
Abstract
Heart failure (HF) is a major deteriorating disease of the myocardium due to weak myocardial muscles. As such, the heart is unable to pump blood efficiently around the body to meet its constant demand. HF is a major global health problem with more than 7 million deaths annually worldwide, with some patients dying suddenly due to sudden cardiac death (SCD). There are several risk factors which are associated with HF and SCD which can negatively affect the heart synergistically. One major risk factor is diabetes mellitus (DM) which can cause an elevation in blood glucose level or hyperglycaemia (HG) which, in turn, has an insulting effect on the myocardium. This review attempted to explain the subcellular, cellular and molecular mechanisms and to a lesser extent, the genetic factors associated with the development of diabetes- induced cardiomyopathy due to the HG which can subsequently lead to chronic heart failure (CHF) and SCD. The study first explained the structure and function of the myocardium and then focussed mainly on the excitation–contraction coupling (ECC) processes highlighting the defects of calcium transporting (SERCA, NCX, RyR and connexin) and contractile regulatory (myosin, actin, titin and troponin) proteins. The study also highlighted new therapies and those under development, as well as preventative strategies to either treat or prevent diabetic cardiomyopathy (DCM). It is postulated that prevention is better than cure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) arises when the organ is unable to pump blood efficiently around the body, especially to the myocardium itself and the brain. The disease is due to several risk factors which can exert lethal synergistic effects on the heart. Figure 1 shows a flow diagram illustrating the different risk factors which can induce HF. If left untreated, the failure becomes chronic with time resulting in CHF. This can lead to cardiac arrhythmias and SCD. One major risk factor for CHF is DM. This review is related to the subcellular, cellular, molecular and genetic factors of diabetes-induced CHF.

Risk factors associated with the development of heart failure and sudden cardiac death
Diabetes mellitus
DM is a chronic metabolic disorder characterized by elevated levels of blood glucose or HG leading over time to serious damage to the heart, blood vessels, eyes, kidneys, nerves as well as other organs of the body [1]. HG typically results from defects in insulin secretion, insulin action or both. Diabetes can be classified into 4 general categories. Type 1 diabetes (T1DM) is due to pancreatic β-cell destruction, usually leading to an absolute insulin deficiency [2]. It only accounts for 5–10% of the worldwide prevalence of the disease [3]. Moreover, 80–90% of diabetic cases in children and adolescents are of T1DM [4]. Type 2 diabetes (T2DM) results from a defect in insulin secretion on the background of insulin resistance [2]. T2DM is the most prevalent type of DM, comprising of 90–95% of the entire diabetic population [5]. Gestational Diabetes Mellitus (GDM) is diagnosed in 2nd or 3rd trimester of pregnancy but may resolve after childbirth [2]. Nonetheless, there is a multiple fold higher risk of developing T2DM later in life [6]. The 4th category is a small subgroup of people with diabetes due to other causes, e.g. monogenic diabetes syndromes (such as neonatal diabetes), chemical- or drug-induced diabetes (such as HIV/AIDS treatment or treatment after organ transplant) and diseases of the exocrine pancreas (such as cystic fibrosis) [2].
Data from the Global Burden of Disease in 2017 estimated that T2DM affected 6.28% of the world’s population with a prevalence rate of 6059 cases per 100 000 — projected to increase to 7079 cases per 100 000 by the year 2030 and 7862 by 2040. However, these values can vary significantly from one country to another. Furthermore, its incidence peaks at 55–59 years of age with males showing a slightly higher prevalence than females (6219 compared to 5898 cases per 100 000) [7]. If left untreated or diagnosed late, diabetes can lead to several long -term macrovascular and microvascular complications. Macrovascular complications include cardiovascular diseases (CVDs), stroke and peripheral vascular disease (PVD). Microvascular complications include nephropathy, neuropathy and retinopathy [8].
A landmark study by Rubler et al. described a new clinical entity in 1972 by reporting of 4 post mortem patients with diabetes-related HF and dilated cardiomyopathy [9]. Termed diabetic cardiomyopathy (DCM), this clinical phenomenon occurs when longstanding DM causes structural and functional changes in the myocardium leading to the development of HF in the absence of microvascular atherosclerotic or myocardial ischaemic disease. This paper will concentrate on the subcellular, cellular, molecular and genetic factors of diabetes induced chronic HF.
Structure and function of the heart
The heart acts as two serial pumps that share several mechanical and electrical components. It is an organ consisting of four chambers namely, 2 atria and 2 ventricles [10]. The right atrium and right ventricle function to pump deoxygenated blood to the lungs. Deoxygenated blood returning from the superior and inferior vena cava enters the right atrium of the heart. From here, it subsequently passes through the tricuspid valve to enter the right ventricle. The right ventricle contracts to push blood through the pulmonary valve into pulmonary arteries to be transported to the lungs [10].
After offloading carbon dioxide and reloading oxygen, blood is directed from the lungs to left atrium of the heart through the pulmonary veins. From the left atrium, blood moves through the mitral valves into the left ventricle. Thereafter, it is pumped through the aortic valve and into the aorta to be distributed around the body. Hence, the function of the left atrium and left ventricle is to pump oxygenated blood throughout the body to maintain normal homeostasis [11].
Of note, the wall of the left ventricle is three times bigger than that of the right ventricle. The interventricular septum bulges into the right cavity possibly due to left ventricular contraction pressure being higher that of the right during systole. A typical cross section therefore shows a circular left ventricular chamber compared to the crescentic shape of the right ventricular chamber.
Despite the naming system used for each chamber, the heart, in fact, lies obliquely in the thorax with its long axis passing downwards and to the left to the apex. In this position, its entire right border consists of the right atrium. The inferior border is made up almost entirely of the right ventricle with a small part of the left ventricle — forming the apex at its inferior and left borders. The left border of the heart consists of almost the entirely the left ventricle with only the auricle of the left atrium forming its uppermost surface. The posterior surface (or base of the heart) is made up almost entirely of the left atrium. The majority of its sternocostal surface consists of right ventricle with a small strip of left ventricle on the left and right atrium on the right. The diaphragmatic surface of the heart is made up of two thirds left ventricle and one third right ventricle [12].
Cardiac conducting system
The heart creates its own electrical impulses and controls the timing and route of those impulses through a conducting pathway [13]. There are 5 parts to this system and they include the sinoatrial (SA) node, the atrioventricular (AV) node, the bundle of His, the right and left bundle branches and the Purkinje fibres. The SA node is located at the junction between the superior vena cava and the right atrium. It generates impulses automatically by spontaneous depolarization of its membrane at a rate quicker than any other cardiac cell type and thus, it is the natural pacemaker of the heart. This depolarization first results in atrial contraction. Next, depolarization continues to conduct slowly at the AV node; situated beneath the right atrial endocardium within the lower interatrial septum. The slow conduction at the AV node facilitates emptying of the atria into the ventricles. The AV node continues as the bundle of His, which splits into the right bundle branch and the main left bundle branch at the crest of the interventricular septum. The right bundle branch continues down the right side of the interventricular septum towards the apex then radiates and divides to form the Purkinje network throughout the right ventricle. The shorter main left bundle branch fans out into the anterior and posterior hemi-bundles. The Purkinje network of the hemi-bundles provides electrical coverage to the left ventricle [13].
Excitation–contraction coupling of the heart
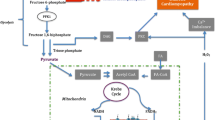
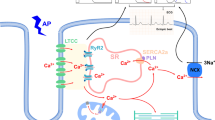
Excitation–contraction coupling (ECC) is the process where an action potential triggers myocyte contraction followed by relaxation. First, action potentials depolarize the cell membrane by travelling along the sarcolemma and down into the transverse tubule (T-tubule). Depolarization causes voltage-sensitive dihydropyridine (DHP) receptors (L-type Ca2+ channels) to open in the T-tubules, allowing for a small amount of Ca2+ (calcium) to enter the cytosol. This occurs during phase 2 of the action potential and contributes to cell depolarization. These “trigger” Ca2+ ions bind to ryanodine receptors (calcium release channels) on the sarcoplasmic reticulum (SR) resulting in Ca2+ release which were stored in the organelle. Intracellular free calcium concentration [Ca2+]i is increased from approximately 10–7 to 10–5 M [14, 15].
Troponin-C (TN-C) is part of the regulatory complex attached to the thin filaments. Ca2+ binding to TN-C induces a conformational change in the regulatory complex such that troponin-I (TN-I) exposes a site on the actin molecule that is able to bind to the myosin ATPase located on the myosin head. Upon binding, the energy provided by ATP hydrolysis causes a conformational change to occur in the actin-myosin complex, resulting in a movement (“ratcheting”) between the acting and heads of myosin. These 2 filaments slide past each other resulting in reduced sarcomere length. As long as cytosolic Ca2+ remains elevated, the movement cycles will continue.
As Ca2+ movement into the cell slows, sarcoplasmic reticulum calcium-ATPase (SERCA), an ATP-dependent calcium pump, sequesters Ca2+ back into the sarcoplasmic reticulum (SR) resulting in lower cytosolic calcium concentration and removal of Ca2+ from TN-C. Some Ca2+ are also transported out of the cell via the sodium-calcium exchanger (NCX) and Ca2+-ATPase pumps on the sarcolemma. Ca2+ unbinding from TN-C reverses the conformational change of the troponin complex to its original state leading to TN-I inhibition of the actin binding site. At the end of the cycle, ATP binds to the myosin head, displacing ADP and the initial sarcomere length is restored. The membrane is repolarized when potassium (K+) exits through the sarcolemma to end the action potential [14, 15]. The ECC process can be disrupted by many insulting risk factors to the heart leading to HF and/or SCD over time (see Fig. 2). One such risk factor is DM.

Influences and changes that occur in cardiomyocytes which predispose the heart to infarction and sudden cardiac death; (DAG diacylglycerol, PKC protein kinase C, NADPH-oxidase nicotinamide adenine dinucleotide phosphate oxidase, ROS reactive oxygen species, Ca.2+ calcium, SERCA sarcoplasmic-endoplasmic calcium -ATPase, NCX sodium calcium exchanger, RyR ryanodine receptor)
Hyperglycaemia on Ca2+ components in the ECC
Prolongation of the action potential duration (APD), slower decay of Ca2+ and modified sensitivity of contractile elements are consistently observed in diabetic cardiomyocytes [16,17,18]. In theory, the reduced rate of Ca2+ removal from the cytosol results in slowed decay in Ca2+ transient in diabetic cardiomyocytes [16]. Reduction of protein levels of sarco-endoplasmic reticulum calcium-ATPase 2a (SERCA2a) was reported in multiple animal model studies of DM [17]. Moreover, increased Ca2+ leakage from the SR also contributes to APD changes. One study showed that under blockades of ryanodine receptors and NCX, Ca2+ leakage was still significantly increased in diabetic mice (32%) compared to nondiabetic mice (12%), leading to elevated diastolic [Ca2+]i [18]. Each component of Ca2+ homeostasis will be discussed in more detail.
Elements that affect intracellular structures involved in Ca2+ homeostasis
Reactive carbonyl species
Reactive carbonyl species (RCS) are tiny electrophilic mono- and di-carbonyl species produced by glucose and lipid autooxidation, triose pathway fluxes, and enzymes such semicarbazide–sensitive amine oxidases and methylglyoxal synthase. 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), which form through lipid peroxidation of polyunsaturated fatty acids (PUFA), and methylglyoxal (MGO) are all members of this category [19]. This process is very common during diabetes. A previous study has shown how MGO plays a significant role in diabetic changes to the heart by increasing mitochondrial ROS generation inside cardiac cells which disrupts intracellular Ca2+ balance and increases oxidative stress [20]. While 4-HNE and MDA play major roles in diabetic complications including neuropathy, retinopathy and arterial damage, research has shown little or no significant effect on cellular Ca2+ homeostasis [21].
Methylglyoxal
MGO is a by-product of glycolysis. Though it has physiological roles [22], MGO is an RCS that can be toxic to the cell by inducing post-transitional modifications through an irreversible non-enzymatic reaction with unprotonated lysine and arginine residues [23]. It is known to be significantly elevated in T1DM and T2DM [24] and has been shown to affect elements in the ECC.
Shao et al. found that while SERCA2a protein remained unchanged, its ability to hydrolyse ATP and transport Ca2+ was significantly reduced in STZ diabetic rats [25]. MGO adduct antibodies were found to be two times higher on SERC2a in diabetic rats with cardiomyopathy compared to control [19]. RyR2 receptors are also affected. MGO was found to increase the openness of an already open RyR2 [25] as well as increase the mean open probability (P0) of low activity RyR2 [26]. Furthermore, these features reduced the conductance of RyR2 by approximately 20% [19].
Alterations are also observed on the contractile apparatus of the heart. Papadaki et al. found that left ventricular (LV) myofilament from patients with diabetes and HF had increased MGO modifications when compared to control. MGO-modification of K293 on actin was speculated to affect actin-tropomyosin interaction via disruption of the position of tropomyosin on actin leading to decreased Ca2+ sensitivity. On myosin, MGO-modification at R370 was thought to prevent a strong bond between myosin and actin which is required for full activation of the filament. Additionally, MGO-modifications were also found on myosin at sites known to be involved with disease-causing mutations: K384 (implicated in hypertrophic cardiomyopathy) and K1899 (familial dilated cardiomyopathy) [27].
Reactive oxygen species and oxidative stress
Oxidative stress is known to play a prominent role in the development and progression of DM. In the heart, reactive oxygen species (ROS) production is mainly produced by the mitochondria, NADPH oxidases, xanthine oxidase and uncoupled nitric oxide synthase (NOS) [28]. ROS directly damages phospholipids and proteins through oxidation, or secondarily by oxidising generating reactive nitrogen species (RNS) from nitric oxide and oxidizing lipids to reactive lipid peroxides [29]. Evidently, it is widely accepted that these molecules play an important part in the pathogenesis of diabetic cardiomyopathy. Lack of insulin-mediated glucose metabolism can cause elevated free fatty acid (FFA) accumulation in cardiomyocytes leading to accumulation of ROS and RNS via activation of NADPH oxidases (NOX) from the mitochondria and modulation of mitochondrial lection chain to generate superoxide (non-mitochondrial source) [30]. Some studies suggest that cytosolic ROS and RNS defence mechanisms might be more impacted than those of the mitochondria. These include modulation of signal transduction pathways that initiate cardiomyocyte hypertrophy [31] and apoptosis [32] as well as extracellular matrix alteration structure via matrix metalloproteinase (MMP) activation [33].
Renin–angiotensin–aldosterone system in DM
Despite a situation of salt and volume excess, inappropriate renin–angiotensin–aldosterone system (RAAS) activation plays a key role in the development of diabetes-related heart changes. It has been shown that increased angiotensin II type 1 receptor and mineralocorticoid receptor signalling in the myocardium boost the adaptive proinflammatory immune response and inflammation [34]. This involves NF-kB activation directly or indirectly by triggering other pathways such as the production of ROS as well as leukocyte adherence, cytokine production, and macrophage infiltration which inevitably contribute to cardiac fibrosis, diastolic dysfunction and heart failure [35].
Inflammation and DM
DM can be described as a systemic inflammatory condition. HG may cause cardiac cells to secrete cytokines, which encourage the migration of monocytes and lymphocytes, resulting in a persistent inflammatory state [36]. Many cytokines and chemokines interact with one another, making it difficult to assess the contribution of specific mediators to the phenotypes seen in DC. TNF-α, IL-6, IL-1β, IL-8 and C-reactive protein (CRP) are some of the most well-known markers of inflammation. Because certain mediators influence the release of other mediators downstream, it is difficult to predict which one of these cytokines causes direct unfavourable cardiac alterations. However, it has been demonstrated that inflammation in diabetes can lead to the activation of nuclear factor kappa-B (NF-κB). This transcription factor causes cytokine-mediated myocardial and vascular damage when expressed, eventually leading to myocyte hypertrophy and myocardial calcium abnormalities [37]. Over time, these changes contribute to HF with eventual ventricular arrythmias leading to SCD [38].
Effect of DM on Ca2+ transporting components
L-type Ca2+ channels
The L-type Ca2+ channel, Cav1.2 is the main pathway of Ca2+ entry into the cell. The channel is more localized in the T-tubule in comparison to the surface sarcolemma [39]. The Cav1.2 channel is a hetero-tetrameric polypeptide complex containing the pore-forming unit, Cavα1c, in addition to other accessory subunits [40]. Some studies have shown a reduction in the L-type Ca2+ current in ventricular myocytes of STZ-induced diabetic rats [18, 41, 42]. Another study done by Bracken et al. showed the L-type Ca2+density was significantly reduced throughout the voltage ranges in myocytes from STZ-treated rats compared to age matched controls. Furthermore, the amplitude of contraction was also found to be significantly lower in STZ-treated rats [43]. Howarth et al. studied the changes in genetic expression associated with ventricular myocyte function and found upregulation of genes encoding the L-type Ca2+ voltage dependant (“Cacn”) channel proteins Cacna1c (alpha 1C subunit), Cacna1g (alpha 1G subunit), Cacna1h (alpha 1H subunit) and Cacna2d1 (alpha2/delta subunit 1) were upregulated in Zucker diabetic fatty (ZDF) rats compared to the control. Upregulation of Cacna1c might alter voltage sensitivity and perhaps activation and/or inactivation properties of the L-type Ca2+channel, which may be a compensatory mechanism for the reduced density and prolonged inactivation of L-type Ca2+ current [44]. Furthermore, altered phosphatidylinositol 3-kinase/ phosphatidylinositol 3,4,5-trisphosphate/ protein kinase B (PI3K/PIP3/Akt) pathway due to insulin or insulin growth factor (IGF-1) is decreased and is possibly the most plausible mechanism postulated to be responsible for the lower L-type Ca2+ current in DCM [45]. Finally, enhanced interleukin-1 activity can also play an important role in inhibiting this channel which contributes to HF [46].
Ryanodine receptor type 2
Ryanodine receptor type 2 (RyR2) is macromolecular homo-tetrameric protein complex that regulates the release of Ca2+ from the SR during the ECC process in the heart [47]. Though the molecular mechanism resulting in RyR2 dysregulation is not fully understood, there is cause for the dysfunctional Ca2+ release from the SR in DM. This may be through oxidation of RyR2 by ROS [48] and/or carbonyl species [49], change in RyR2 sensitivity to Ca2+ activation and functional uncoupling of RyR2 from L-type Ca2+channels on T-tubule membranes [50].
It has been shown that the open probability of cardiac RyR2 was increased by the ROS O2− and H2H/OH− [51, 52]. The opening of a RyR2 cluster causes local, rapid and brief elevations in intracellular free Ca2+ concentration which is termed Ca2+ sparks [53]. Analyses of these sparks can give insight into RyR2 function. Pereira et al. observed less frequency of Ca2+ sparks in db/db (obese T2DM) mice myocytes when compared to the control group, partly because of reduced expression of RyR2 Ca2+ channels [18]. Conversely, other studies found that increased RyR2 oxidation enhanced RyR2 activity and SR Ca2+ leakage [54, 55].
Phosphorylation by protein kinase A (PKA) and calcium/calmodulin-dependent protein kinase II (CaMKII) could result in alterations in sensitivity of RyR2 to Ca2+activation [56, 57]. Marx et al. discussed how PKA phosphorylation of RyR2 resulted in increased Ca2+ sensitivity for activation and elevated channel activity was associated with destabilization of the tetrameric channel complex [58]. Another study found a 1.5-fold increase in phosphorylation sites Ser2808 and Ser2814; both of which are target phosphorylation sites of CaMKII. It was further shown that CaMKII activity was increased by roughly 50% [59]. RyR2 phosphorylation and/or mutations have been linked to deadly ventricular arrhythmias and atrial fibrillation, sinoatrial node and atrioventricular node dysfunction, atrial standstill, dilated cardiomyopathy, HF, and SCD in human cardiac tissue studies. [60, 61]. In a study by Kilfoil et al., they found that in HFpEF, the Ca2+ current, Cav1.2 expression, and phosphorylated RyR were all higher than in controls [62]. It was suggested that increased Ca2+ inflow via L-type Ca2+ channels or leaking of SR Ca2+ into the cytosol via RyR channels could explain the increase in diastolic Ca2+ concentration which is responsible for a delay contraction and relaxation of the myocardium [63].
Sarcoplasmic reticulum calcium-ATPase
SERCA2a is the predominant form responsible for facilitation of Ca2+ storage in cardiac tissue [47]. Studies have shown that SERCA2a expression and activity were decreased in various pathophysiological conditions including diabetes [64]. Kim et al. investigated the diabetic alterations in cardiac sarcoplasmic reticulum calcium-ATPase (SERCA) and phoshpholamban (PLN)—an inhibitor of SERCA2a. They reported increased mRNA and protein levels of PLN while those of SERCA2a were significantly decreased in STZ induced diabetic rats. Additionally, maximal Ca2+ uptake and affinity of SERCA2a for Ca2+ were decreased [65]. Another study described the formation of advanced glycation-end products (AGEs) on SERCA which suggested a novel mechanism by which cardiac relaxation can be slowed during DM [66]. ROS impairs the oligomerization of PLN, altering its inhibitory interaction, thus enhancing SERCA2a transport [48, 67]. However, other data suggest that ROS may induce oxidative modifications on SERCA2a leading to its abnormal function [48, 68]. The protein coding gene Atp2a2, which is responsible for SERCA2, was found to be downregulated in ZDF myocytes [44]. Torre et al. found clear diastolic dysfunction in STZ-induced diabetic rats highlighted by clear mitral inflow changes in conjunction with downregulated SERCA2a expression and activity [69].
Sodium-calcium exchanger
Previous studies of sodium-calcium exchanger (NCX) vary from reduced activity to increased activity in a diabetic environment. Hattori et al. studied the effect of diabetes on the NCX using STZ-induced (T1DM) rats. They found a significantly reduced NCX current in STZ rats compared to the control. Furthermore, there was a 30% decrease in cardiac protein and mRNA levels of NCX1 (the dominant isoform of NCX in the heart) in diabetic rats [70]. Another study found no changes in NCX current density in high-energy (HE) induced obesity rat models compared to control [71]. Increased activity of NCX1 was found in db/db rats (T2DM) by Stølen et al. [72].
Connexin
Connexin 43 (Cx43) is a member of the gap junction family. It is composed of intercellular channels which allows for direct communication between neighbouring myocytes through the exchange of small ions and metabolites [73]. In a normal functioning heart, Cx43 are located at cell poles (intercalated disk) and facilitate proper signal transduction among cells. In progressive heart failure, structural remodelling and lowered cardiac efficiency — through elements such as elevated angiotensin II — promote cardiac wall stress. The result is a migration of Cx43 from cell poles to the lateral membrane leading to dyssynchronous contraction and further cardiac deterioration [74]. Joshi et al. studied connexin in cardiac cardiomyopathy and demonstrated increased tyrosine nitration of Cx43 was linked to impaired functioning of the channels. Furthermore, it was postulated the increase was concurrent with a progressive reduction of phosphorylated tyrosine due to RNS influence on connexin causing the re-localization of Cx43 [75]. Some studies have shown the collagen can affect Cx43 localization and expression [76, 77].
Inflammation, via chemoattractants, enables macrophage recruitment to the myocardium. Along with the already occurring oxidative dysfunction, these processes facilitate a pro-fibrotic function by facilitating fibroblast differentiation into myofibroblasts leading to fibrosis. Furthermore, there is increased metallo-proteinases for extracellular remodelling which plant the seeds for cardiac hypertrophy [78]. Fibrosis is a process that attempts to maintain cardiac structure or remodelling of the myocardium after apoptosis of cardiomyocytes. Fibrotic tissue is unexcitable, which causes disruption and conduction delay between the isolated cardiomyocytes. As a result, electrical propagation is obliged to take on a zigzag and discontinuous pattern throughout cardiac tissue ultimately contributing to arrhythmogenesis [79].
These changes are associated with electrophysiological abnormalities such as QT prolongation — predisposing to arrhythmia, absence of P wave morphology — consistent with arial flutter/fibrillation and widening QRS complexes — associated with impaired atrioventricular (AV) conduction or heart block [75]. In another related study by Zhang et al., they demonstrated that the dysfunction of cardiac conducting system in T1DM rats plays a major role in the development of cardiac arrhythmias due to increases in RR interval, PR interval and QRS complex duration of the ECG. These changes were associated with decreases in rate of the sino-atrial node (SAN) and HCN4 (pacemaker current) as well as downregulation of the gene expression for HCN4 channels, neuro-filament-M and β2-adrenergic receptor within the SAN of the myocardium during T1DM. It is postulated that alterations in the expression of these proteins within the SAN are closely associated with the regulation of the electrical signalling of the heart and as a result adversely affect cardiac action potential generation and propagation which in turn leads to arrythmia [80].
Effect of DM on regulatory contractile proteins
Troponin
Troponin is the sarcomeric Ca2+ regulator for cardiac and skeletal muscle contraction. When troponin binds to Ca2+, it transmits signals, via structural changes, throughout the actin-tropomyosin filaments, activating myosin ATPase activity and muscle contraction [81]. Troponin changes have been seen in inherited cardiomyopathies [82]. Changes in structure have also been seen in DM. Janssens et al. suggested that cardiac troponins may be irreversibly modified by glycation. In their study using diabetic rats, they found advanced glycation end product (AGE) modification of troponin-I (TnI) in diabetic rats but not in the control group [83]. Troponin-I functions to inhibit the actin-myosin interaction and it is tightly activated by phosphorylation. Troponin phosphorylation activity was also seen to be affected in DM. One study found no significant changes in TnI and gene expression of TnI in DM rats but phosphorylation of TnI was 40% higher compared to control. It further indicated that this increased phosphorylation may contribute towards cardiac myofibrillar ATPase activity depression in DM [84]. Conversely, from their study, Greenman et al. deduced that increased myofilament calcium sensitivity is associated with decreased cardiac TnI phosphorylation in diabetic rats [85]. Since TnI is involved in controlling cardiomyocyte contraction, phosphorylation of troponin contributes to decreased cardiac contractility [36].
Myosin
Myosin is a large motor protein that generates force through interaction with actin. It is involved in cellular processes such as muscle contraction, cell migration, cytokinesis and karyokinesis [86]. Jenkins et al. investigated whether changes in cross bridge disposition and myosin inter-filament spacing underline early development in diabetic cardiomyopathy. They found abnormal cross bridge deposition in diabetic hearts but no changes in inter-filament spacing in both groups. However, myosin head transfer by dobutamine was significantly blunted in diabetic rats [87]. Another study found decreased cardiac content of alpha-myosin in STZ rats compared to control [88]. In early diabetes, reduced myosin and myofibrillar ATPase activity is linked to contractile dysfunction [89].
Maholtra et al. found decreased actomyosin ATPase activity in diabetic myocytes (STZ rats) as well as shifts in cardiac myosin heavy chain (V1 to V3) which may play a role in impaired cardiac function [90]. Likewise, Howarth et al. found upregulation of the gene Myh7 (coding for β-myosin heavy chain) and downregulation of Myh6 (α-myosin heavy chain) and Myl2 (myosin light chain 2) in ZDF diabetic rats compared to control group. They deduced that these changes may underlie alterations in the time course of contraction [47]. Other studies have proven the shift of myosin heavy chain forms from α to β [91, 92].
Actin
Actin is a family of multifunctional proteins that form thin filaments in muscle fibrils and microfilaments in the cytoskeleton. The downregulation of α-actin has been observed in myocyte cells in a diabetic environment [92]. Pappritz et al. studied the levels of some contractile proteins in STZ rats at intervals of 6, 9 and 12 weeks. At week 6, they found higher distribution of α-actin, myosin light chain 3, ATP synthase and titin in STZ rats (T1DM) compared to control — which was thought to be a compensatory mechanism to hyperglycaemic insult. However, all these variables, except titin, were decreased in STZ rats, compared to control group, at week 12 [93]. In the cardiac sarcomere, the creation and dissociation of actin-myosin cross-bridges (CBs) is a critical determinant of force development and contractility. Poor cyclic transfer of myosin heads to actin filaments contributes to sarcomere contractile failure. Weddingham et al. found that in diabetic rats, a reduction in the distance between myosin heads and actin filaments during end diastole was linked to a slower rate of LV pressure depreciation [89].
Titin
Titin is the largest sarcomere protein and exists in 2 isoforms: N2BA and N2B. N2BA is the longer more compliant form while N2B is the shorter and stiffer form [94]. A shift in the titin isoform profile or general titin hypo-phosphorylation is linked in the development of cardiac dysfunction. Changes in titin vary in DM ranging from increased [93] to decreased distribution. In another subsequent study, Pappritz et al. found significantly lower titin intensity distribution in db/db mice (T2DM) compared to non-diabetic mice. In addition, protein level evaluation found a lower N2BA/N2B ratio and titin hypo-phosphorylation in db/db mice [95]. These results would conform to the literature that DM is associated with low protein kinase G (PKG) and protein kinase A (PKA) activity and would lead to titin hypo-phosphorylation and cardiomyocyte stiffness [96,97,98,99,100].
Functional ventricular changes in diabetic cardiomyopathy
In addition to factors discussed previously (hyperglycaemia and Ca2+ homeostasis impairment), other pathological elements contribute to ventricular changes and they include increased free fatty acid (FFA) levels, cardiac and systemic insulin resistance, systemic and tissue inflammation, and the activation renin–angiotensin–aldosterone system (RAAS) and the sympathetic nervous system [101].
In the hyperglycaemic environment, extracellular matrix (ECM) protein overproduction causes increased myocardial stiffness and subsequent cardiac dysfunction, eventually leading to cardiac failure [102]. Stimulation of transforming growth factor-beta (TGF-β) induces differentiation of cardiac fibroblasts into myofibroblasts leading to excessive collagen production [103]. Matrix metalloproteinases (MMPs) are a proteolytic enzyme family that plays a significant role in the destruction of EC matrix. Studies have shown a subtype, MMP-2, is downregulated, with reduced activity in diabetic hearts which contributes to fibrosis [104, 105].
In the early stage, diabetic cardiomyopathy is clinically asymptomatic and characterized by increased fibrosis and stiffness. However, there is increase in atrial filling and enlargement, reduction of early diastolic filling as well as an elevated left ventricular end-diastolic pressure [106]. These changes can be detected with magnetic resonance imaging (MRI) and echocardiography.
In the advanced stage of diabetic cardiomyopathy, changes at the cellular level such as impaired autophagy of apoptotic and/or necrotic cells, maladaptive immune response and oxidative stress increase cardiac fibrosis which initially result to impairment of left ventricular diastolic function as well as a slight decrease in ejection fraction. Early systolic dysfunction also begins to occur at this stage [91, 101]
Late-stage myocardial fibrosis can further impair diastolic and systolic function. It also begins to affect coronary microcirculation [107]. Increases in ROS and inflammation promote interstitial collagen deposition and crosslinking which is associated with interstitial fibrosis and impaired myocardial relaxation [108]. The dysfunction along with hypertrophy, thickened sclerotic small vessels, basement membrane thickening, hyaline arteriolar sclerosis and capillary microaneurysms predispose the heart to eventual failure or diabetic cardiomyopathy [109, 110].
Heart failure
Heart failure can be divided into 2 main groups based on left ventricular function. These are HF with reduced left ventricular ejection fraction (HFrEF) where left ventricular (LV) ejection output is < 40% — also known as systolic dysfunction, and HF with preserved ejection faction (HFpEF) where LV is > 50% — known as diastolic dysfunction [111]. A third group termed HF with mid-range ejection fraction (HFmrEF) has been described covering the grey area between 40 and 50%.
DM may play a role in the development of HFpEF due to an inflammatory reaction in adipose tissues. This results to downstream inflammation from the epicardium to myocardium, causing further changes in myocytes leading to increased stiffness. With combination of several other external factors discussed above, this deranged myocardial metabolism aggregates to HF [112].
Arrhythmias in DM
Ventricular arrhythmias have been found to be more frequent in diabetic patients [113]. QTc prolongation is linked to an increased incidence of ventricular arrhythmias and is a powerful predictor of cardiovascular mortality [114].
Reduction in outward transient K+ (Ito) current plays a major role in delayed action potential duration. Although not fully understood, two hypotheses have been proposed regarding the effects of T1DM on potassium currents in cardiac muscle [115]. The first hypothesis involves changes in gene expression of many proteins (including potassium channel proteins) in the absence of insulin [45]. Shimoni et al. demonstrated this event when diabetic myocytes were incubated with insulin for 6 h to restore Ito values to control levels [116]. The second hypothesis assumes that defective glucose metabolism is the cause of decreased cardiac Ito in DM. Torres-Jacome et al. highlighted this by describing the recovery of Ito current amplitudes in diabetic cardiomyocytes after a 6-h incubation with pyruvate [117].
Furthermore, a functional knockout of Ito leads to prolongation of QT intervals [118]. Sato et al. demonstrated how the fast-recovering component of Ito was found to be significantly reduced in OLETF rats (a rat model for T2DM) compared to LETO rats (control group) in both sub-endocardial and sub-epicardial myocytes. The mRNA level of KCND2 — a gene coding for Kv4.2 (one of the α-subunit subfamilies of the voltage-gated K+ channel) — and KChIP2 (an Ito accessory subunit) were significantly lower in the cardiomyocytes of OLETF rats in comparison to LETO rats. As expected, results of the Kv4.2 protein followed the similar significant trend of KCND2 in cardiomyocytes. Finally, the levels of Irx5 (a transcription factor that negatively regulates Kv4.2) were significantly higher in the cardiomyocytes of OLETF rats [119].
Anderson et al. examined the risk of cardiac arrhythmias in insulin- treated T2DM and control subjects. They found progressively increasing heart rate corrected QT interval prolongations during hypoglycaemia in the group of patients with type 2 diabetes [120]. In acute hyperglycaemia, arrhythmias may originate from persistent alteration of Ca2+/calmodulin-dependent protein kinase II (CaMKII) by O-linked N-acetylglucosamine (O-GlcNAc), which causes enhanced activation of spontaneous sarcoplasmic reticulum Ca2+ release [121]. Interestingly, a study by Zhang et al. [80] demonstrated that diabetes can elicit neuropathy in the conductive tissues of diabetic heart by inducing downregulation of neuro-filament M (NF-M) in the sympathetic nerve and beta- adrenergic receptors. These changes were accompanied by downregulation of the expression of a number of channel proteins in the diabetic heart including RyR2, SERCA2, NCX1, CX40-, CX-43 and CX-45, Cav1.3, Cav3.1 and HCN4, the fancy (fi) potassium current and AChKir-3 (potassium efflux channel). The downregulation of the gene expression of NF-M and β2-adrenergic receptor, as well as cation channel transporting proteins, could be linked to the reduced autonomic control of the heart and myocardial contraction, all leading to arrhythmias which are a major cause of mortality in patients with diabetes [122].
Sudden cardiac death
The natural course of untreated diabetic cardiomyopathy may lead to sudden cardiac death (SCD) which is an unexpected death due to cardiac causes. This lethal process occurs in a short period of time (generally within 1 h of symptom onset) in a person with known or unknown cardiac disease [123]. It is responsible for some 300,000 deaths per year in the USA alone [124] and more than 7 million deaths annually worldwide [125]. Processes described above (see also Fig. 2) contribute to structural and functional changes in the heart such as left ventricular muscle disarray and hypertrophy, interstitial fibrosis, oxidative stress and increased cell death [126]. The result is diastolic and systolic dysfunctions as well cardiac arrhythmias and eventually SCD [127].
Potential future therapeutic areas
Table 1 shows current pharmacological treatment options used for treatment in diabetes in a whole which focus on blood glucose control. In the presence of HF drugs such as beta blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), diuretics and calcium channel blockers (for diastolic dysfunction) are popular choices to add to therapy. In addition to current therapies in use to control the metabolic disease, new potential targets are being studied to target diabetes-induced changes to the heart. They include the following:
Gene therapy
The idea of being able to either up or downregulate the expression of key factors in the development of diabetic cardiomyopathy may be approaching through gene therapy [132]. For instance, reduced heart chamber compliance is a hallmark change in HFpEF associated with T2DM. It is partly due to altered titin phosphorylation leading to increased cardiomyocyte stiffness [133]. Hopf et al. showed how treatment with Neuregulin1 (NRG-1) was able to rescue titin-based cardiomyocyte stiffening in DM rats via increased PKG and ERK1/2 activity and reduced PKCα activity. This in turn reversed DM-induced titin hypo-phosphorylation changes [134]. Currently, other targets which are being studied include forkhead box-containing protein 1, O subfamily (FoxO1) [135] and mitochondrial heat shock protein 70 (mtHsp70) [136]. Troponin and other regulatory cardiac proteins could be useful in phenotypic HFpEF classification or clinical trial selection criteria to target a specific HFpEF subset or a population with a higher-risk profile [132, 133, 137].
Non-coding RNAs as biomarkers
MicroRNAs (miRNAs) are a class of non-coding RNAs that regulate gene expression at the post transcriptional level in both physiological and pathological conditions. There is opportunity to use miRNA as biomarkers for diabetic cardiomyopathy owing the possibility of its detection and stability in plasma [138]. Copier et al. suggested that the miRNAs miR-19b-3p and miR-181b-5p could be suitable biomarkers for diabetic cardiomyopathy in asymptomatic DM patients. They found both of these circulating and cardiac miRNAs were linked to cardiac dysfunction in rats with high fat diet during the development of diabetic cardiomyopathy [139]. Other non-coding RNAs being studied include long non-coding RNAs (lncRNAs), which were found to be independent predictors of diastolic function and remodelling in patients with T2DM [140].
Non-coding RNAs as treatment
Bcl-2 and Pim1 are anti-apoptotic and cardio-protective proteins. They are negatively regulated by the miRNA miR-1 which constantly increases through the stages of diabetic cardiomyopathy. Transfection with anti-miR1 activates pro-survival signals in cardiomyocytes and cardiac progenitor cells in a hyperglycaemic environment [141]. Yin et al. demonstrated the protective role of miR-30c in cardiac metabolism in diabetic rats (db/db) via modulation PGC-1β. They found overexpression of miR-30c improved glucose utilization, reduced excessive ROS production and lipid accumulation and subsequent attenuation of cardiomyocyte apoptosis and cardiac dysfunction in db/db mice [133]. Pathological remodelling of the heart and decreased inflammatory response were reduced by endothelial specific overexpression of miR-146a and hyperglycaemia reduces this miRNA’s expression [142]. Moreover, overexpression of homeobox transcript antisense RNA (HOTAIR), an lncRNA which promoted Akt phosphorylation and improved AC16 Human cardiomyocyte cell line viability may also improve diabetic cardiomyopathy through activation of the PI3K/Akt pathway [143].
Oxidative stress modulation
The potential therapeutic action of nuclear factor-erythroid factor 2-related factor (2 Nrf2), a transcription factor in the treatment of several diseases including diabetic cardiomyopathy, has been suggested [144]. Sulforaphane (SFN) is obtained from cruciferous vegetables (e.g. broccoli, cabbages, brussels sprouts) and shows antidiabetic as well as anti-cancer properties in experiments [145]. In one study, SFN was found to almost completely prevent diabetic cardiomyopathy in STZ rats (T1DM) through upregulation of NrF2 expression and transcription function in the heart. Nrf2 drives antioxidant and detoxifying defence to suppress oxidative stress–mediated cardiac protein injury and subsequent heart dysfunction [146]. Furthermore, Nrf2 promotes autophagic clearance of harmful ubiquitinated protein aggregates, thus protecting the heart from proteo-cytotoxicity [147].
Silencing the Nrf2 expression completely abolished this preventative property of SFN [148]. Similarly, diallyl trisulfide (DATS), a powerful antioxidant, was found to protect against hyperglycaemia-induced ROS-mediated apoptosis, with further protection through increased Nrf2 protein stability and nuclear translocation leading to Nrf2-regulated anti-oxidant enzymes in cardiomyocytes exposed to hyperglycaemia [149]. The proteasome inhibitor MG-132 was found to provide a therapeutic effect in diabetic cardiomyopathy possibly through upregulation of Nrf2-dependent anti-oxidative function and downregulation of NF-κB-mediated inflammation in OVE26 mice (T1DM) [150].
Coenzyme Q10 has also emerged as an effective antioxidant. Huynh et al. investigated its properties in STZ induced type 1 diabetic rats. They found upregulation of LV Nox2 and superoxide production with LV oxidative damage. Coenzyme Q10 was found to almost prevent these changes completely in rats given STZ and coenzyme Q10 simultaneously at the beginning of the study [151].
The development of diabetic cardiomyopathy in STZ-induced type 1 diabetic rats can also be prevented by the natural polyphenolic compounds, extracted from curcumin. Studies have shown this preventative property to be associated with suppression of NOX activation which further alleviates excessive generation of ROS and/or RNS [152, 153]. Other anti-oxidant therapy being studied with potential therapeutic effects on diabetic cardiomyopathy include thioredoxin 1, tempol, metallothionein and resveratrol [154,155,156,157].
Alpha-lipoic acid
Alpha-lipoic acid (ALA) was also found to alleviate changes in heart muscle. Li et al. found that ALA was able to attenuate TGF-β expression. Additionally, in STZ-induced diabetic cardiomyopathy, ALA therapy reduced ventricular dysfunction, decreased LV Type I and III collagen deposition, and enhanced MMP-2 activity [158].
Healthy lifestyle habits to combat DM
Following unique practices can help to slow down the general effects of diabetes. The Indo-Mediterranean-type diet consists of bountiful antioxidants such as omega-3 fatty acids, polyphenolics and flavonoids which can help to maintain oxidative function and reduce damage to myocardial cells [78]. Conversely, Western diets predispose individuals to CVDs. This diet has shown to increase AGE which activates receptor for AGE (RAGE) leading to downstream oxidative stress, inflammation, and eventual cardiomyocyte hypertrophy [159].
Consuming large amounts of alcohol (> 5 drinks per day) seemingly increases the risk for ventricular arrhythmia and SCD. Interestingly, Tu et al. showed that consumption of large amounts of beer, spirits and cider correlated with enhanced SCD risk while red and white wine intake was associated with lower risk [160].
Physical activity, specifically aerobic exercise, has been shown to acutely increase insulin sensitivity in the body which enhances blood glucose for synthesis of glycogen and fat oxidation for storage [161]. Physical activity can also increase pancreatic beta cell mass and function by encouraging the pancreas to produce newly synthesized insulin content, thereby producing a synergistic effect with insulin [162].
Smoking has been known to increase SCD. Aune et al. found a three-fold increase in SCD in people who smoke while there was also a 38% increase in former smokers [163].
Conclusion
In conclusion, this review attempted to explain the cellular and molecular mechanisms and to a lesser extent the genetic factor associated with the development of diabetes-induced cardiomyopathy which can subsequently lead to chronic heart failure and sudden cardiac death. The study focused mainly on the ECC process highlighting the defects of calcium transporting (SERCA, NCX, RyR and connexin) and contractile regulatory (myosin, actin, titin and troponin) proteins in the heart. The study also highlighted new therapies which are currently in development to treat or prevent diabetic cardiomyopathy. Nevertheless, prevention is better than cure and as such Fig. 3 shows a flow diagram illustrating the different ways in either preventing or delaying the development of heart failure whether it is induced by diabetes mellitus, other causes or during a combination of the different factors.

Diagram illustrating the different ways in either preventing or delaying the development of heart failure
Data availability
Not applicable.
References
World Health Organization. (n.d.) Diabetes. Retrieved from https://www.who.int/health-topics/diabetes#tab=tab_1
American Diabetes Association (2015) Classification and diagnosis of diabetes. Diabetes Care 38(Supplement 1):S8–S16
Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ (2010) Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 39(3):481–497
Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J, Hamman RF (2014) SEARCH for Diabetes in Youth Study. Prevalence of type 1 and type 2 diabetes among children and adolescents from (2001) to 2009. JAMA 311(17):1778–1786
CDC. (2019, May 30) Diabetes. Retrieved from Centers for Disease Control and Prevention: https://www.cdc.gov/diabetes/basics/type2.html
Herath H, Herath R, Wickremasinghe R (2017) Gestational diabetes mellitus and risk of type 2 diabetes 10 years after the index pregnancy in Sri Lankan women-a community based retrospective cohort study. PLoS One 12(6):e0179647
Khan MA, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J (2020) Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J Epidemiol Global Health 10(1):107–111
Deshpande AD, Harris-Hayes M, Schootman M (2008) Epidemiology of diabetes and diabetes-related complications. Phys Ther 88(11):1254–1264
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A (1972) New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 30(6):595–602
Newby DE, Grubb NR, Bradbury A (2014) Cardiovascular disease. In: Davidson S, Walker BR, Colledge NR, Ralston SH, Penman ID (eds) Davidson’s Principles & Practice of Medicine, 22nd edn. Elsevier, Edinburgh, pp 528–532
Cardiac Physiology (2013) In L Sherwood, Human Physiology: From Cells to Systems (8th ed. 305–340). Belmont, CA: Brooks/Cole
Sinnatamby CS (2011) Middle mediastinum and heart. In Last's Anatomy - Regional and Applied (12th ed. 197–207). Edinburgh: Elsevier
Cardiovascular disease (2009) In P Kumar, M Clark, Kumar & Clark's Clinical Medicine (7th ed. 681–688). Edinburgh: Elsevier
Widmaier EP, Raff H, Strang KT (2014) Cardiac muscle. In: Widmaier EP, Raff H, Strang KT (eds) Vander’s Human Physiology - The mechanisms of body function, 13th edn. McGraw-Hill, New York, pp 292–294
Klabunde RE (2017, December 20) Cardiac excitation-contraction coupling. Retrieved from Cardiovascular Physiology Concepts: https://www.cvphysiology.com/Cardiac%20Function/CF022
Miki T, Yuda S, Kouzu H, Miura T (2013) Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev 18(2):149–166
Belke DD, Dillmann WH (2004) Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 6(6):424–429
Belke DD, Swanson EA, Dillmann WH (2004) Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 53(12):3201–3208
Tian C, Alomar F, Moore CJ, Shao CH, Kutty S, Singh J, Bidasee KR (2014) Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart Fail Rev 19(1):101–112
Vander Jagt DL (2008) Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol Drug Interact 23(1–2):93–124
Baynes JW, Thorpe SR (1999) Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48(1):1–9
Jakubcakova V, Curzi ML, Flachskamm C, Hambsch B, Landgraf R, Kimura M (2013) The glycolytic metabolite methylglyoxal induces changes in vigilance by generating low-amplitude non-REM sleep. J Psychopharmacol 27(11):1070–1050
Schalkwijk CG, Stehouwer CD (2020) Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev 100(1):407–461
Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS (1999) Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 48(1):198–202
Shao CH, Capek HL, Patel KP, Wang M, Tang K, DeSouza C, Bidasee KR (2011) Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 60(3):947–959
Tian C, Shao CH, Moore CJ, Kutty S, Walseth T, DeSouza C, Bidasee KR (2011) Gain of function of cardiac ryanodine receptor in a rat model of type 1 diabetes. Cardiovasc Res 91(2):300–309
Papadaki M, Holewinski RJ, Previs SB, Martin TG, Stachowski MJ, Li A, Kirk JA (2018) Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight 3(20):e121264
van der Pol A, van Gilst WH, Voors AA, van der Meer P (2019) Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail 21(4):425–435
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57(4):660–671
Olukman M, Orhan CE, Celenk FG, Ulker S (2010) Apocynin restores endothelial dysfunction in streptozotocin diabetic rats through regulation of nitric oxide synthase and NADPH oxidase expressions. J Diabetes Complications 24(6):415–423
Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Anversa P (2001) Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 89(3):279–286
Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I (2003) Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal 5(6):789–794
King MK, Coker ML, Goldberg A, McElmurray JH, Gunasinghe HR, Mukherjee R, Spinale FG (2003) Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res 92(2):177–185
Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, Sowers JR (2015) Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension 66(6):1159–1167
Pedreanez A, Mosquera J, Munoz N, Robalino J, Tene D (2022) Diabetes, heart damage, and angiotensin II. What is the relationship link between them? A minireview. Endocr Regul 56(1):55–65
Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122(4):624–638
Ramesh P, Yeo JL, Brady EM, McCann GP (2022) Role of inflammation in diabetic cardiomyopathy. Ther Adv Endocrinol Metab 13:20420188221083530
Klein L, Hsia H (2014) Sudden cardiac death in heart failure. Cardiol Clin 32(1):135–144
Rougier JS, Abriel H (2016) Cardiac voltage-gated calcium channel macromolecular complexes. Biochim Biophys Acta 1863(7 Pt B):1806–1812
Shaw RM, Colecraft HM (2013) L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res 98(2):177–186
Bracken N, Howarth FC, Singh J (2006) Effects of streptozotocin-induced diabetes on contraction and calcium transport in rat ventricular cardiomyocytes. Ann N Y Acad Sci 1084:208–222
Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC (2015) Dapagliflozin reduces the amplitude of shortening and Ca(2+) transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem 400(1–2):57–68
Bracken NK, Woodall AJ, Howarth FC, Singh J (2004) Voltage-dependence of contraction in streptozotocin-induced diabetic myocytes. Mol Cell Biochem 261(1–2):235–243
Howarth FC, Qureshi MA, Hassan Z, Al Kury LT, Isaev D, Parekh K, Adeghate E (2011) Changing pattern of gene expression is associated with ventricular myocyte dysfunction and altered mechanisms of Ca2+ signalling in young type 2 Zucker diabetic fatty rat heart. Exp Physiol 96(3):325–337
Ozturk N, Uslu S, Ozdemir S (2021) Diabetes-induced changes in cardiac voltage-gated ion channels. World J Diabetes 12(1):1–18
Murphy SP, Kakkar R, McCarthy CP, Januzzi JL (2020) Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol 75(11):1324–1340
Al Kury LT (2020) Calcium homeostasis in ventricular myocytes of diabetic cardiomyopathy. J Diabetes Res 1942086
D’Oria R, Schipani R, Leonardini A, Natalicchio A, Perrini S, Cignarelli A, Giorgino F (2020) The role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid Med Cell Longev 2020:5732956
Shao CH, Tian C, Ouyang S, Moore CJ, Alomar F, Nemet I, Bidasee KR (2012) Carbonylation induces heterogeneity in cardiac ryanodine receptor function in diabetes mellitus. Mol Pharmacol 82(3):383–399
Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H (2006) Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103(11):4305–4310
Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schröder E, Wait R, Eaton P (2006) Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 281(31):21827–21836
Guo X, Yuan S, Liu Z, Fang Q (2014) Oxidation- and CaMKII-mediated sarcoplasmic reticulum Ca(2+) leak triggers atrial fibrillation in aging. J Cardiovasc Electrophysiol 25(6):645–652
Cheng H, Lederer WJ, Cannell MB (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262:740–744
Joseph LC, Subramanyam P, Radlicz C, Trent CM, Iyer V, Colecraft HM, Morrow JP (2016) Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm 13(8):1699–1706
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71(2):310–321
Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H (1995) Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem 270(5):2074–2081
Wehrens XH, Lehnart SE, Reiken SR, Marks AR (2004) Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 94(6):e61–e70
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101(4):365–376
Shao CH, Wehrens XH, Wyatt TA, Parbhu S, Rozanski GJ, Patel KP, Bidasee KR (2009) Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J Appl Physiol 106(4):1280–1292
Foti F, De-Giorgio F, Vetrugno G, Basso C, Pilichou K (2020) A de novo ryanodine receptor 2 gene variant in a case of sudden cardiac death. Int J Legal Med 134(2):619–623
Tester DJ, Bombei HM, Fitzgerald KK, Giudicessi JR, Pitel BA, Thorland EC, Ackerman MJ (2020) Identification of a novel homozygous multi-exon duplication in RYR2 among children with exertion-related unexplained sudden deaths in the Amish Community. JAMA Cardiol 5(3):13–18
Kilfoil PJ, Lotteau S, Zhang R, Yue X, Aynaszyan S, Solymani RE, Goldhaber JI (2020) Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J Physiol 598(22):5091–5108
Eisner DA, Caldwell JL, Trafford AW, Hutchings DC (2020) The control of diastolic calcium in the heart: basic mechanisms and functional implications. Circ Res 126(3):395–412
Ganguly PK, Pierce GN, Dhalla KS, Dhalla NS (1983) Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. Am J Physiol 244(6):E528–E535
Kim HW, Ch YS, Lee HR, Park SY, Kim YH (2001) Diabetic alterations in cardiac sarcoplasmic reticulum Ca2+-ATPase and phospholamban protein expression. Life Sci 70(4):367–379
Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR (2004) Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 53(2):463–473
Sivakumaran V, Stanley BA, Tocchetti CG, Ballin JD, Caceres V, Zhou L, Paolocci N (2013) HNO enhances SERCA2a activity and cardiomyocyte function by promoting redox-dependent phospholamban oligomerization. Antioxid Redox Signal 19(11):1185–1197
Balderas-Villalobos J, Molina-Muñoz T, Mailloux-Salinas P, Bravo G, Carvajal K, Gómez-Viquez NL (2013) Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol 305(9):H1344–H1353
Torre E, Arici M, Lodrini AM, Ferrandi M, Barassi P, Hsu SC, Rocchetti M (2022) SERCA2a stimulation by istaroxime improves intracellular Ca2+ handling and diastolic dysfunction in a model of diabetic cardiomyopathy. Cardiovasc Res 118(4):1020–1032
Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando S, Kanno M (2000) Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J Physiol 527(Pt 1):85–94
Ricci E, Smallwood S, Chouabe C, Mertani HC, Raccurt M, Morel G, Bonvallet R (2006) Electrophysiological characterization of left ventricular myocytes from obese Sprague-Dawley rat. Obesity (Silver Spring) 14(5):778–786
Stølen TO, Høydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Wisløff U (2009) Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res 105(6):527–536
Rodríguez-Sinovas A, Sánchez JA, Valls-Lacalle L, Consegal M, Ferreira-González I (2021) Connexins in the heart: regulation, function and involvement in cardiac disease. Int J Mol Sci 22(9):4413
Muralidaran Y, Viswanathan P (2015) Diabetic cardiomyopathy: a new perspective of mechanistic approach. Jf Diabetes & Metabolism 6(10):1000605
Joshi MS, Mihm MJ, Cook AC, Schanbacher BL, Bauer JA (2015) Alterations in connexin 43 during diabetic cardiomyopathy: competition of tyrosine nitration versus phosphorylation. J Diabetes 7(2):250–259
Jeong MH, Kim HJ, Pyun JH, Choi KS, Lee DI, Solhjoo S, Kang JS (2017) Cdon deficiency causes cardiac remodeling through hyperactivation of WNT/β-catenin signaling. Proc Natl Acad Sci U S A 114(8):E1345–E1354
Viczenczova C, Kura B, Chaudagar KK, Szeiffova B, Egan BB, Knezl V, Slezak J (2017) Myocardial connexin-43 is upregulated in response to acute cardiac injury in rats. Can J Physiol Pharmacol 95(8):911–919
Singh RB, Fedacko J, Pella D, Fatima G, Elkilany MM, Vanova N (2022) High exogenous antioxidant, restorative treatment (heart) for prevention of the six stages of heart failure: the heart diet. Antioxidants 11(8):1464
Heikhmakhtiar AK, Tekle AA, Lim KM (2021) Influence of fibrosis amount and patterns on ventricular arrhythmogenesis and pumping efficacy: computational study. Front Physiol 12:644473
Zhang Y, Yang Z, Yani J, Qureshi MA, Logantha SJ, Kassab S, Dobrzynski H (2019) Electrical conduction system remodelling in streptozotocin-induced diabetes mellitus rat heart. Front Physiol 10:826
Gomes AV, Potter JD, Szczesna-Cordary D (2002) The role of troponins in muscle contraction. IUBMB Life 54(6):323–333
Lu QW, Wu XY, Morimoto S (2013) Inherited cardiomyopathies caused by troponin mutations. J Geriatr Cardiol 10(1):91–101
Janssens JV, Ma B, Brimble MA, Van EJ, Delbridge LM, Mellor KM (2018) Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation. Sci Rep 8(1):16084
Liu X, Takeda N, Dhalla NS (1996) Troponin I phosphorylation in heart homogenate from diabetic rat. Biochim Biophys Acta 1316(2):78–84
Greenman AC, Diffee GM, Power AS, Wilkins GT, Gold OM, Erickson JR, Baldi JC (2021) Increased myofilament calcium sensitivity is associated with decreased cardiac troponin I phosphorylation in the diabetic rat heart. Exp Physiol 106(11):2235–2247
Harrington WF, Rodgers ME (1984) Myosin. Annu Rev Biochem 53:35–73
Jenkins MJ, Pearson JT, Schwenke DO, Edgley AJ, Sonobe T, Fujii Y, Shirai M (2013) Myosin heads are displaced from actin filaments in the in situ beating rat heart in early diabetes. Biophys J 104(5):1065–1072
Rundell VL, Geenen DL, Buttrick PM, de Tombe PP (2004) Depressed cardiac tension cost in experimental diabetes is due to altered myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol 287(1):H408–H413
Waddingham MT, Edgley AJ, Tsuchimochi H, Kelly DJ, Shirai M, Pearson JT (2015) Contractile apparatus dysfunction early in the pathophysiology of diabetic cardiomyopathy. World J Diabetes 6(7):943–960
Malhotra A, Lopez MC, Nakouzi A (1995) Troponin subunits contribute to altered myosin ATPase activity in diabetic cardiomyopathy. Mol Cell Biochem 151(2):165–172
Fang ZY, Prins JB, Marwick TH (2004) Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev 25(4):543–567
Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, Taegtmeyer H (2000) Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol 32(6):985–996
Pappritz K, Grune J, Klein O, Hegemann N, Dong F, El-Shafeey M, Van Linthout S (2020) Speckle-tracking echocardiography combined with imaging mass spectrometry assesses region-dependent alterations. Scientific Reports 10(1):3629
Hamdani N, Paulus WJ (2013) Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation 128(1):5–8
Pappritz K, Klein O, Dong F, Hamdani N, Kovacs A, O’Flynn L, Van Linthout S (2021) MALDI-IMS as a tool to determine the myocardial response to syndecan-2-selected mesenchymal stromal cell application in an experimental model of diabetic cardiomyopathy. Proteomics Clin Appl 15(1):e2000050
Eyster CA, Matsuzaki S, Newhardt MF, Giorgione JR, Humphries KM (2020) Diabetes induced decreases in PKA signalling in cardiomyocytes: the role of insulin. PLoS One 15(8):e0231806
Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, Paulus WJ (2013) Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6(6):1239–1249
Paulus WJ, Tschöpe C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodelling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62(4):263–271
van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, Paulus WJ (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126(7):830–839
Hamdani N, Hervent AS, Vandekerckhove L, Matheeussen V, Demolder M, Baerts L, De Keulenaer GW (2014) Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc Res 104(3):423–431
Jia G, DeMarco VG, Sowers JR (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 12(3):144–153
Li CJ, Lv L, Li H, Yu DM (2012) Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha-lipoic acid. Cardiovasc Diabetol 11:73
Petrov VV, Fagard RH, Lijnen PJ (2002) Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 39(2):258–263
Westermann D, Rutschow S, Jäger S, Linderer A, Anker S, Riad A, Tschöpe C (2007) ontributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes 56(3):641–646
Van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, Tschöpe C (2008) Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 103(4):319–327
Westermeier F, Riquelme JA, Pavez M, Garrido V, Díaz A, Verdejo HE, Lavandero S (2016) New molecular insights of insulin in diabetic cardiomyopathy. Front Physiol 7(125):125
Adeghate E, Singh J (2014) Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail Rev 19(1):15–23
Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA (2010) Diabetic cardiomyopathy: mechanisms and therapeutic targets. Drug Discov Today Dis Mech 7(2):e135–e143
Mytas DZ, Stougiannos PN, Zairis MN, Foussas SG, Pyrgakis VN, Kyriazis IA (2009) Diabetic myocardial disease: pathophysiology, early diagnosis and therapeutic options. J Diabetes Complications 23(4):273–282
Wang J, Song Y, Wang Q, Kralik PM, Epstein PN (2006) Causes and characteristics of diabetic cardiomyopathy. Rev Diabet Stud 3(3):108–117
Agarwal G, Singh SK (2017) Arrhythmias in type 2 diabetes mellitus. Indian J Endocrinol Metab 21(5):715–718
Swedberg K (2021) Heart failure subtypes: pathophysiology and definitions. Diabetes Res Clin Pract 175:108815
Packer M (2020) Do most patients with obesity or type 2 diabetes, and atrial fibrillation, also have undiagnosed heart failure? A critical conceptual framework for understanding mechanisms and improving diagnosis and treatment. Eur J Heart Fail 22(2):214–227
Cox AJ, Azeem A, Yeboah J, Soliman EZ, Aggarwal SR, Bertoni AG, Bowden DW (2014) Heart rate-corrected QT interval is an independent predictor of all-cause and cardiovascular mortality in individuals with type 2 diabetes: the Diabetes Heart Study. Diabetes Care 37(5):1454–1461
Andersen A, Bagger JI, Baldassarre MP, Christensen MB, Abelin KU, Faber J, Vilsbøll T (2021) Acute hypoglycemia and risk of cardiac arrhythmias in insulin-treated type 2 diabetes and controls. Eur J Endocrinol 185(2):343–353
O’Brien RM, Granner DK (1996) Regulation of gene expression by insulin. Physiol Rev 76(4):1109–1161
Shimoni Y, Ewart HS, Severson D (1999) Insulin stimulation of rat ventricular K+ currents depends on the integrity of the cytoskeleton. J Physiol 514(Part 3):735–745
Torres-Jacome J, Gallego M, Rodríguez-Robledo JM, Sanchez-Chapula JA, Casis O (2013) Improvement of the metabolic status recovers cardiac potassium channel synthesis in experimental diabetes. Acta Physiol (Oxf) 207(3):447–459
Barry DM, Xu H, Schuessler RB, Nerbonne JM (1998) Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res 83(5):560–567
Sato T, Kobayashi T, Kuno A, Miki T, Tanno M, Kouzu H, Tohse N (2014) Type 2 diabetes induces subendocardium-predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. Am J Physiol Heart Circ Physiol 306(7):H1054–H1065
Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Bers DM (2013) Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502(7471):372–376
Nakou ES, Mavrakis H, Vardas PE (2012) Are diabetic patients at increased risk of arrhythmias? Hellenic J Cardiol 53:335–339
Sovari AA (2020, 12 13) Sudden cardiac death. Retrieved from Medscape: https://emedicine.medscape.com/article/151907-overview
Zhang L, Narayanan K, Suryadevara V, Teodorescu C, Reinier K, Uy-Evanado A, Chugh SS (2015) Occupation and risk of sudden death in a United States community: a case-control analysis. BMJ Open 5(12):e009413
Mehra R (2007) Global public health problem of sudden cardiac death. J Electrocardiol 40(6 Suppl):S118–S122
Poornima IG, Parikh P, Shannon RP (2006) Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 98(5):596–605
Smail MM, Howarth FC, Singh J, Rupee S, Rupee K, Hanoman C, Bidasee K (2020) Mechanisms of diabetes mellitus-induced sudden cardiac death. In P Magnusson, JA LeQuang (Eds) Suden Cardiac Death. IntechOpen. Retrieved from https://www.intechopen.com/chapters/73520
Alomar FA, Al Rubaish A, Al-Muhanna F, Al-Ali AA, McMillan JE, Singh J, Bidasee K (2020) Adeno-associated viral transfer of glyoxalase-1 blunts carbonyl and oxidative stress in heart of type 1 diabetic rats Antioxidants 9(7) Article 592 (22 pages)
Kenny HC, Abel ED (2019) Heart failure in type 2 diabetes mellitus: impact of glucose lowering agents, heart failure therapies and novel therapeutic strategies. Circ Res 124(1):121–141
Dahlén AD, Dashi G, Maslov I, Attwood MM, Jonsson J, Trukhan V, Schiöth HB (2022) Trends in antidiabetic drug discovery: FDA approved drugs, new drugs in clinical trials and global sales. Front Pharmacol 12:807548
Takebayashi K, Aso Y, Inukai T (2010) Role of bile acid sequestrants in the treatment of type 2 diabetes. World J Diabetes 1(5):146–152
Shivaprasad C, Kalra S (2011) Bromocriptine in type 2 diabetes mellitus. Indian J Endocrinol Metab 15(Suppl 1):S17–S24
Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M (2018) Diabetic cardiomyopathy: current and future therapies. Beyond Glycemic Control. Front Physiol 9:1514
Hopf AE, Andresen C, Kötter S, Isić M, Ulrich K, Sahin S, Krüger M (2018) Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can be modulated by neuregulin-1. Circ Res 123(3):342–355
Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Hill JA (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 122(3):1109–1118
Shepherd DL, Hathaway QA, Nichols CE, Durr AJ, Pinti MV, Hughes KM, Hollander JM (2018) Mitochondrial proteome disruption in the diabetic heart through targeted epigenetic regulation at the mitochondrial heat shock protein 70 (mtHsp70) nuclear locus. J Mol Cell Cardiol 119:104–115
Mentz RJ, Greene SJ (2018) Adding troponin to the puzzle of heart failure with preserved ejection fraction: marker or mediator? J Am Coll Cardiol 72(1):41–44
de Lucia C, Komici K, Borghetti G, Femminella GD, Bencivenga L, Cannavo A, Rengo G (2017) microRNA in cardiovascular ageing and age-related cardiovascular diseases. Front Med (Lausanne) 4:74 (an abstract)
Copier CU, León L, Fernández M, Contador D, Calligaris SD (2017) Circulating miR-19b and miR-181b are potential biomarkers for diabetic cardiomyopathy. Sci Rep 7(1):13514
de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, Thum T (2016) Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep 6:37354
Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, Madeddu P (2011) Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ Res 108(10):1238–1251
Feng B, Chen S, Gordon AD, Chakrabarti S (2017) miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol 105:70–76
Qi K, Zhong J (2018) LncRNA HOTAIR improves diabetic cardiomyopathy by increasing viability of cardiomyocytes through activation of the PI3K/Akt pathway. Exp Ther Med 16(6):4817–4823
Cho HY, Reddy SP, Kleeberger SR (2006) Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal 8(1–2):76–87
Fahey JW, Talalay P (1999) Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol 37(9–10):973–979
Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L, Cui T (2016) Nrf2-mediated cardiac maladaptive remodeling and dysfunction in a setting of autophagy insufficiency. Hypertension 67(1):107–117
Wang W, Li S, Wang H, Li B, Shao L, Lai Y, Cui T (2014) Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J Mol Cell Cardiol 72:305–315
Bai Y, Cui W, Xin Y, Miao X, Barati MT, Zhang C, Cai L (2013) Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol 57:82–95
Tsai CY, Wang CC, Lai TY, Tsu HN, Wang CH, Liang HY, Kuo WW (2013) Antioxidant effects of diallyl trisulfide on high glucose-induced apoptosis are mediated by the PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int J Cardiol 168(2):1286–1297
Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y, Cai L (2013) Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. Am J Physiol Heart Circ Physiol 304(4):H567–H578
Huynh K, Kiriazis H, Du XJ, Love JE, Gray SP, Jandeleit-Dahm KA, Ritchie RH (2013) Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med 60:307–317
Soetikno V, Sari FR, Sukumaran V, Lakshmanan AP, Mito S, Harima M, Watanabe K (2012) Curcumin prevents diabetic cardiomyopathy in streptozotocin-induced diabetic rats: possible involvement of PKC-MAPK signaling pathway. Eur J Pharm Sci 47(3):604–614
Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D, Liu C (2012) Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS One 7(12):e52013
Adluri RS, Thirunavukkarasu M, Zhan L, Akita Y, Samuel SM, Otani H, Maulik N (2011) Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice. J Mol Cell Cardiol 20(1):239–247
Taye A, Abouzied MM, Mohafez OM (2013) Tempol ameliorates cardiac fibrosis in streptozotocin-induced diabetic rats: role of oxidative stress in diabetic cardiomyopathy. Naunyn Schmiedebergs Arch Pharmacol 386(12):1071–1080
Liu Q, Wang S, Cai L (2014) Diabetic cardiomyopathy and its mechanisms: role of oxidative stress and damage. J Diabetes Investig 5(6):623–634
Zhang H, Morgan B, Potter BJ, Ma L, Dellsperger KC, Ungvari Z, Zhang C (2010) Resveratrol improves left ventricular diastolic relaxation in type 2 diabetes by inhibiting oxidative/nitrative stress: in vivo demonstration with magnetic resonance imaging. Am J Physiol Heart Circ Physiol 299(4):H985–H994
Hegazy SK, Tolba OA, Mostafa TM, Eid MA, El-Afify DR (2013) Alpha-lipoic acid improves subclinical left ventricular dysfunction in asymptomatic patients with type 1 diabetes. Rev Diabet Stud 10(1):58–67
Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Liepinsh E (2016) Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp Clin Endocrinol Diabetes 124(4):251–256
Tu SJ, Gallagher C, Elliott AD, Linz D, Pitman BM, Hendriks JM, Wong CX (2022) Alcohol consumption and risk of ventricular arrhythmias and sudden cardiac death: an observational study of 408,712 individuals. Heart Rhythm 19(2):177–184
Colberg SR, Sigal RJ, Fernhall B, Regensteiner JG, Blissmer BJ, Rubin RR, Braun B (2010) Exercise and type 2 diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care 33(12):e147–e167
Curran M, Drayson MT, Andrews RC, Zoppi C, Barlow JP, Solomon TPJ, Narendran P (2020) The benefits of physical exercise on the health of the pancreatic β‐cell: a review of the evidence. Exp Physiology 105:579–589. https://doi.org/10.1113/EP088220
Aune D, Schlesinger S, Norat T, Riboli E (2018) Tobacco smoking and the risk of sudden cardiac death: a systematic review and meta-analysis of prospective studies. Eur J Epidemiol 33(6):509–521
Author information
Authors and Affiliations
Contributions
The idea and design of the paper came from Ram B. Singh, Manal Smail and Jaipaul Singh. Sunil Rupee, Khemraj Rupee, Carlin Hanoman and Abla Mohammed Ahmed Ismail did the literature search, drew the diagrams and did the first version of the paper. Ram B. Singh, Manal Smail and Jaipaul Singh finally reviewed the paper for corrections and made additions. Sunil Rupee made the final version. Subsequent reviews and changes were done by Sunil Rupee, Jaipaul Singh and Ram B. Singh. Each author contributed more or less the same except for Sunil Rupee, who did more, and for this reason, he is the first/main author.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rupee, S., Rupee, K., Singh, R.B. et al. Diabetes-induced chronic heart failure is due to defects in calcium transporting and regulatory contractile proteins: cellular and molecular evidence. Heart Fail Rev 28, 627–644 (2023). https://doi.org/10.1007/s10741-022-10271-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-022-10271-5