revealed most of the genetic variation being apportioned within populations.
MtDNA detected a close relationship between the Gozo island (GOZ) and Latium (LFM) populations (which is missing in the microsatellite data set) and clustered most of the Sardinian haplotypes in the same part of the haplotype network. Adriatic and Ionian haplotypes, on the other hand, are not only divergent from haplotypes found at geographically distant locations but are also scattered in the network and separated from each other by many intervening missing haplotypes. This implies that coalescence of haplotypes is relatively shallow––i.e., they converge earlier into their last common ancestor in the Western, Tyrrhenian and Eastern basins than in the Adriatic and Ionian ones. The high number of missing intermediates necessary to connect Adriatic and Ionic haplotypes suggests extended back-in-time loss of genetic diversity. The partial isolation of these two basins from the rest of the Mediterranean Sea is also evident with microsatellites, where structure clustered PMP and PSF together and away from the rest. While it seems relatively feasible to envision population crashes in a species confined to an environment as ephemeral as accumulations of decaying seagrass wrack, our data do not provide conclusive evidence to explain why these phenomena would be occurring more frequently at the Adriatic and Ionian locations than in the other populations included in the study. A variety of factors (either acting in isolation or synergistically) could be responsible for the observed inter-area differences. First, seagrasses could be stranded with a lower frequency along Adriatic and Ionian shores and/or such accumulations might be persisting for a shorter period of time along such coastlines. Differences in the tourism impact and in the efficacy and intensity of mechanical beach grooming performed on the beaches might also play a role in this context. Second, O. montagui might have a more scattered distribution along the Adriatic and Ionian coasts than in the other basins; this would imply that along such coasts, the species would rely chiefly on long-distance migrants to re-colonize locations where populations had been completely extirpated. Finally, surface sea currents might not be dispersing banquettes in the area of study at the same rate as in other areas of the Mediterranean Sea. Interestingly, Serra et al. (2010) identified a major genetic discontinuity between the Western and Eastern Mediterranean basins in the seagrass P. oceanica; the genetic break in the seagrass corresponds to the barrier we have found in this study between the same basins (Fig. 1a). We take the fact that the same recurrent pattern is observed in taxonomically unrelated groups as good evidence that the same force (sea currents) is responsible.
A straightforward geographic pattern does emerge, neither at the mtDNA nor at the nuclear level. Nonetheless, pairwise F ST values consistently revealed a highly significant subdivision among populations, especially at the microsatellite level. MtDNA identified a probable barrier to gene flow between the Tyrrhenian and the Sardinian populations. The result is consistent with F ST data, where LFM was significantly differentiated from SAR and SNG only. Both markers (mtDNA and microsatellites) identified barriers to gene flow within the Western basin (SAR vs. SNG), between the Western basins and the other basins, and between the Eastern basin and the Ionian and Adriatic Seas. It is worth noting that all three identified barriers correspond to areas where year-round gyres are known to occur (Fig. 1, see also: Blondel et al. 2010; Robinson et al. 2001). Steady gyres are thought to generate genetic discontinuities in marine species that rely on passive transport to disperse (Sala-Bozano et al. 2009). The identified barriers are, however, only moderately supported by the associated bootstrap values.
If gyres were to be considered the main responsible for the observed barriers, one should then conclude that they would act as semi-permeable obstacles to gene flow at best. This is particularly valid for the SNG vs. GOZ comparison. The deep separation at the mtDNA level between the Maltese (GOZ) and Southern Sardinian (SNG) populations could be traced back to the (historical) repeated isolation of the Eastern basin from the Western Mediterranean Sea during Pleistocene ice cycles, when the sea level repeatedly dropped and the Siculo-Tunisian strait (i.e., the area comprised between Porto Palo in Sicily and Cap Bon in Tunisia) was either considerably narrowed or almost totally dried up (Boissin et al. 2010). A very rough estimate of divergence time between SNG and GOZ (based on the formula T = D/2μ where D is the uncorrected genetic distance and μ is the crustacean COI mutation rate of Knowlton et al. 1993) resulted in 0.3–0.5 million years, thus spanning the Riss and Mindel glacial events.
On the other hand, SNG and GOZ have relatively similar microsatellite genotypic compositions (see Fig. 1a), which translates into a low (yet significant) F ST value. To explain the difference between the two classes of markers we should envision gene flow being mostly mediated by males. Mediterranean talitrids tend to have a female-biased sex ratio (Fallaci et al. 2003; Pavesi and De Matthaeis 2009) even though precise data on O. montagui are unfortunately not available. Fallaci et al. (2003) showed that in the sandhopper Talitrus saltator, males spend considerably more time than females on the surface, while females tend to stay close to their burrowing zone and to move less, hence reducing their chances to be dragged away by waves. If this was the case also for O. montagui, we could hypothesize that male contribution to the genetic make-up of populations might not be negligible. This is an interesting avenue of research but at the present stage available field data on O. montagui are insufficient to address the issue properly.
With reference to the Western Mediterranean basin, north (SAR) and south (SNG) Sardinia populations are not strongly affiliated. In fact, there is a putative barrier between these two populations, which is moderately supported at microsatellite level (62%) but highly supported at mtDNA level (98%). Also, the two populations share just a single haplotype (out of a total of seven), and SNG result to be connected by single or few mutational steps to SAR. The lack of significance of the high F ST value between them at the COI level (0.487) is likely due to our relative small sample size (in terms of both number of populations and loci), which translates in large confidence intervals of estimates (see Table 3). This reconciles the mtDNA-based F ST data with the outcomes of Barrier indicating a very strong barrier to gene flow between SNG and SAR. De Matthaeis et al. (1995) analysed populations from the same localities with allozymes and found them to be genetically quite homogeneous, but allozymes notoriously detect only a small fraction of the existing genetic variation and could have not been able to reveal the genetic discontinuity we found with our markers.
More generally, previous allozyme-based studies on O. montagui revealed high or very high levels of gene flow at the scale of the Mediterranean Sea (De Matthaeis et al. 2000a). Although our sampling was currently limited to a relatively low number of locations, it is worth noting that, whatever geographic scale is considered, we almost invariably retrieved a significant divergence among sampled populations. These results suggest that these preceding studies in literature were based on genetic markers not variable enough to detect the differences evidenced in the current study.
Additionally, even though a substantial phylogeographic break between the Adriatic and Ionian populations and all the other populations was identified in the present study, this was not as pronounced as that found in the talitrid Macarorchestia remyi. This is a cryptozoic species strictly associated with stranded rotting logs, which was screened for COI sequence variation by Pavesi et al. (2011). For this species, a strong divergence between Adriatic and Tyrrhenian populations (with no shared haplotypes between the two basins and with F ST being statistically significant and equal to 0.978) was reported. Genetic differentiation was also pronounced on a local scale, as no evidence of gene flow between two populations located a few kilometres apart from one another along the Apulian Adriatic coast was recorded (F ST = 0.348 and statistically significant). In O. montagui, one haplotype co-occurs at the Latium, Maltese and Ionian locations, whilst two Adriatic haplotypes differ by just a single mutation from Southern Sardinian haplotypes. The species seems thus able to passively move migrants over long distances, although sporadically, as attested by the generally significant F ST values.
Considering the results presented here on O. montagui and those described above for M. remyi, we invoke differences in the kind of environment colonized by the two species to explain their observed contrasting patterns of genetic structuring. O. montagui, by living within layers of dead leaves of heaps of banquette and in close proximity to the water line, has higher probabilities to be dragged away by currents and to be transported to other shores than M. remyi. This would in turn lead to a higher exchange of individuals and to a less geographically structured pattern of population differentiation. This scenario would also explain why a pattern of IBD was not identified in the current study, even though it is evident that we need a geographically denser sampling to be conclusive on this point. According to results emerging in this study and adopting the generalizations of Slatkin (1993), one can tentatively conclude that O. montagui, at the scale considered in this study, has not yet reached an equilibrium between gene flow and genetic drift since the populations studied tended to be genetically differentiated from one another but the observed divergence was not a function of the geographic distance between them (no IBD).
Conclusions
Results emerging in this study depict quite a complex scenario. Populations tend to differentiate locally, even though the extent at which such differentiation is discerned may vary according to the markers employed. The overall genetic structure of the species, as De Matthaeis et al. (2000a) suggested, may be determined by the pattern of sea surface currents. An additional role may also be played by the possibility of individuals to survive in water for prolonged periods of time, and hence to be transported over long distances through drifting seagrass wrack. It is evident that additional ecological data is necessary to exhaustively understand the mosaic pattern of genetic divergence.
In view of the relatively low number of populations sampled, results reported in this study do not provide conclusive evidence of hypothesized processes, even though populations analysed cover a relatively wide geographical scale. Nonetheless, this preliminary study is useful in unveiling the population genetic structure of a species that colonizes a transitional coastal environment. It will be followed up by more comprehensive sampling pursuant to obtain a more detailed description of genetic connectivity within O. montagui, and to ultimately address the issue of the effect of the geographic scale on its population genetic structure.
References
Angelini E, Longo N, Zane L, Bisol PM (1996) Biodiversità in anfipodi Talitridi da un punto di vista ecogenetico. Biol Mar Medit 3(1):115–119
Blondel J, Aroson J, Bodiou J-Y, Boeuf G (2010) The Mediterranean region: biological diversity in space and time. Oxford University Press, Oxford
Boissin E, Hoareau TB, Berrebi P (2010) Effects of current and historic habitat fragmentation on the genetic structure of the sand goby Pomatoschistus minutus (Osteichthys, Gobiidae). Biol J Linn Soc 102:175–198
Bonizzoni M, Bourjea J, Chen B, Crain BJ, Cui L, Fiorentino V, Hartmann S, Hendricks S, Ketmaier V, Ma X, Muths D, Pavesi L, Pfautsch S, Rieger MA, Santonastaso T, Sattabongkot J, Taron CH, Taron DJ, Tiedemann R, Yan G, Zheng B, Zhong D (2011) Permanent genetic resources added to molecular ecology resources database 1(April), pp 2011–31, May 2011. Mol Ecol Res 11:935–936
Cervelli M, Bortolomei A, Linertini A (2006) Differenziazione genetica tra popolazioni di Orchestia montagui delle coste sarde. Biol Mar Medit 13(1):10675–11071
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–631
Chapuis MP, Loiseau A, Michalakis Y, Lecoq M, Estoup A (2005) Characterization and PCR multiplexing of polymorphic microsatellite loci for the locust Locusta migratoria. Mol Ecol Notes 5:554–557
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
De Matthaeis E, Cobolli M, Davolos D, Mattoccia M (1995) Stime di flusso genico tra popolazioni di Orchestia montagui (Amphipoda, Talitridae) delle isole circumsarde. Biogeographia 23:249–260
De Matthaeis E, Davolos D, Cobolli M, Ketmaier V (2000a) Isolation by distance in equilibrium and nonequilibrium populations of four talitrid species in the Mediterranean Sea. Evolution 54(5):1606–1613
De Matthaeis E, Ketmaier V, Davolos D, Schembri PJ (2000b) Patterns of genetic diversity in Mediterranean supralittoral amphipods (Crustacea, Amphipoda). Pol Arch Hydrobiologii 47:473–487
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software Structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Estoup A, Cornuet JM (2005) Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169:1727–1738
Fallaci M, Colombini I, Lagar M, Scapini F, Chelazzi L (2003) Distribution patterns of different age classes and sexes in a Tyrrhenian population of Talitrus saltator (Montagu). Mar Biol 142:101–110
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Goudet J (1995) Fstat (vers. 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Goudet J (2001) Fstat, a program to estimate and test gene diversities and fixation indices (version 2.9.3) Updated from Goudet (1995) http://www2.unil.ch/popgen/softwares/fstat.htm
Henzler CM, Ingólfsson A (2007) The biogeography of the beachflea, Orchestia gammarellus (Crustacea, Amphipoda, Talitridae), in the North Atlantic with special reference to Iceland: a morphometric and genetic study. Zool Scripta 37:57–70
Ketmaier V, Pirollo D, De Matthaeis E, Tiedemann R, Mura G (2008) Large scale mitochondrial phylogeography in the halophilic fairy schrimp Phallocryptus spinosa (Milne-Edwards 1840) (Branchiopoda: Anostraca). Aquat Sci 70:65–76
Knowlton N, Weigt LA, Solorzano LA, Mills DK, Bermingham E (1993) Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science 260:1629–1631
Manni F, Guérard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum Biol 76(2):173–190
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Meglecz E, Petenian F, Danchin E, Coeur d’Acier A, Rasplus JY, Faure E (2004) High similarity between flanking regions of different microsatellites detected within each of two species of lepidoptera: Parnassius apollo and Euphydryas aurinia. Mol Ecol 13:1693–1700
Pavesi L, De Matthaeis E (2009) Life history of the amphipod Macarorchestia remyi (Schellenberg, 1950) on a Tyrrhenian sandy beach, Italy. Hydrobiologia 635:171–180
Pavesi L, De Matthaeis E, Tiedemann R, Ketmaier V (2011) Temporal population genetics and COI phylogeography of the sandhopper Macarorchestia remyi (Amphipoda: Talitridae). Zool Stud 50(2):220–229
Peakall R, Smouse PE (2006) GenAlex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly PJ (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Robinson AR, Leslie WG, Theocharis A, Lascaratos A (2001) Mediterranean Sea circulation encyclopedia of ocean sciences, Academic Press 1689–1706
Sala-Bozano M, Ketmaier V, Mariani S (2009) Contrasting signals from multiple markers illuminate population connectivity in a marine fish. Mol Ecol 18:4811–4826
Serra IA, Innocenti AM, Di Maida G, Calvo S, Migliaccio M, Zambianchi E, Pizzigalli C, Arnaud-Haond A, Duarte CM, Serrao EA, Procaccini G (2010) Genetic structure in the Mediterranean seagrass Posidonia oceanica: disentangling past vicariance events from contemporary patterns of gene flow. Mol Ecol 19:557–568
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Swofford D (2002) Paup* ß-version 4.10 Sunderland, MA: Sinauer Associates
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
UCLA (2011) Academic Technology Services, Statistical Consulting Group. From: http://www.ats.ucla.edu/stat/R/
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Microchecker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Villacorta C, Cánovas F, Oromí P, Juan C (2009) Isolation and characterization of microsatellite loci for the cavehopper Palmorchestia hypogaea (Amphipoda: Talitridae). Conserv Genet Resour 1(1):401–404
Wildish DJ (1988) Ecology and natural history of aquatic Talitroidea. Can J Zool 66:2340–2359
Acknowledgments
This study was financed by a financial grant from the Research Committee of the University of Malta, to which the authors are indebted. Four anonymous reviewers are acknowledged for useful criticisms on a first version of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pavesi, L., Deidun, A., De Matthaeis, E. et al. Mitochondrial DNA and microsatellites reveal significant divergence in the beachflea Orchestia montagui (Talitridae: Amphipoda). Aquat Sci 74, 587–596 (2012). https://doi.org/10.1007/s00027-012-0250-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00027-012-0250-y