
Abstract
Almost half of all heart failure (HF) disease burden is due to HF with preserved ejection fraction (HFpEF). The primary symptom in patients with HFpEF, even when well compensated, is severe exercise intolerance and is associated with their reduced quality of life. Recently, studies showed that HFpEF patients have multiple skeletal muscle (SM) abnormalities, and these are associated with decreased exercise intolerance. The SM abnormalities are likely intrinsic to the HFpEF syndrome, not a secondary consequence of an epiphenomenon. These abnormalities are decreased muscle mass, reduced type I (oxidative) muscle fibers, and reduced type I-to-type II fiber ratio as well as a reduced capillary-to-fiber ratio, abnormal fat infiltration into the thigh SM, increased levels of atrophy genes and proteins, reduction in mitochondrial content, and rapid depletion of high-energy phosphate during exercise with markedly delayed repletion of high-energy phosphate during recovery in mitochondria. In addition, patients with HFpEF have impaired nitric oxide bioavailability, particularly in the microvasculature. These SM abnormalities may be responsible for impaired diffusive oxygen transport and/or impaired SM oxygen extraction. To date, exercise training (ET) and caloric restriction are some of the interventions shown to improve outcomes in HFpEF patients. Improvements in exercise tolerance following aerobic ET are largely mediated through peripheral SM adaptations with minimal change in central hemodynamics and highlight the importance of targeting SM to improve exercise intolerance in HFpEF. Focusing on the abnormalities mentioned above may improve the clinical condition of patients with HFpEF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a known cause of significant mortality and morbidity worldwide in middle-aged and older adults. In the USA, the lifetime risk of HF is estimated to be 1 in 5 at age 40 [1], and is projected to increase by 46% by 2030 [2]. Almost half of all HF disease burden is due to HF with preserved ejection fraction (HFpEF) [2]. In the highest age decile (≥ 90 years old), nearly all patients with HF have preserved EF. HFpEF is a clinical syndrome associated with poor health-related quality of life (HRQOL), substantial healthcare resource utilization, and mortality, in large part related to high rates of hospitalizations in patients with HF [3]. After HF hospitalization, the 5-year survival of HFpEF is a dismal 35%, worse than many cancers [4], although HFpEF was initially considered a hemodynamic disorder characterized by hypertension, cardiac hypertrophy, and diastolic dysfunction, which is now recognized as a systemic syndrome involving the heart, lungs, kidneys, skeletal muscle (SM), adipose tissue, and vascular system [5].
The primary symptom in patients with HFpEF is reduced exercise tolerance (peak exercise oxygen uptake, VO2peak) and is associated with their reduced HRQOL [6, 7]. In addition, declines in VO2peak in older HF patients are compounded by comorbidities, aging, sarcopenia, and myosteatosis (increased muscle fat infiltration), malnutrition, and physical inactivity [6,7,8]. VO2peak is defined as the highest achievable rate at which oxygen can be transported from air to tissues and utilized by the mitochondria during maximal exercise. In accordance with the Fick principle [VO2 = Cardiac output (CO) × Arterial-venous oxygen difference (a-vO2Diff)], the decreased VO2peak in HFpEF may be due to abnormalities in convective and diffusive O2 transport and/or impaired SM extraction and utilization [9].
It has traditionally been assumed that reduced exercise CO was the primary factor limiting exercise intolerance in HFpEF. Later, other investigators found that the blunted CO was secondary to chronotropic incompetence (CI) [7, 10, 11]. Indeed, approximately 30 to 50% of patients with HFpEF are thought to have CI manifested by a lower than predicted maximal HR during symptom-limited exercise [7, 10, 11]. Although VO2peak has been observed to correlate with both changes in CO and a-vO2Diff, recent studies showed that reduced O2 extraction accounts for at least 50% of the reduction in VO2peak and is a stronger independent predictor of VO2peak than exercise CO (Fig. 1) [6, 12, 13]. Moreover, Haykowsky et al. has shown that the improvement in peak a-vO2Diff accounted for the nearly all of the increase VO2peak following exercise training (ET) [14]. The mechanisms responsible for this impaired ability to augment a-vO2Diff during peak exercise might relate to impaired diffusive oxygen transport due to peripheral/microvascular dysfunction and/or SM abnormalities that result in impaired oxygen extraction and utilization (Fig. 2) [6, 9, 12,13,14,15,16,17,18,19,20,21].

Comparison at seated rest, 12 W, 25 W, and peak exercise between HFpEF patients and HCs. A Oxygen consumption, B arteriovenous oxygen content difference, C heart rate, D cardiac output, E systemic vascular resistance (SVR), and F systolic blood pressure. All variables adjusted for sex (*p < 0.05). The p-value at the upper left of each panel represents the group-by-intensity interaction. Dashed lines represent healthy controls (HC), and solid lines represent patients with heart failure with preserved ejection fraction (HFpEF) (reproduced from JACC with permission. J Am Coll Cardiol. 2011; 58:265–74)

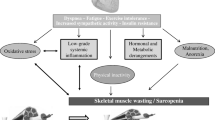
Potential causes for the skeletal muscle abnormalities in HFpEF. HFpEF, heart failure with preserved ejection fraction; RAAS, renin angiotensin aldosterone system; NO, nitric oxide; GH, growth hormone; ROS, reactive oxygen species; cGMP, cyclic guanosine monophosphate; PKG, protein kinase; SM, skeletal muscle; VO2, oxygen consumption; ATP, adenosine triphosphate; Pcr, phosphocreatine
This review combines current clinical knowledge and fundamental biological mechanisms to address the essential and emerging issue of SM abnormalities in HFpEF. We will briefly discuss the role of ET and novel treatment strategies to improve SM morphology and function.
Impaired SM blood flow in HFpEF
SM is one of the largest organs in the human body, accounting for approximately 40% of total body mass, and acts as a major site for protein storage and glucose disposal. Muscle-specific blood flow can increase almost 100-fold from rest to maximal exercise [22, 23]. The overall regulation of SM blood flow is achieved through sympathetic-mediated redistribution of blood from non-exercising regions to the working muscles coupled with metabolic-mediated vasodilation “autoregulation” in the exercising muscles [24, 25]. Within SM, blood flow regulation and oxygen delivery result from integrating several stimuli, including the mechanical effects of contraction, local metabolic and endothelium-derived substances, vasoactive factors associated with erythrocytes, and the sympathetic nervous system.
Prior animal studies showed that obese-HFpEF rats demonstrated an abnormal leg blood flow response to contractions with fiber type-specific structural capillary loss [26, 27]. Supporting this finding, clinical studies found that non-invasive (ultrasound) measurements of leg blood flow and vascular conductance are markedly decreased in patients with HFpEF during exercise [28,29,30,31]. This suggests that impaired autoregulation in the exercising SM vasculature may play a key role in exercise intolerance. However, abnormal blood flow response to exercise is inconsistent in HFpEF studies [15, 21, 32, 33]. This may be due to differences in the muscles studied or different measurement techniques or heterogeneity.
Additionally, Houstis et al. found that the deficit in a-vO2Diff was related to a 36% reduction in the patient’s SM diffusion capacity, and improved diffusion capacity resulted in increased VO2peak [15]. Regional vasodilation in SM is mediated in part by nitric oxide (NO) and prostaglandin-induced vasodilation. Patients with HFpEF have impaired NO availability, particularly in the microvasculature, which might contribute to SM diffusion capacity (Fig. 3) [7, 34, 35]. Borlaug et al. found that systemic vascular conductance and microvascular reserve were positively related to VO2peak in HFpEF [7]. Similarly, microvascular endothelial dysfunction was an independent predictor of poorer prognosis, mainly readmission, in patients with HFpEF [36]. A consequence of the blunted microvascular reserve is that it may be associated with decreased diffusive oxygen transport to the exercising muscle, which would reduce exercise tolerance. Indeed, peripheral endothelial dysfunction might impair matching of perfusion to regional demand in SM microcirculation [37].

Role of nitric oxide in skeletal muscle autoregulation. eNOS, nitric oxide synthase; NO, nitric oxide; sGC, soluble guanylate cyclase; GTP, guanosine-5′-triphosphate; cGMP, cyclic guanosine monophosphate; HFpEF, heart failure with preserved ejection fraction; O2-, superoxide; ONOO-, peroxynitrite
In addition, Kitzman’s group reported that compared with healthy controls (HCs), older HFpEF patients had a reduced capillary-to-fiber ratio (1.35 ± .32 versus 2.53 ± 1.37, p = .006) (Fig. 4) [18] and were a significant independent predictor of VO2peak (partial r = .34, p = .02). Capillary rarefaction disrupts the microvascular oxygenation dynamics in SM, and one of the mechanisms that could contribute to such rarefaction is impaired NO-mediated vasodilation [38, 39]. Of note, the reduced microvascular density in SM in older HFpEF patients matches a similar finding in cardiac muscle as reported by Mohammed et al. [40]. If a systemic process is responsible, then adverse effects on striated muscle in both cardiac and SM compartments would be expected [41]. In addition, HFpEF patients are commonly obese. The obligate perfusion to excess adipose tissues might diminish proper flow matching to metabolism, contributing to a lower peak a-vO2Diff [42, 43].

Relationship of capillary-to-fiber ratio (A) and percentage of type I muscle fibers (B) with peak O2 uptake (VO2) in older patients with heart failure with preserved ejection fraction (■) and age-matched healthy control subjects (▲) (reproduced from Heart Failure Clinic with permission. Heart Fail Clin. 2017; 13: 485–502)
Abnormalities in SM mass and composition
Most of the oxygen consumed during exercise occurs in the active muscles; therefore, a loss in metabolically active tissue (sarcopenia) may contribute to exercise intolerance in HFpEF patients. Animal models of HFpEF (hypertensive or cardiometabolic) have shown decreased muscle mass [26, 27]. A recent cardiometabolic obese-HFpEF rat model induced multiple SM alterations in the rat hindlimb, including impaired muscle mechanics related to shortening velocity, fiber atrophy, and the capillary loss that implies a perfusive oxygen delivery limitation [26]. Haykowsky et al. measured lean body mass and VO2peak in older HFpEF patients and age-matched HCs using dual-energy X-ray absorptiometry and maximal exercise testing [17]. Older HFpEF patients had significantly reduced total and lean leg mass and decreased VO2peak indexed to lean body mass versus HCs. Also, the change in VO2peak with increasing percent leg lean mass was blunted in HFpEF compared to HCs (the slope of the relationship of peak VO2 with percent leg lean mass, HFpEF (11 ± 5 ml/min) versus HCs (36 ± 5 ml/min; p < .001)), suggesting that SM hypoperfusion or impaired O2 utilization by the active muscles may play an important role in limiting exercise performance in older HFpEF patients. Haykowsky et al., using phase-contrast magnetic resonance imaging, extended these results by directly characterizing thigh muscle composition and found that older patients with HFpEF had increased thigh intramuscular fat (IMF), whether expressed as absolute area or as a proportion of the thigh compartment (TC) despite the similar amount of subcutaneous fat. Furthermore, the ratio of IMF/SM was increased, and both IMF area (partial r = −.51, p = .002) and IMF/SM ratio (partial r = −.45, p = .006) were significant independent predictors of peak exercise VO2 (HFpEF versus HC group, IMF area (35.6 ± 11.5 versus 22.3 ± 7.6 cm2, p = .01), percent IMF/TC (26 ± 5 versus 20 ± 5%, p = .005), and the ratio of IMF/SM (.38 ± .10 versus 0.28 ± .09, p = .007)) (Fig. 5) [20]. Increased myosteatosis is inversely related to the mitochondrial density and suppresses mitochondrial biogenesis [44]. Weiss et al. also found markedly increased intermuscular adipose tissue in HFpEF compared to HF with reduced ejection fraction (HFrEF) patients (SM fat fraction was increased almost threefold in HFpEF patients as compared to HCs, in contrast, nonsignificantly increased in HFrEF patients) [45].

reproduced from Heart Failure Clinic with permission. Heart Fail Clin. 2017; 13: 485–502)
Magnetic resonance imaging axial image of the mid-thigh in a patient with heart failure with preserved ejection fraction (HFpEF) and healthy controls (HC) (
Myosteatosis may reduce VO2peak in patients with HFpEF through several mechanisms described previously. Heinonen et al. using positron emission tomography found that adipose tissue blood flow adjacent to the active muscles increased sevenfold during continuous isometric knee-extension exercise in nonobese younger healthy sedentary women [46]. Interestingly, Zamani et al. recently found that body composition (measured by whole-body DEXA), particularly the degree of adiposity, was correlated with a-vO2Diff, with increasing fat associated with decreased a-vO2Diff (correlation coefficient − .61. p < .001) [21]. Thus, increased thigh IMF in older patients with HFpEF may “steal” the blood usually delivered to the active muscles during exercise, thereby reducing oxygen delivery to active muscles. Adipose within the SM is also metabolically active and can impair oxidative metabolism and mitochondrial function. Inflammatory cytokines produced by adipocytes also have direct catabolic effects on SM [41, 44].
SM is divided into two broad types based on fiber types—type I (slow-twitch “oxidative”) and type II (fast-twitch “glycolytic”) muscle fibers. At the microscopic level, SM biopsies (vastus lateralis muscle) from HFpEF patients showed a reduced percentage of type I fibers and reduced type I-to-type II fiber ratio as well as a reduced capillary-to-fiber ratio (in HFPEF versus HC patients, the percentage of type I fibers (39.0 ± 11.4% versus 53.7 ± 12.4%, p < .001), type I-to-type II fiber ratio (.72 ± .39 versus 1.36 ± .85, p = .001)) (Fig. 4) [18]. The lower type 1 fibers correlate to decreased VO2peak (partial r = .40, p = .004). Similarly, recently, Zamani et al. identified a marked difference in myofibre type present in HFpEF subjects, with a much lower percentage of type I fibers than either HCs or hypertensive subjects of similar age (70% in HCs versus 50% in HFpEF (p < 0.01) [47]. Compared with type II fibers, type I fibers have the greater oxidative capacity and mitochondrial density and contribute disproportionately to the ability to perform sustained aerobic exercise. While speculative, a reduction in the percentage of type I fibers could be associated with reduced oxidative capacity and mitochondrial density and contribute to prolonged oxygen uptake kinetics and reduced VO2peak in HFpEF [48].
Recently, a study demonstrated increased levels of SM atrophy genes and proteins (transforming growth factor-β1, cathepsin L, myostatin-2, F-box only protein-32) in stable outpatients with HFpEF compared with HF HFrEF and HCs [49]. They also showed reduced gene expression of Akt-2, which is a rate limiting and a crucial step in protein synthesis. Cathepsin L plays a significant role in autophagy, a further important mechanism of muscle atrophy. Myostatin is a highly conserved member of the transforming growth factor-beta superfamily that signals through the activin receptor type IIB. The activation of the myostatin pathway was shown to negatively regulate muscle size primarily by inhibiting the Akt pathway leading to reduced protein synthesis and increased protein degradation [50].
Abnormalities in SM oxidative function in HFpEF
There are multiple SM abnormalities in HFpEF that impair oxygen utilization and appear to contribute to reduced VO2peak (Fig. 2). Among these, growing evidence indicates that impaired mitochondrial function may be among the most consequential. When a muscle repeatedly contracts for long periods, the ATP supply needs to be constantly replenished through mitochondrial oxidative phosphorylation. If the intricate metabolic pathways within the mitochondria were to become altered and less efficient, endurance within the SM would decrease. As the sole mechanism for utilizing oxygen and fuel substrate to produce energy, mitochondrial health is obviously a critical determinant of VO2peak. The rate of breakdown and resynthesis of high-energy phosphates during and following exercise are fundamental determinants of whole-body VO2 during exercise and recovery.
In an animal model of HFpEF, Bowen et al. found multiple abnormalities, including reduced in situ mitochondrial respiratory reserve capacity, a key measure of SM oxidative phosphorylation that correlates well with VO2peak in humans [27]. Among the patients with mitochondrial myopathies, VO2peak is decreased despite normal cardiac function. These patients suffer from impaired oxidative metabolism in SM. SM relies more on substrate-level phosphorylation for energy production during exercise, leading to exaggerated circulatory and ventilatory responses (decreased VO2 and increased CO response to exercise) [51]. Using [30] Phosphate magnetic resonance spectroscopy, Bhella and colleagues found the abnormal hemodynamic response to exercise, similar to that observed in patients with mitochondrial myopathies; however, only two patients were studied [13]. They suggested that HFpEF patients might display a hyperdynamic cardiac response to exercise with CO higher than expected for a given VO2. This impairment may limit functional capacity by two mechanisms: (1) early SM fatigue and (2) metabolic signals to increase the CO response to exercise, which a left ventricle may poorly tolerate with elevated filling pressure [13]. Due to the relatively smaller sample size, these results cannot be generalized to broader patient populations with HFpEF. These preliminary results were confirmed by Weiss et al., who performed serial magnetic resonance spectroscopy measurements of creatinine phosphate during calf extensor exercise to exhaustion and recovery in HFpEF patients compared with HFrEF patients and HCs. HFpEF patients had severe exercise intolerance associated with rapid high-energy phosphate depletion, which was observed early during exercise [45]. Furthermore, HFpEF patients had markedly delayed repletion of high-energy phosphate during recovery [45]. There was a strong correlation between the average rate of phosphocreatine decline during exercise and the maximum exercise time (R2 = .83, p < .001).
Analyzing muscle biopsies from 20 HFpEF patients and 17 age-matched HCs, Molina et al. measured the expression of mitofusins 1 and 2 (Mfn1 and Mfn2), proteins localized to the mitochondrial outer membrane that plays an essential role in the fusion of these organelles (Mfn2 plays an important role in mitochondrial quality control by mediating complementation of organelles and the elimination of dysfunctional mitochondria by autophagy and citrate synthase is the key enzyme regulating oxidative metabolism). Protein expression of porin, Mfn2 (normalized to porin), and citrate synthase was significantly lower (p = .01, p = < .001, and p = .01 respectively) in SM tissue of patients with HFpEF compared to HCs (Fig. 6) [19]. In a recent study, Zamani et al. showed (mass spectrometry in muscle biopsy samples from 13 HFpEF participants) broad reductions in the proteins and complexes involved in energy fuel metabolism, including tricarboxylic acid cycle enzymes and the mitochondrial complexes that make up the electron transport chain in patients with HFpEF SM that correlated with exercise capacity, independent of peak oxygen delivery [47]. Bekfani et al. identified smaller mitochondria and reduced mitochondrial volume density in HFpEF SM compared with similarly aged controls [49]. They also described reductions in gene expression of key proteins involved in fatty acid oxidation and carbohydrate metabolism, alongside increased gene expression of proteins associated with muscle atrophy [49].

Representative western blot bands from 3 patients with HFpEF and 3 healthy controls (HCs). For each protein, images were obtained from the same blot and exposure. A potential difference in skeletal muscle mitochondrial content was determined by analysis of porin expression. The samples were electrophoretically transferred to nylon polyvinyl difluoride (PVDF) membrane and the blots were incubated with commercially available primary antibodies to Mfn1 (1:1000), Mfn2 (1:1000), porin (1:1000), and GAPDH (1:2000) (Abcam, Cambridge, MA). Densitometry values for Mfn1 and Mfn2 were normalized to porin in order to account for differences in mitochondrial content. Measurement of porin was normalized to GAPDH. Normalization of mitofusins to porin, rather than GAPDH, was appropriate because these proteins reside on the mitochondrial outer membrane (reproduced from JACC with permission. JACC Heart Fail. 2016; 4:636–645)
These observations suggest that HFpEF patients relied less on oxidative pathways during exercise, as evidenced by decreased oxidative phosphorylation ATP production rates, and more on anaerobic metabolism, as evidenced by increased anaerobic glycolysis ATP production rates [13, 15, 17,18,19, 45, 47, 49]. In addition, a previous study showed that AMP-activated protein kinase/glucose transporter-4 signaling is suppressed in SM in obese/hypertensive HFpEF rats and patients with metabolic syndrome (but not HFpEF), but might suggest that SM glucose metabolism is diminished in HFpEF [52]. It has also been described that increased circulating lipid metabolites, particularly the long-chain acylcarnitine metabolites derived from β-oxidation of free fatty acids (FFA) in HFpEF patients, may indicate that FFA metabolism is predominant in HFpEF [53]. Interestingly, and in contrast to HFrEF or HCs, patients with HFpEF cannot lower venous PO2 during exercise and therefore demonstrate a blunted peripheral O2 extraction response [13, 15, 171819, 45, 47, 49]. The extent of this impaired muscle O2 extraction in HFpEF is likely explained, at least in part, by the significant mitochondrial abnormalities reported in patients with HFpEF [13, 15, 171819, 45, 47, 49]. Clearly, more studies are warranted to clarify the role of limitations to muscle O2 diffusion in HFpEF. Potential causes for the SM abnormalities in HFpEF are shown in Fig. 2.
In addition to locomotory muscle, studies also linked respiratory muscle dysfunction to exercise intolerance in HFpEF, as shown by direct diaphragm contractility measures in experimental models. Multiple alterations to the diaphragm have been reported in the HFpEF rat model, including in vitro muscle weakness and fatigue alongside a type II-to-I fiber-type shift, fiber atrophy, and impaired in situ mitochondrial respiration [27]. These diaphragm alterations and dysfunction are not reversed following 8 weeks of aerobic ET [54]. Inspiratory (i.e., diaphragm) muscle weakness is evident and closely associated with symptoms of dyspnea and poor prognosis in patients with HFpEF [55,56,57,58].
Interventions to improve exercise intolerance and SM function in HFpEF
Exercise interventions
To date, ET, in addition to caloric restriction (CR), is one of the interventions shown to improve outcomes in HFpEF patients [59,60,61]. Several randomized controlled trials have examined the efficacy of ET to improve VO2peak, 6-min walk distance, and HRQOL in patients with HFpEF [14, 61,62,63,64,65,66,67,68,69,70,71]. It appears that structured and supervised moderate continuous training, high-intensity interval training, and resistance training can benefit the patients with HFpEF [59]. Currently, no studies have examined the role of ET on SM morphology or function in HFpEF. However, studies suggested that peripheral mechanisms, such as improved SM perfusion and metabolism, likely play a major role in adapting ET in HFpEF [59, 60]. Specifically, Kitzman group demonstrated that 84% of the improvement in VO2peak following 16 weeks of aerobic ET was attributed to increases in peak exercise a-vO2Diff [14]. This is further supported by Fu et al. who reported that 12 weeks of high-intensity interval ET significantly increased VO2 with the improvements in VO2peak driven by increases in estimated peak exercise a-vO2Diff and leg muscle oxygenation, with little or no change in peak exercise CO [67]. In fact, Bhella et al. reported that ET could favorably shift to more efficient muscle O2 utilization in older HFpEF patients [13]. In addition, studies showed that ET in patients with HFpEF is associated with an improvement in VO2 and HRQOL without significant changes in LV systolic or diastolic function [72, 73]. Even small muscle mass exercises, single-leg knee extensor exercises where the limiting role of the heart is minimized have been shown to induce various peripheral structural and functional adaptations improving VO2 without changing CO in HF patients [74]. The possible mechanisms responsible for these exercise-mediated peripheral adaptations that underlie improvements in peak exercise a-vO2Diff may be related to improved peripheral muscle perfusion and enhanced mitochondrial function. SM oxidative capacity and efficiency conceivably improved by ET in HFpEF patients since ET increased capillary and mitochondrial density, changed muscle fiber subtypes distribution, leg oxygen delivery, and diffusive conductance, and increased red blood cell capillary transit time through the SM vasculature in an animal model of HFpEF [26, 27, 54]. Bowen et al. showed that in Dahl salt-sensitive HFpEF rats, ET could prevent SM contractile dysfunction in the diaphragm and soleus, associated with preserved mitochondrial function [27]. Theoretically, improvement in SM mitochondrial function may significantly contribute to training-related improvements in VO2peak in human HFpEF, which is known to be the case for HFrEF [60]. This can make a strong case for targeting SM, particularly mitochondrial function, to improve exercise intolerance in HFpEF.
Nutritional interventions
In contrast to nutritional supplements, CR has been demonstrated to trigger vital subcellular benefits in older adults through molecular signaling pathways (e.g., mTor and AMP kinase) that are suppressed or stimulated, with downstream clinical benefits [75]. In older, obese individuals without HF, CR has improved SM function. Kitzman et al. showed that CR improved muscle leg muscle quality and reduced abdominal and thigh subcutaneous fat in older HFpEF patients. In addition, the change in VO2peak was positively correlated with both the change in percent lean mass (r = 0.32; p = 0.003) and the change in thigh SM to IMF ratio (r = .27; p = .02) [61].
Novel pharmacological interventions
Pharmacological approaches to SM growth remain an active area of research. The randomized clinical trial INDIE-HFpEF (Inorganic Nitrite Delivery to Improve Exercise Capacity in Heart Failure With Preserved Ejection Fraction) showed that inhaled inorganic nitrite (NO donor) did not improve VO2peak and exercise capacity in HFpEF patients. However, inadequate drug delivery from the nebulizer was raised as an issue [76]. Recently, the same group showed the beneficial effects of inhaled and intravenous sodium nitrite on SM O2 conductance, VO2 kinetics, O2 utilization during submaximal exercise, and alveolar-capillary membrane O2 conductance HFpEF patients [77]. Several promising biologic and small molecule interventions are currently developing to rejuvenate SM, including myostatin inhibitors, selective androgen receptor modulators, and an activator of the fast SM troponin complex [78]. Nonetheless, trials of myostatin inhibitors have revealed many side effects that heretofore have diminished enthusiasm for clinical application.
There are now multiple agents in phase 2 clinical trials, primarily of older patients with physical disability associated with sarcopenia, targeting a variety of SM abnormalities, including mitochondrial dysfunction. Neladenoson bialanate is a partial adenosine A1 receptor agonist that has been shown in preclinical models to improve SM mitochondrial function, enhance sarco/endoplasmic reticulum 2a activity, and optimize energy substrate utilization [77]. Among the HFpEF patients, the Partial AdeNosine A1 Receptor Agonist in Patients With Chronic Heart Failure and Preserved Ejection Fraction (PANACHE) study showed no significant dose–response relationship detected for neladenoson with regard to the change in exercise capacity from baseline to 20 weeks [79].
Key knowledge gaps
-
1.
How much do aging and non-cardiac comorbidities contribute to the SM abnormalities in HFpEF?
-
2.
Are SM alterations a consequence of the disease process?
-
3.
In future studies, do we need to study the SM morphology and function of much older, sedentary, and older active non-HF controls?
-
4.
Are there overarching, systemic processes in HFpEF that trigger SM impairments?
-
5.
Do pre-existing SM characteristics determine responses to HF?
-
6.
Does exercise training ameliorate SM alterations in HFpEF, and what are the improvement mechanisms?
Conclusions
HFpEF is associated with multiple SM abnormalities, including (1) decreased muscle mass, reduced type I fibers, and type I-to-type II fiber ratio; (2) reduced capillary-to-fiber ratio; (3) myosteatosis; (4) reduction in mitochondrial content; (5) rapid depletion of high-energy phosphate during exercise with markedly delayed repletion of high-energy phosphate during recovery in mitochondria; (6) reductions in gene expression of key proteins involved in fatty acid oxidation and carbohydrate metabolism; and (7) increased gene expression of proteins associated with muscle atrophy. These abnormalities may be responsible for impaired diffusive oxygen transport and utilization by the active muscles and contribute significantly to exercise intolerance. To date, ET, in addition to CR, is one of the interventions shown to improve outcomes in HFpEF patients. In patients with HFpEF, improvements in VO2peak following aerobic ET are largely mediated through peripheral “non-cardiac” factors with minimal change in CO. Specifically, SM oxidative capacity and efficiency can be improved by ET, which increases capillary and mitochondrial density, changes muscle fiber subtypes distribution, and increases red blood cell capillary transit time through the SM vasculature. Accordingly, SM may be an important target of therapy to improve HFpEF patients’ aerobic endurance and VO2peak.
References
Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D’Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D (2002) Framingham Heart Study. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation 106:3068–3072
Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Piña IL, Trogdon JG (2013) American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 6:606–619
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128:e240–e327
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355:251–259
Upadhya B, Kitzman DW (2020) Heart failure with preserved ejection fraction: new approaches to diagnosis and management. Clin Cardiol 43:145–155
Haykowsky MJ, Brubaker PH, John JM, Stewart KP, Morgan TM, Kitzman DW (2011) Determinants of exercise intolerance in elderly heart failure patients with preserved ejection fraction. J Am Coll Cardiol 58:265–274
Borlaug BA, Olson TP, Lam CSP, Flood KS, Lerman A, Johnson BD et al (2010) Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol 56:845–854
Forman DE, Arena R, Boxer R, Dolansky MA, Eng JJ, Fleg JL, Haykowsky M, Jahangir A, Kaminsky LA, Kitzman DW, Lewis EF, Myers J, Reeves GR, Shen WK (2017) American Heart Association Council on Clinical Cardiology; Council on Cardiovascular and Stroke Nursing; Council on Quality of Care and Outcomes Research; and Stroke Council. Prioritizing functional capacity as a principal end point for therapies oriented to older adults with cardiovascular disease: a scientific statement for healthcare professionals from the American Heart Association 135:e894–e918
Poole DC, Richardson RS, Haykowsky MJ, Hirai DM, Musch TI (1985) Exercise limitations in heart failure with reduced and preserved ejection fraction. J Appl Physiol 2018(124):208–224
Borlaug BA, Melenovsky V, Russell SD et al (2006) Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation 114:2138–2147
Brubaker PH, Kitzman DW (2011) Chronotropic incompetence: causes, consequences, and management. Circulation 123:1010–1020
Dhakal BP, Malhotra R, Murphy RM, Pappagianopoulos PP, Baggish AL, Weiner RB, Houstis NE, Eisman AS, Hough SS, Lewis GD (2015) Mechanisms of exercise intolerance in heart failure with preserved ejection fraction: the role of abnormal peripheral oxygen extraction Circ Heart Fail 8:286–294
Bhella PS, Prasad A, Heinicke K, Hastings JL, Arbab-Zadeh A, Adams-Huet B, Pacini EL, Shibata S, Palmer MD, Newcomer BR, Levine BD (2011) Abnormal haemodynamic response to exercise in heart failure with preserved ejection fraction. Eur J Heart Fail 13:1296–1304
Haykowsky MJ, Brubaker PH, Stewart KP, Morgan TM, Eggebeen J, Kitzman DW (2012) Effect of endurance training on the determinants of peak exercise oxygen consumption in elderly patients with stable compensated heart failure and preserved ejection fraction. J Am Coll Cardiol 60:120–128
Houstis NE, Eisman AS, Pappagianopoulos PP, Wooster L, Bailey CS, Wagner PD, Lewis GD (2018) Exercise intolerance in heart failure with preserved ejection fraction: diagnosing and ranking its causes using personalized O2 pathway analysis. Circulation 137:148–161
Kinugawa S, Takada S, Matsushima S, Okita K, Tsutsui H (2015) Skeletal muscle abnormalities in heart failure. Int Heart J 56:475–484
Haykowsky MJ, Brubaker PH, Morgan TM, Kritchevsky S, Eggebeen J, Kitzman DW (2013) Impaired aerobic capacity and physical functional performance in older heart failure patients with preserved ejection fraction: role of lean body mass. J Gerontol A Biol Sci Med Sci 68:968–975
Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM, Haykowsky MJ (2014) Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol 306:H1364–H1370
Molina AJ, Bharadwaj MS, Van Horn C, Nicklas BJ, Lyles MF, Eggebeen J, Haykowsky MJ, Brubaker PH, Kitzman DW (2016) Skeletal muscle mitochondrial content, oxidative capacity and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail 4:636–645
Haykowsky M, Kouba EJ, Brubaker PH, Nicklas BJ, Eggebeen J, Kitzman DW (2014) Skeletal muscle composition and its relation to exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Cardiol 113:1211–1216
Zamani P, Proto EA, Mazurek JA, Prenner SB, Margulies KB, Townsend RR, Kelly DP, Arany Z, Poole DC, Wagner PD, Chirinos JA (2020) Peripheral determinants of oxygen utilization in heart failure with preserved ejection fraction: central role of adiposity. JACC Basic Transl Sci 5:211–225
Saltin B (2007) Exercise hyperaemia: magnitude and aspects on regulation in humans. J Physiol 583:819–823
Kwee BJ, Mooney DJ (2017) Biomaterials for skeletal muscle tissue engineering. Curr Opin Biotechnol 47:16–22
Beere PA, Russell SD, Morey MC, Kitzman DW, Higginbotham MB (1999) Aerobic exercise training can reverse age-related peripheral circulatory changes in healthy older men. Circulation 100:1085–1094
Katz SD, Zheng H (2002) Peripheral limitations of maximal aerobic capacity in patients with chronic heart failure. J Nucl Cardiol 9:215–225
Espino-Gonzalez E, Tickle PG, Benson AP, Kissane RWP, Askew GN, Egginton S, Bowen TS (2021) Abnormal skeletal muscle blood flow, contractile mechanics and fibre morphology in a rat model of obese-HFpEF. J Physiol 599:981–1001
Bowen TS, Rolim NP, Fischer T, Baekkerud FH, Medeiros A, Werner S, Bronstad E, Rognmo O, Mangner N, Linke A, Schuler G, Silva GJ, Wislolf U, Adams V (2015) on behalf of the Optimex Study Group Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail 17:263–272
Lee JF, Barrett-O’Keefe Z, Garten RS, Nelson AD, Ryan JJ, Nativi JN, Richardson RS, Wray DW (2016) Evidence of microvascular dysfunction in heart failure with preserved ejection fraction. Heart 102:278–284
Weavil JC, Thurston TS, Hureau TJ, Gifford JR, Kithas PA, Broxterman RM, Bledsoe AD, Nativi JN, Richardson RS, Amann M (2020) Heart failure with preserved ejection fraction diminishes peripheral hemodynamics and accelerates exercise-induced neuromuscular fatigue. Am J Physiol Heart Circ Physiol 320:H338–H351
Maréchaux S, Samson R, van Belle E, Breyne J, de Monte J, Dédrie C, Chebai N, Menet A, Banfi C, Bouabdallaoui N, Le Jemtel TH, Ennezat PV (2016) Vascular and microvascular endothelial function in heart failure with preserved ejection fraction. J Card Fail 22:3–11
Kishimoto S, Kajikawa M, Maruhashi T, Iwamoto Y, Matsumoto T, Iwamoto A, Oda N, Matsui S, Hidaka T, Kihara Y, Chayama K, Goto C, Aibara Y, Nakashima A, Noma K, Higashi Y (2017) Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. Int J Cardiol 231:181–187
Hundley WG, Bayram E, Hamilton CA, Hamilton EA, Morgan TM, Darty SN, Stewart KP, Link KM, Herrington DM, Kitzman DW (2007) Leg flow-mediated arterial dilation in elderly patients with heart failure and normal left ventricular ejection fraction. Am J Physiol Heart Circ Physiol 292:H1427–H1434
Haykowsky MJ, Herrington DM, Brubaker PH, Morgan TM, Hundley WG, Kitzman DW (2013) Relationship of flow-mediated arterial dilation and exercise capacity in older patients with heart failure and preserved ejection fraction. J Gerontol A Biol Sci Med Sci 68:161–167
Hellsten Y, Nyberg M, Jensen LG, Mortensen SP (2012) Vasodilator interactions in skeletal muscle blood flow regulation. J Physiol 590:6297–6305
Dyakova EY, Kapilevich LV, Shylko VG, Popov SV, Anfinogenova Y (2015) Physical exercise associated with NO production: signaling pathways and significance in health and disease. Front Cell Dev Biol 3:19
Matsue Y, Suzuki M, Nagahori W, Ohno M, Matsumura A, Hashimoto Y, Yoshida K, Yoshida M (2013) Endothelial dysfunction measured by peripheral arterial tonometry predicts prognosis in patients with heart failure with preserved ejection fraction. Int J Cardiol 168:36–40
Poole DC, Hirai DM, Copp SW, Musch TI (2012) Muscle oxygen transport and utilization in heart failure: implications for exercise (in) tolerance. Am J Physiol Heart Circ Physiol 302:H1050–63
Tickle PG, Hendrickse PW, Degens H, Egginton S (2020) Impaired skeletal muscle performance as a consequence of random functional capillary rarefaction can be restored with overload-dependent angiogenesis. J Physiol 598:1187–1203
Hirai DM, Tabuchi A, Craig JC et al (2020) Skeletal muscle capillary hemodynamics in rats with heart failure with preserved ejection fraction. FASEB J 34(S1):1–11
Mohammed SF, Hussain S, Mirzoyev SA et al (2015) Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131:550–559
Upadhya B, Haykowsky MJ, Eggebeen J, Kitzman DW (2015) Exercise intolerance in heart failure with preserved ejection fraction: more than a heart problem. J Geriatr Cardiol 12:294–304
Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA (2017) Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 136:6–19
Reddy YNV, Melenovsky V, Redfield MM, Nishimura RA, Borlaug BA (2016) High-output heart failure: a 15-year experience. J Am Coll Cardiol 68:473–482
Correa-de-Araujo R, Addison O, Miljkovic I, Goodpaster BH, Bergman BC, Clark RV, Elena JW, Esser KA, Ferrucci L, Harris-Love MO, Kritchevsky SB, Lorbergs A, Shepherd JA, Shulman GI, Rosen CJ (2020) Myosteatosis in the context of skeletal muscle function deficit: an interdisciplinary workshop at the National Institute on Aging. Front Physiol 11:963
Weiss K, Shar M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, Golozar A, Steinberg A, Gerstenblith G, Russel SD, Weiss RG (2017) Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail 10:e004129
Heinonen I, Bucci M, Kemppainen J, Knuuti J, Nuutila P, Boushel R, Kalliokoski KK (1985) Regulation of subcutaneous adipose tissue blood flow during exercise in humans. J Appl Physiol 2012(112):1059–1063
Zamani P, Proto EA, Wilson N, Fazelinia H, Ding H, Spruce LA, Davila A Jr, Hanff TC, Mazurek JA, Prenner SB, Desjardins B, Margulies KB, Kelly DP, Arany Z, Doulias PT, Elrod JW, Allen ME, McCormack SE, Schur GM, D’Aquilla K, Kumari D, Thakuri D, Prabhakaran K, Langham MC, Poole DC, Seeholzer SH, Reddy R, Ischiropoulos H, Chirinos JA (2021) Multimodality assessment of heart failure with preserved ejection fraction skeletal muscle reveals differences in the machinery of energy fuel metabolism. ESC Heart Fail 8:2698–2712
Hearon CM Jr, Sarma S, Dias KA, Hieda M, Levine BD (2019) Impaired oxygen uptake kinetics in heart failure with preserved ejection fraction. Heart 105:1552–1558
Bekfani T, Bekhite Elsaied M, Derlien S, Nisser J, Westermann M, Nietzsche S, Hamadanchi A, Fröb E, Westphal J, Haase D, Kretzschmar T, Schlattmann P, Smolenski UC, Lichtenauer M, Wernly B, Jirak P, Lehmann G, Möbius-Winkler S, Schulze PC (2020) Skeletal muscle function, structure, and metabolism in patients with heart failure with reduced ejection fraction and heart failure with preserved ejection fraction. Circ Heart Fail 13:e007198
Egerman MA, Glass DJ (2014) Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol 49:59–68
Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG (2003) The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain 126:413–423
Lai YC, Tabima DM, Dube JJ et al (2016) SIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation 133:717–731
Hunter WG, Kelly JP, McGarrah RW 3rd et al (2016) Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: evidence for shared metabolic impairments in clinical heart failure. J Am Heart Assoc 5:e003190
Bowen TS, Brauer D, Rolim NPL, Bækkerud FH, Kricke A, Ormbostad Berre AM, Fischer T, Linke A, da Silva GJ, Wisloff U, Adams VJ (2017) Exercise training reveals inflexibility of the diaphragm in an animal model of patients with obesity-driven heart failure with a preserved ejection fraction. Am Heart Assoc 6:e006416
Lavietes MH, Gerula CM, Fless KG, Cherniack NS, Arora RR (2004) Inspiratory muscle weakness in diastolic dysfunction. Chest 126:838–844
Yamada K, Kinugasa Y, Sota T, Miyagi M, Sugihara S, Kato M, Yamamoto KJ (2016) Inspiratory muscle weakness is associated with exercise intolerance in patients with heart failure with preserved ejection fraction: a preliminary study. Card Fail 22:38–47
Hamazaki N, Kamiya K, Yamamoto S, Nozaki K, Ichikawa T, Matsuzawa R, Tanaka S, Nakamura T, Yamashita M, Maekawa E, Meguro K, Noda C, Yamaoka-Tojo M, Matsunaga A, Ako J (2020) Changes in respiratory muscle strength following cardiac rehabilitation for prognosis in patients with heart failure. J Clin Med 9:952
Hamazaki N, Kamiya K, Matsuzawa R, Nozaki K, Ichikawa T, Tanaka S, Nakamura T, Yamashita M, Maekawa E, Noda C, Yamaoka-Tojo M, Matsunaga A, Masuda T (2020) Ako Prevalence and prognosis of respiratory muscle weakness in heart failure patients with preserved ejection fraction. J Respir Med 161:105834
Amjad A, Brubaker PH, Upadhya B (2021) Exercise training for prevention and treatment of older adults with heart failure with preserved ejection fraction. Exp Gerontol 155:111559
Tucker WJ, Haykowsky MJ, Seo Y, Stehling E, Forman DE (2018) Impaired exercise tolerance in heart failure: role of skeletal muscle morphology and function. Curr Heart Fail Rep 15:323–331
Kitzman DW, Brubaker P, Morgan T, Haykowsky M, Hundley G, Kraus WE, Eggebeen J, Nicklas BJ (2016) Effect of caloric restriction or aerobic exercise training on peak oxygen consumption and quality of life in obese older patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 315:36–46
Gary RA, Sueta CA, Dougherty M, Rosenberg B, Cheek D, Preisser J, Neelon V, McMurray R (2004) Home-based exercise improves functional performance and quality of life in women with diastolic heart failure. Heart Lung 33:210–218
Kitzman DW, Brubaker PH, Morgan TM, Stewart KP, Little WC (2010) Exercise training in older patients with heart failure and preserved ejection fraction. Circ Heart Fail 3:659–667
Kitzman DW, Brubaker PH, Herrington DM, Morgan TM, Stewart KP, Hundley WG, Abdelhamed A, Haykowsky MJ (2013) Effect of endurance exercise training on endothelial function and arterial stiffness in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. J Am Coll Cardiol 62:584–592
Edelmann F, Gelbrich G, Dungen H, Fröhling S, Wachter R, Stahrenberg R, Binder L, Töpper A, Lashki DJ, Schwarz S, Herrmann-Lingen C, Löffler M, Hasenfuss G, Halle M, Pieske B (2011) Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex-DHF (Exercise training in Diastolic Heart Failure) pilot study. J Am Coll Cardiol 58:1780–1791
Smart NA, Haluska B, Jeffriess L, Leung D (2012) Exercise training in heart failure with preserved systolic function: a randomized controlled trial of the effects on cardiac function and functional capacity. Congest Heart Fail 18:295–301
Fu TC, Yang NI, Wang CH, Cherng WJ, Chou SL, Pan TL, Wang JS (2016) Aerobic interval training elicits different hemodynamic adaptations between heart failure patients with preserved and reduced ejection fraction. Am J Phys Med Rehabil 95:15–27
Angadi SS, Mookadam F, Lee CD, Tucker WJ, Haykowsky MJ, Gaesser GA (2014) High-intensity interval training vs. moderate-intensity continuous exercise training in heart failure with preserved ejection fraction: a pilot study. J Appl Physiol 95:15–27
Alves AJ, Ribeiro F, Goldhammer E, Rivlin Y, Rosenschein U, Viana JL, Duarte JA, Sagiv M, Oliveira J (2012) Exercise training improves diastolic function in heart failure patients. Med Sci Sports Exerc 44:776–785
Donelli da Silveira A, Beust de Lima J, da Silva Piardi D et al (2020) High-intensity interval training is effective and superior to moderate continuous training in patients with heart failure with preserved ejection fraction: a randomized clinical trial. Eur J Prev Cardiol 27:1733–1743
Mueller S, Winzer EB, Duvinage A, Gevaert AB, Edelmann F, Haller B, Pieske-Kraigher E, Beckers P, Bobenko A, Hommel J, Van de Heyning CM, Esefeld K, von Korn P, Christle JW, Haykowsky MJ, Linke A, Wisløff U, Adams V, Pieske B, van Craenenbroeck EM, Halle M (2021) Optim Ex-Clin Study Group. Effect of high-intensity interval training, moderate continuous training, or guideline-based physical activity advice on peak oxygen consumption in patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 325:542–551
Fujimoto N, Prasad A, Hastings JL, Bhella PS, Shibata S, Palmer D, Levine BD (2012) Cardiovascular effects of 1 year of progressive endurance exercise training in patients with heart failure with preserved ejection fraction. Am Heart J 164:869–877
Pandey A, Parashar A, Kumbhani DJ, Agarwal S, Garg J, Kitzman D, Levine B, Drazner M, Berry J (2015) Exercise training in patients with heart failure and preserved ejection fraction: meta-analysis of randomized control trials. Circ Heart Fail 8:33–40
Esposito F, Reese V, Shabetai R, Wagner PD, Richardson RS (2011) Isolated quadriceps training increases maximal exercise capacity in chronic heart failure: the role of skeletal muscle convective and diffusive oxygen transport. J Am Coll Cardiol 58:1353–1362
Lopez-Otin C, Galluzzi L, Freije JMP, Madeo F, Kroemer G (2016) Metabolic control of longevity. Cell 166:802–821
Borlaug BA, Anstrom KJ, Lewis GD et al (2018) Effect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE-HFpEF randomized clinical trial. JAMA 320:1764–1773
Reddy YNV, Stewart GM, Obokata M, Koepp KE, Borlaug BA (2021) Peripheral and pulmonary effects of inorganic nitrite during exercise in heart failure with preserved ejection fraction. Eur J Heart Fail 23:814–823
Jasuja R, LeBrasseur NK (2014) Regenerating skeletal muscle in the face of aging and disease. Am J Phys Med Rehabil 93:S88-96
Shah SJ, Voors AA, McMurray JJV, Kitzman DW, Viethen T, Bomfim Wirtz A, Huang E, Pap AF, Solomon SD (2019) JAMA 321:2101–2112
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Upadhya reported receiving honoraria from Novartis. Dr. Brubaker reported receiving honoraria from Merck and Boehringer Ingelheim.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
MD, M.A., Parrott, C.F., Ph.D., M.J.H. et al. Skeletal muscle abnormalities in heart failure with preserved ejection fraction. Heart Fail Rev 28, 157–168 (2023). https://doi.org/10.1007/s10741-022-10219-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-022-10219-9