
Abstract
Cardiac amyloidosis (CA) is an infiltrative restrictive cardiomyopathy caused by accumulation in the heart interstitium of amyloid fibrils formed by misfolded proteins. Most common CA types are light chain amyloidosis (AL) caused by monoclonal immunoglobulin light chains and transthyretin amyloidosis (ATTR) caused by either mutated or wild-type transthyretin aggregates. Previously considered a rare disease, CA is increasingly recognized among patients who may be misdiagnosed as undifferentiated heart failure with preserved ejection fraction (HFPEF), paradoxical low-flow/low-gradient aortic stenosis, or otherwise unexplained left ventricular hypertrophy. Progress in diagnosis has been due to the refinement of cardiac echocardiographic techniques (speckle tracking imaging) and magnetic resonance (T1 mapping) and mostly due to the advent of bone scintigraphy that has enabled noninvasive diagnosis of ATTR, limiting the need for endomyocardial biopsy. Importantly, proper management of CA starts from early recognition of suspected cases among high prevalence populations, followed by advanced diagnostic evaluation to confirm diagnosis and typing, preferentially in experienced amyloidosis centers. Differentiating ATTR from other types of amyloidosis, especially AL, is critical. Emerging targeted ATTR therapies offer the potential to improve outcomes of these patients previously treated only palliatively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyloidosis is a systemic disease caused by the deposition of misfolded protein aggregates that form amyloid fibrils in the interstitial space of various organs [1]. Cardiac amyloidosis (CA) results from infiltration of the heart interstitium by amyloid, which causes stiffening of the heart muscle and progressive cardiac dysfunction. Amyloidoses are classified according to the type of the primary misfolded protein, with over 30 different proteins being etiologically implicated. However, two types of proteins cause the majority of CA cases: monoclonal immunoglobulin light chains, which are produced from a plasma cell clone (light chain amyloidosis, AL), and transthyretin (TTR), a protein synthesized primarily in the liver (causing transthyretin amyloidosis, ATTR). Transthyretin normally serves as a carrier for thyroxine and holo-retinol binding protein and can form amyloid fibrils either when the TTR gene is mutated or if the wild type, nonmutated TTR becomes misfolded [2].
CA was previously considered a rare disease, with AL being the most common type. However, with the recent adoption of noninvasive diagnostic techniques that can accurately diagnose ATTR without an absolute need for cardiac biopsy—most importantly myocardial scintigraphy with certain bone-avid tracers—it was realized that ATTR is far more common than previously appreciated, especially among elderly patients [3]. Importantly, it has also been realized that CA is frequently misdiagnosed as HFPEF, low-flow low-gradient aortic stenosis, or otherwise unexplained left ventricular (LV) hypertrophy [4,5,6]. The introduction of specific therapies for ATTR CA has further increased the need to accurately diagnose these patients, early in the course of the disease. Thus, there is a need to develop appropriate tools to assist clinicians in recognizing possible ATTR cases among populations with high prevalence and then properly evaluate and confirm diagnosis. The present manuscript constitutes a consensus opinion document developed by a panel of CA and hereditary amyloidosis experts in Greece, aiming to provide practical tools for the early recognition and proper diagnostic confirmation of ATTR CA. Moreover, a brief overview of recently approved and emerging ATTR disease-modifying therapies is provided.
Classification and epidemiology of CA
The two most common types of CA are AL and ATTR. In AL, amyloid fibrils derived from clonal immunoglobulin light chains infiltrate various organs. It is a multisystemic disease with cardiac involvement observed in 50–75% of cases, but the kidneys, liver, lungs, autonomic and peripheral nervous systems and soft tissues are also involved quite commonly [7]. However, isolated cardiac AL is not uncommon, occurring in about 20% of patients (data: Department of Clinical Therapeutics). The prevalence of AL has been reported between 8 and 12 cases per million person-years; it affects slightly more frequently men, and its peak incidence is between the 5th and 7th decades [8]. Treatment of AL consists of cytotoxic chemotherapy aiming to eliminate the clonal plasma cells and therefore the production of the precursor protein [9, 10]. If untreated, prognosis of cardiac AL is dismal, with median survival of approximately 6 months from onset of heart failure (HF) [11]. Even with current therapies, mortality of high-risk patients with elevated cardiac biomarkers exceeds 40% at 1 year after diagnosis [12].
ATTR can be either hereditary or wild type. Hereditary or mutant ATTR (mATTR) is caused by mutations of TTR gene that result in TTR protein with an amyloidogenic potential. mATTR is considered a rare disease, with currently estimated prevalence of 50,000 affected individuals worldwide [13]. It has a worldwide distribution but it is endemic in areas of Portugal, northern Italy, Japan, Sweden and Cyprus [14]. mATTR is inherited in an autosomal-dominant manner with variable penetrance. Over 120 mutations of TTR gene have been described with varying phenotypic expression depending on the mutation; not all mutations are amyloidogenic. Most common phenotypes are dominant neuropathic, dominant cardiomyopathic, or mixed. The most common mutation worldwide is V122I, which is detected in approximately 3–4% of African-Americans and is associated with predominantly cardiac manifestations [15, 16]; other cardiomyopathic TTR mutations are L111M and I68L. The most common neuropathic mutation is V30M, which, however, may be associated with a mixed phenotype in late-onset disease. Other less common manifestations of mATTR include ocular abnormalities (including vitreous opacities that can cause gradual visual loss and trabecular obstruction causing chronic open-angle glaucoma), renal involvement (nephrotic syndrome and progressive renal failure), and rarely central nervous system involvement [17]. Prognosis of mATTR varies significantly according to the type of mutation, fibril type, and also within families. However, the main prognostic determinant is the presence of cardiac involvement, as median survival of patients with HF at diagnosis is 2.5–3.5 years versus 8–10 years in patients presenting only with polyneuropathy [18].
Although the cause for misfolding of wild-type TTR (wtATTR) has not been delineated, abnormal protein kinetics and aging-related post-translational modifications have been hypothesized [19]. wtATTR affects predominantly older individuals, usually over the age of 70–75 years, of whom > 90% are men. Although accurate estimates of ATTR prevalence are not available, it appears quite common in certain patient populations. Recent data report rates of wtATTR as high as 13% in patients with HFPEF, 16% in patients with aortic stenosis undergoing transcatheter valve replacement, 5% in those undergoing surgical replacement, and 5% in patients with presumed hypertrophic cardiomyopathy [4,5,6]. Furthermore, in asymptomatic patients undergoing bone scintigraphy (BS) for other reasons, ATTR has been detected in 1–3% as incidental finding [20, 21]. Cardiac manifestations predominate in wtATTR. However, musculoskeletal involvement may also be observed, that manifests clinically as carpal tunnel syndrome, spinal stenosis, and biceps tendon rupture. In most cases, wtATTR progresses slowly, but symptoms of advanced, refractory HF may develop abruptly after an initial slowly progressing phase. Overall, median survival is approximately 3.5 years after diagnosis, depending on the severity of HF [22,23,24].
Diagnostic workup of cardiac ATTR
Clinical presentation
Clinical manifestations of cardiac ATTR are not specific. Most commonly, patients present with symptoms of HF, including dyspnea on exertion, paroxysmal nocturnal dyspnea, orthopnea, fatigue, exercise intolerance, and peripheral edema that combined with the finding of concentric LV remodeling, usually lead to misdiagnosis of generic, undifferentiated HFPEF. Moreover, diagnosis of ATTR can be concealed in patients with co-existing comorbidities that are associated with ventricular hypertrophy, such as arterial hypertension and aortic stenosis in older individuals [25]. Likewise, in younger individuals with mATTR, LV hypertrophy may be misdiagnosed as hypertrophic cardiomyopathy [26]. However, several clinical clues may raise the suspicion of ATTR in ambiguous cases (Table 1). These may include an apparent “cure” of pre-existing hypertension or a need to down-titrate hypertensive medications such as ACE inhibitors; intolerance of beta-blockers in patients with symptoms of HF; severe aortic stenosis with low-flow, low-gradient pattern, that is attributed to the restrictive physiology of the amyloid-infiltrated LV; and a history of or concurrent conduction system abnormalities necessitating pacemaker implantation. Of note, pacemaker implantation may have preceded HF manifestations by several years [2]. Co-existent extra-cardiac abnormalities that raise suspicion of ATTR include peripheral sensorimotor neuropathy (lower limb hypo/dysesthesias and motor dysfunction) and dysautonomia (orthostatic hypotension, erectile dysfunction, sweating and alternating bowel pattern), occurring most commonly in mATTR, and orthopedic manifestations due to ligament infiltration, including bilateral carpal tunnel syndrome (50% in wtATTR) [23], biceps tendon rupture (33% in wtATTR) [27], and lumbar spinal stenosis [28]. Interestingly, carpal tunnel syndrome usually precedes development of symptoms of cardiac ATTR by approximately 5–15 years [29].
Biomarkers
There are no specific biomarkers available for the diagnosis of any type of CA. Serum TTR levels (pre-albumin) have no diagnostic or prognostic value in ATTR. However, certain biomarkers, most importantly natriuretic peptides and troponins, are helpful in the diagnostic workup and as prognostic markers in ATTR. Regarding diagnosis, very high levels of natriuretic peptides that are disproportionate to the severity of cardiac dysfunction, together with persistently increased troponin levels in patients with presumed HFPEF or LV hypertrophy, should alert clinicians towards a possible diagnosis of CA (Table 1) [19, 30]. In addition, the assessment of NT-proBNP may also be of value for the early identification of cardiac involvement in patients with initially neuropathic mATTR [31]. Natriuretic peptides and troponins are incorporated in clinical prognostic scoring systems for cardiac ATTR developed from independent cohorts [24, 32]. According to the Mayo clinic prognostic score, serum NT-proBNP > 3000 pg/ml and troponin T > 0.05 ng/ml can discriminate patients with very high 4-year mortality rate (57% versus 18% in patients with both biomarkers below the cutoffs) [24]. Serum creatinine and eGFR are also of prognostic value in cardiac ATTR (both wtATTR and mATTR), with the poorest outcome observed in patients with eGFR < 45 ml/min, especially when combined with high NT-proBNP (> 3000 pg/ml) [32].
In all cases with suspected CA, laboratory assessment should include detailed assessment for the presence of a monoclonal immunoglobulin, by using serum and urine immunofixation and quantification of serum free kappa and lambda immunoglobulin light chains, which could be suggestive of AL [1, 19]. Immunofixation electrophoresis should be performed instead of simple protein electrophoresis because the concentration of kappa or lambda chains may be too low to be detected by the latter. Free light chain (FLC) assay measures serum concentrations of kappa and lambda light chains. Normal concentrations are 3.3 to 19.4 mg/l for kappa and 5.7 to 26.3 mg/l for lambda chains. Normal range of kappa:lambda ratio is 0.26–1.65. Monoclonal paraprotein testing is considered positive if a monoclonal paraprotein is detected in immunofixation and FLC kappa:lambda ratio is abnormal (if the ratio is < 0.26 a clonal lambda chain is suggested, whereas a ratio > 1.65 is suggestive of a kappa chain) [33]. However, clinicians should be aware that the presence of monoclonal immunoglobulins or abnormally elevated immunoglobulin light chains is not specific for AL, as monoclonal gammopathy of undetermined significance (MGUS) is common among elderly individuals, affecting at least 5% of the general population over 65 years old [34,35,36,37]. Another confounding factor is the relatively high incidence of MGUS among patients with ATTRwt, which can be present in up to 25% of cases [38,39,40]. In addition, serum light chains may be elevated in patients with renal dysfunction who otherwise have no evidence of monoclonal paraprotein or amyloid organ infiltration, since light chains are cleared through the kidneys [41]. Moreover, kappa and lambda chains are differentially cleared by the kidneys, leading to skewed kappa to lambda ratio in renal dysfunction. Thus, a wider normal range of kappa/lambda ratio is used in patients with renal dysfunction compared with normal renal function [41]. Thus, careful evaluation of the tests for monoclonal gammopathy is required, preferentially in experienced referral centers.
Electrocardiography
The most frequent electrocardiographic (ECG) finding in ATTR is poor R-wave progression (or pseudo-infarction patterns) in precordial/anterior leads, observed in up to 60–70% of cases [6, 42]. This pattern is most common in patients with intermediate or severe LV hypertrophy but less common in normal or mildly hypertrophied LV [43].
Low QRS voltage (QRS amplitude ≤ 0.5 mV in limb leads or ≤ 1 mV in precordial leads; Sokolow index < 1.5 mV), although considered a classic feature of CA, has low sensitivity, especially in ATTR (30% of wtATTR cases and 15–45% in mATTR), although is more common in AL (45% of cases) [22, 23, 25, 42, 44,45,46]. Recently, markers that correct low QRS voltage by echocardiographically assessed LV mass have been proposed as more sensitive to detect CA, including the ratio of Sokolow index to the cross-sectional area of the LV wall (values < 1.5 indicative of CA) and the ratio of total QRS score (sum of QRS voltages in all leads) to LV mass index [47, 48]. However, it should be noted that up to 20% of patients with wtATTR may show ECG criteria of LV hypertrophy [1, 42].
Arrhythmias are frequent in CA. Most common are conduction abnormalities and atrial fibrillation (AF). Conduction abnormalities include 1st-degree atrioventricular block (up to 33% in mATTR, 45% in wtATTR) and intraventricular blocks (LBBB or RBBB) in 32% of wtATTR [6, 42]. Up to 18–23% of patients with wtATTR have an implanted pacemaker due to conduction abnormalities, which is less commonly seen in AL (5%) or mATTR. AF is reported in up to 55% of wtATTR, but in 17% of mATTR. Lastly, QTc prolongation is present in > 60% in ATTR. Importantly, none of the aforementioned ECG findings is specific to diagnose type of CA.
Echocardiography
Echocardiography is the most widely used, readily available imaging modality to raise the suspicion of CA in everyday clinical practice. Caveats include a low sensitivity in the early stages of CA and limited discriminatory ability between CA and other causes of LV hypertrophy [49]. Characteristic echocardiographic findings in CA include LV hypertrophy with restrictive LV physiology as well as other structural and functional abnormalities resulting from the indiscriminate interstitial amyloid infiltration. Echocardiographic findings vary according to amyloid disease progression. As such, they range from mild LV hypertrophy with atrioventricular valve thickening and impairment of longitudinal LV function in early/subclinical stages to marked LV hypertrophy with severe diastolic dysfunction (restrictive physiology) and ultimately development of biventricular systolic dysfunction in the advanced stage [50].
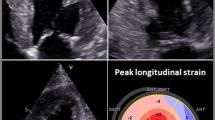
Left ventricular hypertrophy is a common morphological finding across all types of CA (Fig. 1a). However, it is more severe in wtATTR compared with other types, even after adjustment for differences in demographics and disease stage [46]. Even so, up to 40% of ATTR patients have no or mild LV hypertrophy [43]. LV hypertrophy is most frequently symmetrical but less commonly may be asymmetrical, affecting preferentially the interventricular septum (IVS). Asymmetric hypertrophy has been reported in up to 23% of wtATTR, while it may be more frequent in late-onset V30M mATTR [6, 43]. In advanced stages, hypertrophy can even affect the papillary muscles [50, 51]. Elderly wtATTR patients with aortic stenosis treated with transcutaneous aortic valve replacement (TAVR) show more severe LV hypertrophy and diastolic dysfunction (restrictive pattern), compared with non-ATTR counterparts [4]. Right ventricular (RV) free wall hypertrophy (> 7 mm) is a very frequent finding in ATTR with at least moderate LV hypertrophy (76% of patients with IVS > 14 mm). Ventricular dilatation is uncommon, except for RV dilatation in cases with pulmonary hypertension [46].

Typical echocardiographic findings of cardiac amyloidosis and their diagnostic accuracy (sensitivity and specificity). a Apical four-chamber view shows severe left ventricular (LV) hypertrophy, dilated left atrium (LA) and thickened interatrial septum (IAS) and atrioventricular (AV) valves. b Transmitral pulsed wave inflow velocities showing E/A ratio > 2, suggestive of Grade III diastolic dysfunction (DD). c Tissue Doppler imaging (TDI) of the basal interventricular septum, showing depressed systolic (s’), early diastolic (e’), and late diastolic (a’) velocities. d Bull’s eye map of LV longitudinal strain obtained with speckle-tracking imaging, showing the typical relative sparing of the apical strain compared with basal and mid segments. Data regarding sensitivity and specificity were obtained from ref. [64]. Diagnostic utility of qualitative findings is reported using a descriptive term [84]. Relative apical sparing is calculated as the ratio of apical longitudinal/sum of base and mid longitudinal LV strain [64]. GLS, global longitudinal strain
With nonharmonic imaging, hypertrophied LV walls may show a “granular sparkling” appearance that is suggestive, but not specific, of CA as a result of the higher echogenicity of the amyloid fibrils compared with the surrounding normal myocardium. Granular sparkling is observed more frequently (up to 65%) in patients with at least moderate hypertrophy [43]. In harmonic imaging, hypertrophied LV shows a speckled appearance [50].
Amyloid infiltration of atrial and valvular tissue appears as thickening of interatrial septum and atrioventricular valves, respectively, with associated valvular regurgitation (Fig. 1a) [52]. Combination of AV valve thickening, RV hypertrophy, and sparkling myocardium should raise suspicion of ATTR in patients with increased IVS thickness [43]. Small pericardial effusions are common in CA (> 40% in ATTR). However, larger effusions and tamponade are relatively rare [53].
As an archetypic restrictive cardiomyopathy, CA is characterized by LV diastolic dysfunction (DD), the severity of which parallels disease stage. Pseudonormal pattern is most common (43% of ATTR), while restrictive pattern is found in 35% of ATTR (Fig. 1b) [54]. Abnormal relaxation or no DD can be seen in early stage (4.5% of wtATTR and 29% with mATTR) [49]. Early mitral annular diastolic velocity (E′) decreases early in AL (even without overt hypertrophy), worsens as hypertrophy progresses, and becomes severely depressed in the advanced stage (Fig. 1c) [49, 54]. Atrial dilatation with low or absent atrial transmitral velocity classically co-exists, due to the elevated ventricular filling pressure and atrial amyloid infiltration (Fig. 1a). Overall, no differences are observed as to the severity of DD between different CA etiologies [46].
Left ventricular ejection fraction is usually preserved up to the advanced stages. However, subtle systolic dysfunction is documented using myocardial deformation imaging (tissue Doppler-TDI or speckle tracking-STE) early in the disease course, even in the asymptomatic stage [55,56,57,58]. Deformation imaging has been shown to allow differentiation of CA from other causes of LV hypertrophy. Compared with hypertensive and hypertrophic cardiomyopathy, longitudinal, radial, and circumferential strains are reduced in CA. However, in CA, there is a typical sparing of longitudinal strain in the apex compared with mid and basal LV segments (apical sparing, Fig. 1d) [50]. Deformation abnormalities are similarly observed in AL and ATTR and are proportional to the severity of LV hypertrophy and amyloid infiltration [59]. However, this association is less prominent in AL than in ATTR, underscoring the importance of other contributors to the pathophysiology of LV systolic dysfunction in AL, including direct myocardial toxicity by the circulating immunoglobulin light chains [60, 61]. Quantitative indexes of deformation abnormalities have been proposed to discriminate CA from other of causes of LV hypertrophy. Relative apical longitudinal strain (RALS) index is calculated as average apical LS/(average basal LS + average mid-LS) [62]. RALS index > 2.1 showed high discriminative capacity for the identification of CA among other hypertrophic etiologies [63]. Ejection fraction strain ratio (EFSR) is calculated as LVEF/global LS and was higher in CA compared with hypertrophic cardiomyopathy and hypertensive heart disease. EFSR > 4.1 had 89.7% sensitivity and 91.7% specificity to discriminate CA in a mixed population of 25 AL/15 ATTR patients [64]. Besides its diagnostic utility, STE strain is an independent predictor of clinical outcomes in CA, both ATTR and AL, with worse strain values associated with poorer outcomes [59, 65]. Deformation abnormalities of the atria and RV are also observed [66]. A similar pattern of apical sparing is observed by STE in the RV, which may also assist in the differential diagnosis of CA [67]. Table 2 summarizes the most common echocardiographic findings in cardiac amyloidosis.
Cardiac magnetic resonance
In addition to anatomic assessment of cardiac walls/chambers, cardiac magnetic resonance (CMR) with late gadolinium enhancement (LGE) and newer extracellular volume (ECV) techniques allows characterization of tissue composition, in particular amyloid infiltration. The two most typical LGE patterns in CA are either a diffuse subendocardial enhancement in a noncoronary artery territory distribution or transmural enhancement [1, 68]. However, atypical patterns might be observed, especially in early disease stages [69]. T1 mapping allows the quantification of myocardial extracellular volume either before (native T1) or after injection of contrast medium. Native T1 time is increased in CA and correlates with amyloid burden [70] and is higher in AL than ATTR, whereas post-contrast ECV is higher in ATTR than AL. Diagnostic accuracy of T1 mapping for CA of any subtype has been examined in a recently published study that included 436 patients with histologically confirmed CA among 800 suspected cases [71]. It was shown that native T1 time had 98% negative prognostic value at a cutoff < 1.036 ms and 98% positive predictive value at a cutoff > 1.164 ms to correctly identify CA among suspected cases. At intermediate native T1 values (1.036–1.164 ms), post-contrast ECV had a very high diagnostic accuracy at a cutoff > 0.37. However, T1 technique could not differentiate between CA subtypes. Advantages of native T1 mapping include no need for contrast injection, allowing its use in patients with renal dysfunction as well as serial evaluations. Limitations include platform-dependent variation of normal ranges, confounding by other pathologies such as tissue edema, inability to discriminate between amyloid subtypes or other infiltrative cardiac disorders, relative contraindication in patients with implanted pacemakers/defibrillators, and contraindication in patients with claustrophobia [1].
Myocardial scintigraphy with bone-seeking radiotracers
The observation in the 1980s that certain 99mTechnetium (99mTc)-phosphate derivative radionuclide tracers originally developed for bone scintigraphy (BS) accumulated in the amyloid-infiltrated heart has led to an extensive evaluation of the role of BS in diagnosing CA, although several decades later [72,73,74]. Originally applied for the detection of acute myocardial necrosis, 99mTc-pyrophosphate (99mTc-PYP) was the first radiotracer shown to be of diagnostic potential in CA. Subsequently, several studies have shown that bone tracers including 99mTc-PYP, 99mTc-3,3-dicarboxypropane-2,1-diphosphonate (99mTc-DPD), and 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP) bind with high specificity to ATTR amyloid fibrils. In AL CA, there is usually limited or no uptake of the tracer, but in up to 10% of patients with advanced AL CA, there may be significant uptake of the tracer [40, 75,76,77,78,79]. Importantly, not all bone radiotracers are suitable for detection of ATTR CA [80]. In 2005, Perugini and colleagues published their seminal study, which demonstrated the clinical utility of 99mTc-DPD BS to discriminate ATTR from AL or negative controls [80]. Researchers introduced a visual semi-quantitative 4-point score to evaluate cardiac radiotracer uptake: score 0, absent cardiac uptake and normal bone uptake; score 1, mild cardiac uptake, that is less than bone uptake; score 2, moderate cardiac uptake equal to bone uptake that appears to be attenuated; and score 3, strong cardiac uptake with mild or absent bone uptake. In this study, all ATTR patients showed cardiac uptake of at least Perugini score 2 (in 20%) or score 3 (in 80%), while all AL and negative controls showed no bone tracer cardiac uptake (score 0). A large multi-institutional study further evaluated the role of BS for the diagnosis of ATTR [40]. In this study by Gillmore et al., 718 patients with histologically confirmed CA out of 1217 cases with suspected CA were included. Confirmed cases consisted of 181 (25%) AL and 530 (74%) ATTR patients. Bone tracers that were used included 99mTc-DPD in 72% of patients, 99mTc-PYP in 16%, and 99mTc-HMDP in 12%. Monoclonal paraprotein was investigated with serum and urine immunofixation and serum free light chain assay in all patients. This study showed that positive BS (Perugini grade 1–3) was > 99% sensitive and 86% specific in correctly diagnosing ATTR but when Perugini grade 2 or 3 scans were considered, then the sensitivity was 90% with a specificity of 97%. The majority of false positive scans were seen in patients with AL and were graded as Perugini grade 1 on 99mTc-DPD. When combined with negative monoclonal paraprotein testing, Perugini grade 2 or 3 scans had 100% specificity and positive predictive value for ATTR. However, in cases with either positive paraprotein testing or grade 0 or 1 scans, the diagnostic yield of BS was low [38,39,40]. Therefore, it was concluded that biopsy is required to differentiate between AL and ATTR in patients with a detected monoclonal immunoglobulin. A recent bivariate meta-analysis with 529 patients reported a pooled sensitivity and specificity of 99mTc-PYP, 99mTc-DPD, or 99mTc-HMDP BS of 92.2% (95% CI 89–95%) and 95.4% (95% CI 77–99%), respectively, for ATTR [81]. Based on the above data, diagnosis of ATTR CA is established in patients with a grade 2 or 3 positive BS in the absence of a monoclonal immunoglobulin assessed by serum and urine investigations that include immunofixation and serum free light chains assay.
However, careful evaluation of BS for the diagnosis of ATTR CA is required, preferentially in an experienced center. The pattern of myocardial uptake, i.e., diffuse versus focal/multifocal/regional, should guide the diagnosis towards ATTR versus other underlying cardiac pathologies such as myocardial infarction [82]. Regarding intensity of uptake, other diagnostic parameters have been evaluated to assess bone radiotracer retention in ATTR, including heart-to-contralateral hemithorax (H/CL) uptake ratio and heart-to-whole-body (H/WB) retention ratio [78, 79]. For H/CL, a region of interest (ROI) is drawn over the heart which is corrected for background and ribs by a mirror ROI over the contralateral chest [78, 82]. H/CL ratio presents the fraction of mean uptake in the heart ROI to contralateral chest ROI. An H/CL ratio ≥ 1.5 (corresponding to Perugini grade 2 or 3) had a 97% sensitivity and 100% specificity for diagnosing ATTR [78]. The H/WB ratio is positively correlated with LV wall thickness, being higher in patients with LV wall ≥ 1.2 cm compared with no hypertrophy [83].
BS protocol for CA requires no specific patient preparation or fasting and is also applicable in patients reluctant or unable to undergo a CMR study [84]. A standard dose of 10–25 mCi (370–925 MBq) of 99mTc-PYP, 99mTc-DPD, or 99mTc-HMDP is injected intravenously at rest. Image acquisition is performed at 1 h and is preferentially repeated at 3 h post-injection for 99mTc-PYP and at 2 or 3 h for 99mTc-DPD or 99mTc-HMDP. Planar, anterior, and left anterior oblique/left lateral imaging is conducted in the supine position using low-energy, high-resolution collimators for a total of 500,000 counts on a 128 × 128 or 256 × 256 matrix, with the heart centered in the field of view and photopeak at 140 keV, with a symmetric 10% window, and allows for direct visual assessment and quantification of the degree of myocardial uptake by optical comparison with rib uptake. Single photon emission tomographic (SPET) imaging in a 64 × 64 matrix, 32 frames at 20 s/frame, is necessary in all cases, especially with positive planar scintigraphy, to avoid overlap of bone uptake and assess the regional distribution of myocardial radiotracer retention, including uptake in the IVS that is commonly affected by amyloidosis. To summarize, ATTR is scintigraphically confirmed by diffuse, intense myocardial radiopharmaceutical retention, specifically grade ≥ 2 upon optical assessment and/or H/CL ≥ 1.5 on semi-quantitative analysis in conjunction with diffusely intense depiction upon SPET processing.
Iodine-123-metaiodobenzylguanidine (123I-MIBG) scintigraphy that assesses myocardial autonomic sympathetic innervation may allow early detection of cardiac involvement in mixed mATTR with late-onset cardiac involvement (such as late onset V30M) or cardiomyopathic phenotype (V122I) [85,86,87] (Table 3). In a series of 142 individuals with confirmed V30M of whom 53% had polyneuropathy, reduced 123I-MIBG cardiac uptake was an independent predictor of 5-year mortality [87].
Positron emission tomography
Positron emission tomography (PET) may potentially allow direct tagging of amyloid fibrils by PET radiotracers including 18F-florbetapir [88, 89], 18F-florbetaben [90], and 11C-PiB (Pittsburgh B compound) [91, 92] and has given promising results for early diagnosis and quantification of the cardiac and extracardiac amyloid burden [88, 90]. In pilot studies, PET with the aforementioned compounds correctly identified CA patients from patients with hypertensive heart disease or healthy controls [90, 93]. However, it could not differentiate between CA subtypes.
Biopsy
The gold standard for diagnosis of amyloidosis is tissue biopsy. The presence of amyloid fibrils is confirmed by positive Congo red staining, which produces a pathognomonic green birefringence when viewed under cross-polarized light. A specialized pathology laboratory that performs high volumes of Congo red testing, by an experienced pathologist, is required for accurate diagnosis. Tissue is obtained either from the affected organ (heart, kidney, liver) or from peripheral tissue, such as abdominal fat pad, salivary glands, gingiva, skin, or gastrointestinal tract, which is a less invasive alternative. Although abdominal fat aspirate is the preferred peripheral tissue in most cases with suspected AL amyloidosis, with 69–88% sensitivity and 97% specificity for AL [94], it has low sensitivity for ATTR (below 30%); therefore, its role in the diagnostic assessment of patients with suspected CA and negative serum and urine paraprotein testing is limited [60, 95].
Tissue biopsy allows for the typing of amyloid fibrils. Immunohistochemistry (IHC) is the most widely available method for fibril typing, but has low sensitivity and specificity in fat tissue. In EMB, its diagnostic accuracy for ATTR is higher but still limited for AL, due to the nonspecific binding of multiple antisera (usually for ATTR and kappa/lambda chains) to circulating proteins that are present in histopathological samples [96]. Immunoelectron microscopy can be very helpful as it identified the amyloid fibrils and also uses specific gold-labeled antibodies to stain the fibrils.
The gold standard for amyloid fibril typing is mass spectrometry (MS), which detects peptides derived from amyloid abundant regions and after comparisons to reference databases, confirms amyloid subtype with high specificity [97]. However, MS is not widely available and only very few centers in the world can perform MS routinely. In our clinical practice, mass spectrometric analysis is considered in cases where amyloid typing cannot be established with first-line assays (IFE/FLC and BS) and includes patients with (a) confirmed CA with no other organ involvement, clonal immunoglobulin present and indeterminate or positive BS (i.e., grades 1–3) and (b) confirmed CA of no obvious type (negative paraprotein analyses, negative BS and negative genetic testing).
With the advent of BS for noninvasive diagnosis of ATTR, histologic evaluation is reserved for cases with suspected CA that fall within the following scenarios: (a) abnormal monoclonal paraprotein tests, irrespective of scintigraphic results. Here, initial preferred tissue source is abdominal fat. If fat biopsy is negative but clinical suspicion remains high, either another peripheral tissue or endomyocardial biopsy (EMB) is recommended; (b) high clinical and/or echocardiographic/CMR suspicion of CA, no evidence of plasma cell dyscrasia and indeterminate scintigraphic results (Perugini grade 0 or 1), where EMB is recommended; (c) high suspicion of AL despite a negative abdominal fat biopsy result, where EMB should be considered; and (d) negative paraprotein testing, if myocardial scintigraphy with 99mTc-PYP/99mTc-DPD is not available.
Genetic analysis (wtATTR versus mATTR)
All cases with confirmed ATTR should undergo genetic testing to differentiate between wtATTR and mATTR, even if no family history of amyloidosis or polyneuropathy is present, because the penetrance of mATTR is variable among mutations, within families and country of origin. There is no age limit for genetic testing and some mutations may cause symptoms for the first time well over the age of 70. In our experience (Department of Clinical Therapeutics), 4% of ATTR patients > 70 years old carried a TTR mutation. If mATTR is diagnosed, genetic counseling and screening of relatives for identification and follow-up of asymptomatic carriers are indicated. Identification of ATTR subtype may impact therapeutic options. Currently, approved therapies for cardiac ATTR, mutant and wild type, include only amyloid stabilizers, while in mATTR with polyneuropathy (with or without cardiomyopathy) TTR silencers (patisiran and inotersen) are also approved.
Diagnostic management of cardiac amyloidosis
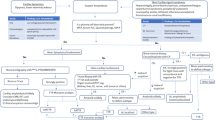
The basis for effective diagnostic and therapeutic management of CA patients is the early recognition and suspicion of possible cases based on clinical red flags, cardiac imaging, and biomarkers. We propose that all patients with suspected CA should then be referred for further diagnostic workup to experienced centers in amyloidosis so as to avoid common diagnostic pitfalls, such as misinterpretation/misuse of BS and incomplete/misinterpreted immunoglobulin testing (Fig. 2). Moreover, based on previous literature and our centers’ experience, we propose an algorithm for the advanced diagnostic evaluation of patients with suspected CA. In all patients, diagnostic workup should start with serum and urine immunofixation and free light chain assays. If monoclonal paraprotein is detected, further hematologic workup should be performed to confirm or exclude AL. If no monoclonal immunoglobulin is detected, the next step is to perform BS with 99mTc-PYP/DPD/HMDP. If BS is strongly positive (Perugini grade 2 or 3); then, ATTR is confirmed. If BS is indeterminate, EMB is required to exclude or confirm CA and its type. In all confirmed ATTR cases, genetic testing should follow to establish ATTR subtype (wild type or mutant) (Fig. 3).

Proposed integrated diagnostic management strategy of patients with cardiac amyloidosis

Proposed algorithm for the diagnostic workup of patients with suspected cardiac amyloidosis
Current and emerging therapies for cardiac ATTR
Treatment of cardiac ATTR consists of symptomatic cardiovascular management and disease-modifying therapies, including pharmacologic agents targeting the TTR amyloidogenic pathway and liver transplantation.
Management of cardiovascular manifestations of CA
Loop diuretics are prescribed to alleviate congestion, frequently combined with mineralocorticoid receptor antagonists, especially in patients with RV dysfunction and peripheral edema. However, aggressive use of diuretics is discouraged and a stepwise approach is preferred, as these patients depend heavily on preload. Other HF recommended therapies are generally not well tolerated by CA patients. Vasodilators including angiotensin converting enzyme inhibitors and angiotensin receptor antagonists may cause hypotension. Beta-blockers may aggravate HF symptoms due to the dependence of cardiac output on heart rate because of an essentially fixed stroke volume due to restrictive physiology. Patients with amyloidosis may also exhibit autonomic dysfunction, further reducing tolerance to common HF drugs. The use of oral vasopressor/antihypotensive therapies, such as midodrine (an α1-receptor agonist) may be helpful, especially in mixed phenotype mATTR.
In patients with AF, ventricular rate is usually naturally controlled due to amyloid infiltrated atrioventricular conduction system. However, rapid ventricular rates may occur, which are poorly tolerated due to shortening of the diastolic period that reduces stroke volume. Rate control drugs in CA are problematic. Apart from previously discussed beta-blockers, nondihydropyridine calcium channel blockers are contraindicated because they can cause atrioventricular heart block and worsening HF and even cardiogenic shock due to their negative inotropic effect. Digoxin has been controversial due to hypothesized increased susceptibility to toxicity because of digoxin binding to amyloid fibrils, although this has not been confirmed in a recent series [98]. In two recent case series, significant arrhythmias occurred in up to 12% of CA patients treated with digoxin, however not clearly linked to digoxin toxicity [98, 99]. Expert opinion recommends that if digoxin is trialed for rate control or advanced HF, it should be started in low doses, with frequent monitoring of hemodynamics, renal function, electrolytes and drug plasma levels [100]. Rhythm control is considered if ventricular rate is poorly controlled with pharmacologic agents [101]. Pharmacologic cardioversion is performed with amiodarone, which may also be used for rate control. Direct electric cardioversion may be used, with acute success rate similar to non-CA AF (up to 90–94% of patients) [102]. However, it is associated with higher complication rates, including ventricular arrhythmias, bradyarrhythmias requiring pacemaker implantation, and thromboembolic events. Cardiac amyloidosis is associated with increased thrombogenicity, even without AF [103]. In AF particularly, there is increased incidence of LA thrombi even in low-risk patients such as those with recent onset AF (< 48 h) and anticoagulation for > 3 weeks before AF onset [102]. Therefore, transesophageal echocardiogram should be strongly considered in all CA patients before AF cardioversion, while therapeutic anticoagulation should be commenced in all patients with AF irrespective of CHADS-VASc score. The choice between warfarin and novel oral anticoagulants (NOACs) should be based on specific patient characteristics as there are no relevant data from randomized studies. However, in a recent retrospective analysis, thrombotic and bleeding event rates were comparable between patients who received warfarin or NOACs, although labile INRs were detected in up to half of warfarin-treated patients, predisposing to increased thrombotic and hemorrhagic risk [104].
Complex ventricular arrhythmias are frequent in CA [100]. Nonsustained ventricular tachycardia (NSVT) has been reported in 18–74%, sustained VT or ventricular fibrillation (VF) in 8–19% and appropriate implantable cardioverter-defibrillator (ICD) shocks in 6.5–32% in small patient series. Sudden death occurs in up to half of CA patients. However, underlying rhythm may not be VT/VF and, indeed, historically electro-mechanical dissociation was thought to be implicated in most cases. Currently, the use of ICDs in CA remains controversial, due to lack of data showing mortality benefit with ICDs, combined with the previously considered dismal disease prognosis. European guidelines recommend ICD implantation for secondary prevention in cardiac AL or mATTR patients with ventricular arrhythmia causing hemodynamic instability who are expected to survive > 1 year with good functional status (Class IIa, Level of Evidence C) [105]. However, with the recent introduction of disease-modifying targeted therapies, recommendations for ICD implantation in CA will probably need to be re-examined.
There is limited experience regarding the safety and efficacy of cardiac resynchronization therapy (CRT) in patients with CA. RV pacing, which is frequently indicated in CA patients, may aggravate their functional class and left ventricular function [106]. Data from single-center retrospective studies have suggested that CRT may be associated with improved NYHA class, LVEF and severity of MR in CA patients with indication of RV pacing or reduced LVEF and wide QRS complex [106, 107]. No data are available yet regarding cardiac contractility modulation therapy in patients with CA, reduced LVEF and narrow QRS, although this therapy has been shown to improve functional status and quality of life in patients with HF and reduced LVEF of common etiologies.
Disease-modifying therapies
During the past decade, ATTR-specific therapies that target several checkpoints of amyloid formation have been developed, including TTR tetramer stabilization and liver TTR RNA silencing. Therapies aiming towards TTR amyloid fibril-specific extraction from tissues are currently under investigation.
Currently available therapies
Stabilizers
Stabilizers act by binding to the TTR tetramer, inhibiting their dissociation and not allowing amyloid formation by misfolded, amyloidogenic, TTR monomers [108]. Tafamidis is a selective TTR stabilizer, a benzoxazole derivative that lacks nonsteroidal anti-inflammatory activity. It binds specifically to the thyroxine binding site of TTR tetramer, stabilizing kinetically wtTTR, and several TTR variants in vitro [109]. It is administered orally and is available in two formulations, tafamidis meglumine (20 mg tablets) and tafamidis free salt (61 mg tablets), which are not substitutable on a per mg basis. Evidence from randomized clinical studies, and subsequent open-label extension studies showed that tafamidis slowed deterioration of neurological function, and maintained health-related quality of life in patients with early-stage V30M polyneuropathy [110,111,112,113]. Tafamidis has been approved for the treatment of stage 1 familial amyloid polyneuropathy in Europe and Japan (but not in US). Efficacy and safety of tafamidis in cardiac ATTR were examined in a large phase 3, randomized, placebo-controlled trial (ATTR-ACT), in which 441 cardiac ATTR (mutant or wild-type) patients were randomized in a 2:1:2 ratio to receive tafamidis 80 mg, tafamidis 20 mg, or placebo for 30 months [114]. Overall mortality at 30 months was reduced in the two tafamidis arms combined by 30% (29.5% vs 42.9%, an absolute decrease of 13.4%). Moreover, cardiovascular hospitalizations were reduced by 32% with tafamidis. In addition, tafamidis reduced the rate of decline in exercise capacity and quality of life compared with placebo. Adverse events did not differ between pooled tafamidis and placebo arms. Tafamidis is currently the only approved disease-modifying specific therapy for cardiac ATTR in Europe and the USA, based on evidence from ATTR-ACT trial.
Diflunisal, a nonsteroidal anti-inflammatory drug, binds in vitro to the thyroxine binding site of TTR, kinetically stabilizing TTR tetramers [115], and has been evaluated in a small randomized, placebo-controlled trial in patients with neuropathic mATTR [116]. However, as a nonsteroidal anti-inflammatory drug, it is associated with side effects [117], while there is no data from randomized clinical trials in cardiac ATTR [118,119,120,121]. Diflunisal may be considered for off-label use only in very selected patients with cardiac ATTR.
Doxycycline and tauroursodeoxycholic acid
Doxycycline and tauroursodeoxycholic acid (TUDCA) are used “off-label” in CA, due to their suggested disruption of deposited TTR fibrils, both mutant and wild type, according to in vitro data [122,123,124]. The combination of doxycycline (100 mg BID) and TUDCA (250 mg TID) administered for up to 12 months has been examined in a phase II study in a mixed mATTR and wtATTR cohort, where it stabilized cardiac and neurological disease progression with an acceptable toxicity profile [125].
Emerging therapies for cardiac ATTR
TTR gene silencers are antisense oligonucleotides or small RNA interference molecules that inhibit expression of TTR gene by deactivating its messenger RNA (mRNA). Currently approved silencers include patisiran, a small-interfering RNA molecule (siRNA) and inotersen, an antisense oligonucleotide. Both patisiran and inotersen have been approved for their use only in patients with stage 1 or 2 mATTR polyneuropathy; however, some improvement in parameters of cardiac function and natriuretic peptides was also noted with both drugs in patients with mATTR and cardiac involvement [126,127,128,129,130,131,132]. Vutrisiran is an investigational siRNA that is administered subcutaneously every 3 months and is currently being tested in phase 3 trials in mATTR. Currently, the use of these drugs in wtATTR is not approved and studies evaluating their role in this population are ongoing.
Investigational therapies
PRX004 is a monoclonal antibody that has been designed to specifically bind to and extract TTR amyloid fibrils from affected organs [133]. Tolerability and pharmacological properties of PRX004 are currently being investigated in a phase 1 clinical trial in mATTR (NCT03336580).
AG10 is an investigational TTR stabilizer that mimics the effect of a stabilizing variant, T139M, on TTR tetramers [134, 135]. Safety and TTR stabilizing effect of AG10 in cardiac ATTR were examined in a phase 2, placebo-controlled study, where it demonstrated effective, dose-dependent TTR stabilization, with acceptable safety profile [136]. AG10 has entered phase 3 clinical testing in cardiac ATTR (ATTRIBUTE-CM trial, NCT03860935).
Liver and heart transplantation
Liver transplantation (LT) has been considered a potentially curative therapy for mATTR, as the liver is the main source of amyloidogenic TTR [137]. It may be considered in patients with early onset neuropathic Val30Met mATTR, in whom it has been shown to prevent progression of polyneuropathy [138]. However, cardiac amyloidosis has been observed after LT, possibly resulting from deposition of wild-type TTR upon pre-existing variant fibrils that serve as matrix for newly deposited ones [139]. Combined heart and liver transplantation (CHLT) could be considered in young patients with advanced mATTR neuropathy and end-stage heart failure [140]. Prognosis after CHLT does not differ significantly from patients who received heart transplants for other etiologies, with 75.8% 5-year survival in a series from Mayo Clinic, comparable with single heart transplantation [141]. Single heart transplantation (HT) has been less commonly performed in mATTR patients with isolated cardiac involvement. In wtATTR, single heart transplantation may be considered in appropriate individuals with advanced heart failure due to CA [140]. However, as wtATTR affects quite elderly persons, they are often ineligible for HT. In a small series of 7 relatively young wtATTR patients (mean age 66 ± 9 years) who underwent HT, 3-year survival was 100%, although subsequent amyloid involvement of the gastrointestinal tract and peripheral nerves was observed in 2/7 patients [142].
Conclusions
Cardiac amyloidosis is a previously under-recognized entity. Phenotypically, it is characterized by LV hypertrophy with advanced diastolic dysfunction and preserved systolic function. It may commonly be misdiagnosed as undifferentiated HFPEF, low-flow low-gradient aortic stenosis, and hypertensive heart disease in elderly patients or hypertrophic cardiomyopathy in younger individuals. Several clinical and cardiac imaging findings can assist in raising field physicians’ suspicion for CA, prompting patient referral to experienced centers for further diagnostic workup to establish diagnosis and subtyping. Etiologic confirmation is of crucial importance for prognostic assessment and proper therapeutic management, as AL is a malignant disease requiring urgent therapy while ATTR is a more benign entity albeit limiting longevity and impairing quality of life. Recent advent of specific therapies could improve outcomes of ATTR patients, previously managed only palliatively.
References
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C (2017) Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 135(14):1357–1377
Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S (2009) Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 120(13):1203–1212
Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM (2014) Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2(2):113–122
Castano A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, Rubin J, Chiuzan C, Nazif T, Vahl T, George I, Kodali S, Leon MB, Hahn R, Bokhari S, Maurer MS (2017) Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 38(38):2879–2887
Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, Bornstein B, Salas C, Lara-Pezzi E, Alonso-Pulpon L, Garcia-Pavia P (2015) Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 36(38):2585–94
Gonzalez-Lopez E, Gagliardi C, Dominguez F, Quarta CC, de Haro-Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo-Marcos M, Lorenzini M, Lara-Pezzi E, Foffi S, Alonso-Pulpon L, Rapezzi C, Garcia-Pavia P (2017) Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 38(24):1895–1904
Falk RH, Alexander KM, Liao R, Dorbala S (2016) AL (light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol 68(12):1323–1341
Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, Dungu J, Banypersad SM, Wechalekar AD, Whelan CJ, Hawkins PN, Gillmore JD (2013) Systemic amyloidosis in England: an epidemiological study. Br J Haematol 161(4):525–532
Kastritis E, Dialoupi I, Gavriatopoulou M, Roussou M, Kanellias N, Fotiou D, Ntanasis-Stathopoulos I, Papadopoulou E, Ziogas DC, Stamatelopoulos K, Manios E, Ntalianis A, Eleutherakis-Papaiakovou E, Papanikolaou A, Migkou M, Papanota AM, Gakiopoulou H, Psimenou E, Tselegkidi MI, Tsitsilonis O, Kostopoulos I, Terpos E, Dimopoulos MA (2019) Primary treatment of light-chain amyloidosis with bortezomib, lenalidomide, and dexamethasone. Blood Adv 3(20):3002–3009
Kastritis E, Dimopoulos MA (2016) Recent advances in the management of AL amyloidosis. Br J Haematol 172(2):170–186
Kastritis E, Dimopoulos MA (2011) Prognosis and risk assessment in AL amyloidosis–state of the art. Amyloid 18(Suppl 1):89–91
Jaccard A, Comenzo RL, Hari P, Hawkins PN, Roussel M, Morel P, Macro M, Pellegrin JL, Lazaro E, Mohty D, Mercie P, Decaux O, Gillmore J, Lavergne D, Bridoux F, Wechalekar AD, Venner CP (2014) Efficacy of bortezomib, cyclophosphamide and dexamethasone in treatment-naive patients with high-risk cardiac AL amyloidosis (Mayo Clinic stage III). Haematologica 99(9):1479–1485
Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante-Bordeneuve V, Schmidt HHJ, Merlini G (2015) Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 66(21):2451–2466
Adams D, Koike H, Slama M, Coelho T (2019) Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 15(7):387–404
Jacobson DR, Pastore R, Pool S, Malendowicz S, Kane I, Shivji A, Embury SH, Ballas SK, Buxbaum JN (1996) Revised transthyretin Ile 122 allele frequency in African-Americans. Hum Genet 98(2):236–238
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med 336(7):466–473
Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, Biagini E, Lorenzini M, Grigioni F, Leone O, Cappelli F, Palladini G, Rimessi P, Ferlini A, Arpesella G, Pinna AD, Merlini G, Perlini S (2013) Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 34(7):520–528
Ruberg FL, Berk JL (2012) Transthyretin (TTR) cardiac amyloidosis. Circulation 126(10):1286–1300
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS (2019) Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 73(22):2872–2891
Longhi S, Guidalotti PL, Quarta CC, Gagliardi C, Milandri A, Lorenzini M, Potena L, Leone O, Bartolomei I, Pastorelli F, Salvi F, Rapezzi C (2014) Identification of TTR-related subclinical amyloidosis with 99mTc-DPD scintigraphy. JACC Cardiovasc Imaging 7(5):531–532
Mohamed-Salem L, Santos-Mateo JJ, Sanchez-Serna J, Hernandez-Vicente A, Reyes-Marle R, Castellon Sanchez MI, Claver-Valderas MA, Gonzalez-Vioque E, Haro-Del Moral FJ, Garcia-Pavia P, Pascual-Figal DA (2018) Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int J Cardiol 270:192–196
Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ (2013) Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2(2):e000098
Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC (2016) Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation 133(3):282–290
Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A (2016) Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 68(10):1014–1020
Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, Helio T, Linhart A, Mogensen J, Pinto Y, Ristic A, Seggewiss H, Sinagra G, Tavazzi L, Elliott PM (2013) Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 34(19):1448–1458
m. Authors, Task Force, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H, (2014) ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 35(39):2733–79
Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH (2017) Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA 318(10):962–963
Yanagisawa A, Ueda M, Sueyoshi T, Okada T, Fujimoto T, Ogi Y, Kitagawa K, Tasaki M, Misumi Y, Oshima T, Jono H, Obayashi K, Hirakawa K, Uchida H, Westermark P, Ando Y, Mizuta H (2015) Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 28(2):201–207
Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, Shapiro D, Evans PJ, Maschke S, Kilpatrick SE, Tan CD, Rodriguez ER, Monteiro C, Tang WHW, Kelly JW, Seitz WH Jr, Hanna M (2018) Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol 72(17):2040–2050
Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, Falk RH, Cheung KN, Patel AR, Pano A, Packman J, Grogan DR (2012) Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J 164(2):222–228 e1
Damy T, Deux JF, Moutereau S, Guendouz S, Mohty D, Rappeneau S, Guellich A, Hittinger L, Loric S, Lefaucheur JP, Plante-Bordeneuve V (2013) Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid 20(4):212–220
Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez-Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN (2018) A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 39(30):2799–2806
Abraham RS, Katzmann JA, Clark RJ, Bradwell AR, Kyle RA, Gertz MA (2003) Quantitative analysis of serum free light chains A new marker for the diagnostic evaluation of primary systemic amyloidosis. Am J Clin Pathol 119(2):274–278
Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, Dispenzieri A, Katzmann JA, Melton LJ 3rd (2006) Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 354(13):1362–1369
Landgren O, Graubard BI, Kumar S, Kyle RA, Katzmann JA, Murata K, Costello R, Dispenzieri A, Caporaso N, Mailankody S, Korde N, Hultcrantz M, Therneau TM, Larson DR, Cerhan JR, Rajkumar SV (2017) Prevalence of myeloma precursor state monoclonal gammopathy of undetermined significance in 12372 individuals 10–49 years old: a population-based study from the National Health and Nutrition Examination Survey. Blood Cancer J 7(10):e618
Singh A, Geller HI, Alexander KM, Padera RF, Mitchell RN, Dorbala S, Castillo JJ, Falk RH (2018) True, true unrelated? Coexistence of Waldenstrom macroglobulinemia and cardiac transthyretin amyloidosis. Haematologica 103(8):e374–e376
Sidiqi MH, Dasari S, McPhail ED, Buadi FK, Warsame R, Lacy MQ, Dingli D, Gonsalves WI, Kapoor P, Kourelis T, Leung N, Muchtar E, Grogan M, Lust JA, Kumar S, Kyle RA, Rajkumar VS, Gertz M, Dispenzieri A (2019) Monoclonal gammopathy plus positive amyloid biopsy does not always equal AL amyloidosis. Am J Hematol 94(5):E141–E143
Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, Sarosiek S (2018) Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 25(1):62–67
Maleszewski JJ, Murray DL, Dispenzieri A, Grogan M, Pereira NL, Jenkins SM, Judge DP, Caturegli P, Vrana JA, Theis JD, Dogan A, Halushka MK (2013) Relationship between monoclonal gammopathy and cardiac amyloid type. Cardiovasc Pathol 22(3):189–194
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PN (2016) Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412
Hutchison CA, Harding S, Hewins P, Mead GP, Townsend J, Bradwell AR, Cockwell P (2008) Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 3(6):1684–1690
Cyrille NB, Goldsmith J, Alvarez J, Maurer MS (2014) Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 114(7):1089–1093
Damy T, Maurer MS, Rapezzi C, Plante-Bordeneuve V, Karayal ON, Mundayat R, Suhr OB, Kristen AV (2016) Clinical, ECG and echocardiographic clues to the diagnosis of TTR-related cardiomyopathy. Open Heart 3(1):e000289
Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante-Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, Investigators T (2016) Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 68(2):161–72
Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, Rowczenio D, Pflugfelder PW, Fox Z, Lachmann HJ, Wechalekar AD, Hawkins PN, Gillmore JD (2012) Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 33(9):1120–1127
Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, Gagliardi C, Milandri A, Rapezzi C, Falk RH (2014) Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation 129(18):1840–1849
Carroll JD, Gaasch WH, McAdam KP (1982) Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol 49(1):9–13
Quarta C, Perlini S, Longhi S, Berardini A, Musca F, Salinaro F, Obici L, Milandri A, Gallo P, Gagliardi C, Biagini E, Mingardi F, Pazzi C, Merlini G, Rapezzi C (2012) A simple voltage/mass index improves diagnosis of cardiac amyloidosis: an electrocardiographic and echocardiographic study of 570 patients with left ventricular hypertrophy. J Am Coll Cardiol 59(13):E1586
Di Bella G, Pizzino F, Minutoli F, Zito C, Donato R, Dattilo G, Oreto G, Baldari S, Vita G, Khandheria BK, Carerj S (2014) The mosaic of the cardiac amyloidosis diagnosis: role of imaging in subtypes and stages of the disease. Eur Heart J Cardiovasc Imaging 15(12):1307–1315
Di Nunzio D, Recupero A, de Gregorio C, Zito C, Carerj S, Di Bella G (2019) Echocardiographic findings in cardiac amyloidosis: inside two-dimensional, Doppler, and strain imaging. Curr Cardiol Rep 21(2):7
Cacciapuoti F (2015) The role of echocardiography in the non-invasive diagnosis of cardiac amyloidosis. J Echocardiogr 13(3):84–89
Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, Jaccard A (2013) Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis 106(10):528–540
Siddiqi OK, Ruberg FL (2018) Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 28(1):10–21
Al-Zahrani GB, Bellavia D, Pellikka PA, Dispenzieri A, Hayman SR, Oh JK, Miyazaki C, Miller FA Jr (2009) Doppler myocardial imaging compared to standard two-dimensional and Doppler echocardiography for assessment of diastolic function in patients with systemic amyloidosis. J Am Soc Echocardiogr 22(3):290–298
Koyama J, Ray-Sequin PA, Falk RH (2003) Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue Doppler echocardiography in patients with AL (primary) cardiac amyloidosis. Circulation 107(19):2446–2452
Bellavia D, Abraham TP, Pellikka PA, Al-Zahrani GB, Dispenzieri A, Oh JK, Bailey KR, Wood CM, Novo S, Miyazaki C, Miller FA Jr (2007) Detection of left ventricular systolic dysfunction in cardiac amyloidosis with strain rate echocardiography. J Am Soc Echocardiogr 20(10):1194–1202
Porciani MC, Lilli A, Perfetto F, Cappelli F, Massimiliano Rao C, Del Pace S, Ciaccheri M, Castelli G, Tarquini R, Romagnani L, Pastorini T, Padeletti L, Bergesio F (2009) Tissue Doppler and strain imaging: a new tool for early detection of cardiac amyloidosis. Amyloid 16(2):63–70
Sun JP, Stewart WJ, Yang XS, Donnell RO, Leon AR, Felner JM, Thomas JD, Merlino JD (2009) Differentiation of hypertrophic cardiomyopathy and cardiac amyloidosis from other causes of ventricular wall thickening by two-dimensional strain imaging echocardiography. Am J Cardiol 103(3):411–415
Ternacle J, Bodez D, Guellich A, Audureau E, Rappeneau S, Lim P, Radu C, Guendouz S, Couetil JP, Benhaiem N, Hittinger L, Dubois-Rande JL, Plante-Bordeneuve V, Mohty D, Deux JF, Damy T (2016) Causes and consequences of longitudinal LV dysfunction assessed by 2D strain echocardiography in cardiac amyloidosis. JACC Cardiovasc Imaging 9(2):126–138
Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, Apstein CS, Liao R (2004) Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 94(8):1008–1010
Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, Connors LH, Sawyer DB, Semigran MJ, Macgillivray TE, Seldin DC, Falk R, Liao R (2010) Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A 107(9):4188–4193
Phelan D, Collier P, Thavendiranathan P, Popovic ZB, Hanna M, Plana JC, Marwick TH, Thomas JD (2012) Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 98(19):1442–1448
Liu D, Hu K, Niemann M, Herrmann S, Cikes M, Stork S, Gaudron PD, Knop S, Ertl G, Bijnens B, Weidemann F (2013) Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circ Cardiovasc Imaging 6(6):1066–1072
Pagourelias ED, Mirea O, Duchenne J, Van Cleemput J, Delforge M, Bogaert J, Kuznetsova T, Voigt JU (2017) Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging 10(3):e005588
Senapati A, Sperry BW, Grodin JL, Kusunose K, Thavendiranathan P, Jaber W, Collier P, Hanna M, Popovic ZB, Phelan D (2016) Prognostic implication of relative regional strain ratio in cardiac amyloidosis. Heart 102(10):748–754
Nochioka K, Quarta CC, Claggett B, Roca GQ, Rapezzi C, Falk RH, Solomon SD (2017) Left atrial structure and function in cardiac amyloidosis. Eur Heart J Cardiovasc Imaging 18(10):1128–1137
Arvidsson S, Henein MY, Wikstrom G, Suhr OB, Lindqvist P (2018) Right ventricular involvement in transthyretin amyloidosis. Amyloid 25(3):160–166
Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, Rosmini S, Quarta CC, Whelan CJ, Kellman P, Gillmore JD, Moon JC, Hawkins PN, Fontana M (2017) Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 70(4):466–477
Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, Klingel K, Kandolf R, Sechtem U (2008) Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 51(10):1022–1030
Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, Pica S, Castelletti S, Piechnik SK, Robson MD, Gilbertson JA, Rowczenio D, Hutt DF, Lachmann HJ, Wechalekar AD, Whelan CJ, Gillmore JD, Hawkins PN, Moon JC (2014) Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging 7(2):157–165
Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, Gritti M, Quarta C, Knight DS, Wechalekar AD, Lachmann HJ, Perlini S, Pontone G, Moon JC, Kellman P, Gillmore JD, Hawkins PN, Fontana M (2020) Noncontrast magnetic resonance for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 13(1 Pt 1):69–80
Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM (1982) Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 103(4 Pt 1):468–473
Falk RH, Lee VW, Rubinow A, Hood WB Jr, Cohen AS (1983) Sensitivity of technetium-99m-pyrophosphate scintigraphy in diagnosing cardiac amyloidosis. Am J Cardiol 51(5):826–830
Gertz MA, Brown ML, Hauser MF, Kyle RA (1987) Utility of technetium Tc 99m pyrophosphate bone scanning in cardiac amyloidosis. Arch Intern Med 147(6):1039–1044
Chacko L, Martone R, Cappelli F, Fontana M (2019) Cardiac amyloidosis: updates in imaging. Curr Cardiol Rep 21(9):108
Bourogianni O, Papadaki E, Foukarakis E, Koukouraki S (2019) Isolated cardiac amyloidosis. Utility of bone seeking tracers scintigraphy in differentiating the subtype of amyloid: a case report, J Nucl Cardiol 26(1):337–341
Galat A, Rosso J, Guellich A, Van Der Gucht A, Rappeneau S, Bodez D, Guendouz S, Tissot CM, Hittinger L, Dubois-Rande JL, Plante-Bordeneuve V, Itti E, Meignan M, Damy T (2015) Usefulness of (99m)Tc-HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid 22(4):210–220
Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS (2013) (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 6(2):195–201
Papantoniou V, Valsamaki P, Kastritis S, Tsiouris S, Delichas Z, Papantoniou Y, Tsiouma M, Athanasoulis T, Fotopoulos A, Dimopoulos MA (2015) Imaging of cardiac amyloidosis by (99m)Tc-PYP scintigraphy. Hell J Nucl Med 18(Suppl 1):42–50
Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi-Reggiani L, Fallani F, Branzi A, Rapezzi C (2005) Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 46(6):1076–1084
Treglia G, Glaudemans A, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, Ceriani L, Prior JO, Giovanella L, Slart R (2018) Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging 45(11):1945–1955
Valsamaki PN, Zissimopoulos A (2019) Cardiac amyloidosis. Two main subtypes and diagnosis by nuclear medicine: SPET tracer revival. Hell J Nucl Med 22(3):161–164
Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, Ferlini A, Longhi S, Lorenzini M, Reggiani LB, Gagliardi C, Gallo P, Villani C, Salvi F (2011) Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 4(6):659–670
Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans A, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart R, Verberne HJ, Bourque JM (2019) ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol 26(6):2065–2123
Delahaye N, Dinanian S, Slama MS, Mzabi H, Samuel D, Adams D, Merlet P, Le Guludec D (1999) Cardiac sympathetic denervation in familial amyloid polyneuropathy assessed by iodine-123 metaiodobenzylguanidine scintigraphy and heart rate variability. Eur J Nucl Med 26(4):416–424
Tanaka M, Hongo M, Kinoshita O, Takabayashi Y, Fujii T, Yazaki Y, Isobe M, Sekiguchi M (1997) Iodine-123 metaiodobenzylguanidine scintigraphic assessment of myocardial sympathetic innervation in patients with familial amyloid polyneuropathy. J Am Coll Cardiol 29(1):168–174
Coutinho MC, Cortez-Dias N, Cantinho G, Conceicao I, Oliveira A, Bordalo e Sa A, Goncalves S, Almeida AG, de Carvalho M, Diogo AN, (2013) Reduced myocardial 123-iodine metaiodobenzylguanidine uptake: a prognostic marker in familial amyloid polyneuropathy. Circ Cardiovasc Imaging 6(5):627–36
Dorbala S, Vangala D, Semer J, Strader C, Bruyere JR Jr, Di Carli MF, Moore SC, Falk RH (2014) Imaging cardiac amyloidosis: a pilot study using (1)(8)F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging 41(9):1652–1662
Osborne DR, Acuff SN, Stuckey A, Wall JS (2015) A routine PET/CT protocol with streamlined calculations for assessing cardiac amyloidosis using (18)F-florbetapir. Front Cardiovasc Med 2:23
Law WP, Wang WY, Moore PT, Mollee PN, Ng AC (2016) Cardiac amyloid imaging with 18F-florbetaben PET: a pilot study. J Nucl Med 57(11):1733–1739
Antoni G, Lubberink M, Estrada S, Axelsson J, Carlson K, Lindsjo L, Kero T, Langstrom B, Granstam SO, Rosengren S, Vedin O, Wassberg C, Wikstrom G, Westermark P, Sorensen J (2013) In vivo visualization of amyloid deposits in the heart with 11C-PIB and PET. J Nucl Med 54(2):213–220
Lee SP, Lee ES, Choi H, Im HJ, Koh Y, Lee MH, Kwon JH, Paeng JC, Kim HK, Cheon GJ, Kim YJ, Kim I, Yoon SS, Seo JW, Sohn DW (2015) 11C-Pittsburgh B PET imaging in cardiac amyloidosis. JACC Cardiovasc Imaging 8(1):50–59
Dorbala S, Falk R (2019) Imaging cardiac amyloidosis: Patient page. J Nucl Cardiol 26(1):217–221
Guy CD, Jones CK (2001) Abdominal fat pad aspiration biopsy for tissue confirmation of systemic amyloidosis: specificity, positive predictive value, and diagnostic pitfalls. Diagn Cytopathol 24(3):181–185
Fernandez de Larrea C, Verga L, Morbini P, Klersy C, Lavatelli F, Foli A, Obici L, Milani P, Capello GL, Paulli M, Palladini G, Merlini G (2015) A practical approach to the diagnosis of systemic amyloidoses. Blood 125(14):2239–2244
Satoskar AA, Efebera Y, Hasan A, Brodsky S, Nadasdy G, Dogan A, Nadasdy T (2011) Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am J Surg Pathol 35(11):1685–1690
Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, Dogan A (2009) Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 114(24):4957–4959
Muchtar E, Gertz MA, Kumar SK, Lin G, Boilson B, Clavell A, Lacy MQ, Buadi FK, Hayman SR, Kapoor P, Dingli D, Rajkumar SV, Dispenzieri A, Grogan M (2018) Digoxin use in systemic light-chain (AL) amyloidosis: contra-indicated or cautious use? Amyloid 25(2):86–92
Donnelly JP, Sperry BW, Gabrovsek A, Ikram A, Tang WHW, Estep J, Hanna M (2020) Digoxin use in cardiac amyloidosis. Am J Cardiol 133:134–138
Giancaterino S, Urey MA, Darden D, Hsu JC (2020) Management of Arrhythmias in Cardiac Amyloidosis. JACC Clin Electrophysiol 6(4):351–361
Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL (2018) Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Heart Fail 5(5):772–779
El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, Noseworthy PA, Lin G, Pislaru SV, Egbe AC, Grogan M, Nkomo VT (2019) Direct current cardioversion of atrial arrhythmias in adults with cardiac amyloidosis. J Am Coll Cardiol 73(5):589–597
Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, Syed IS, Hughes DA, Lust JA, Jaffe AS, Gertz MA, Klarich KW (2007) Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation 116(21):2420–2426
Mitrani LR, De Los Santos J, Helmke S, Biviano AB, Maurer MS (2019) Anticoagulation with warfarin versus novel oral anticoagulants in atrial fibrillation in amyloid transthyretin amyloidosis cardiomyopathy: a retrospective cohort study. J Cardiac Fail 25(8):S82
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ, E.S.C.S.D. Group (2015) ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 36(41):2793–2867
Donnellan E, Wazni OM, Saliba WI, Baranowski B, Hanna M, Martyn M, Patel D, Trulock K, Menon V, Hussein A, Aagaard P, Jaber W, Kanj M (2019) Cardiac devices in patients with transthyretin amyloidosis: Impact on functional class, left ventricular function, mitral regurgitation, and mortality. J Cardiovasc Electrophysiol 30(11):2427–2432
Donnellan E, Wazni OM, Hanna M, Kanj M, Saliba WI, Jaber WA (2020) Cardiac resynchronization therapy for transthyretin cardiac amyloidosis. J Am Heart Assoc 9(14):e017335
Castano A, Drachman BM, Judge D, Maurer MS (2015) Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 20(2):163–178
Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R (2012) Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 109(24):9629–9634
Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceicao IM, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Wilson A, Grogan DR (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 79(8):785–92
Merlini G, Coelho T, Waddington Cruz M, Li H, Stewart M, Ebede B (2020) Evaluation of mortality during long-term treatment with tafamidis for transthyretin amyloidosis with polyneuropathy: clinical trial results up to 8.5 years. Neurol Ther 9(1):105–115
Merlini G, Plante-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, Packman J, Tripp T, Grogan DR (2013) Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res 6(6):1011–1020
Barroso FA, Judge DP, Ebede B, Li H, Stewart M, Amass L, Sultan MB (2017) Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid 24(3):194–204
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C, Investigators A-AS (2018) Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379(11):1007–1016
Miller SR, Sekijima Y, Kelly JW (2004) Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest 84(5):545–552
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson-Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ, Diflunisal Trial C (2013) Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310(24):2658–67
Emdin M, Aimo A, Rapezzi C, Fontana M, Perfetto F, Seferovic PM, Barison A, Castiglione V, Vergaro G, Giannoni A, Passino C, Merlini G (2019) Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J 40(45):3699–3706
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS (2012) Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 18(6):315–319
Ikram A, Donnelly JP, Sperry BW, Samaras C, Valent J, Hanna M (2018) Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid 25(3):197–202
Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, Vellanki N, Hellawell J, Siddiqi OK, Fox J, Maurer MS, Ruberg FL (2019) Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. J Card Fail
Griffin JM, Maurer MS (2019) Transthyretin cardiac amyloidosis: a treatable form of heart failure with a preserved ejection fraction. Trends Cardiovasc Med
Cardoso I, Merlini G, Saraiva MJ (2003) 4’-iodo-4’-deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: screening for TTR fibril disrupters. FASEB J 17(8):803–809
Macedo B, Batista AR, Ferreira N, Almeida MR, Saraiva MJ (2008) Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of familial amyloidotic polyneuropathy. Biochim Biophys Acta 1782(9):517–522
Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ (2010) Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 8:74
Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, Perlini S, Saraiva MJ, Merlini G (2012) Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 19(Suppl 1):34–36
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba-Vitalo C, Nochur SV, Vaishnaw AK, Sah DW, Gollob JA, Suhr OB (2013) Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369(9):819–829
Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, Schmidt H, Waddington-Cruz M, Campistol JM, Bettencourt BR, Vaishnaw A, Gollob J, Adams D (2015) Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis 10:109
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB (2018) Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379(1):11–21
Solomon SD, Adams D, Kristen A, Grogan M, Gonzalez-Duarte A, Maurer MS, Merlini G, Damy T, Slama MS, Brannagan TH 3rd, Dispenzieri A, Berk JL, Shah AM, Garg P, Vaishnaw A, Karsten V, Chen J, Gollob J, Vest J, Suhr O (2019) Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 139(4):431–443
Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, Kim TW, Jesse Kwoh T, Matson J, Norris D, Yu R, Watt A, Monia BP (2016) Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid 23(3):148–157
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Plante-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceicao I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T (2018) Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 379(1):22–31
Dasgupta NR, Rissing SM, Smith J, Jung J, Benson MD (2020) Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid 27(1):52–58
Galant NJ, Bugyei-Twum A, Rakhit R, Walsh P, Sharpe S, Arslan PE, Westermark P, Higaki JN, Torres R, Tapia J, Chakrabartty A (2016) Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: a potential diagnostic and therapeutic for TTR amyloidoses. Sci Rep 6:25080
Miller M, Pal A, Albusairi W, Joo H, Pappas B, Haque Tuhin MT, Liang D, JampalaR Liu F, Khan J, Faaij M, Park M, Chan W, Graef I, Zamboni R, Kumar N, Fox J, Sinha U, Alhamadsheh M (2018) Enthalpy-driven stabilization of transthyretin by AG10 mimics a naturally occurring genetic variant that protects from transthyretin amyloidosis. J Med Chem 61(17):7862–7876
Hammarstrom P, Schneider F, Kelly JW (2001) Trans-suppression of misfolding in an amyloid disease. Science 293(5539):2459–2462
Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, Selby VN, Jacoby D, Hanna M, Nativi-Nicolau J, Patel J, Rao S, Sinha U, Turtle CW, Fox JC (2019) Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 74(3):285–295
Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, Andersen O, Karlberg I, Norden G, Nakazato M et al (1991) Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 40(3):242–246
Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, Furtado E, Barroso E, Daniel J, Samuel D, Adam R, Karam V, Poterucha J, Lewis D, Ferraz-Neto BH, Cruz MW, Munar-Ques M, Fabregat J, Ikeda S, Ando Y, Heaton N, Otto G, Suhr O (2015) Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation 99(9):1847–1854
Yazaki M, Mitsuhashi S, Tokuda T, Kametani F, Takei YI, Koyama J, Kawamorita A, Kanno H, Ikeda SI (2007) Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 7(1):235–242
Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, Danziger-Isakov L, Kirklin JK, Kirk R, Kushwaha SS, Lund LH, Potena L, Ross HJ, Taylor DO, Verschuuren EAM, Zuckermann A, International Society for Heart Lung Transplantation Infectious Diseases P, Heart F, Transplantation C (2016) The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: a 10-year update. J Heart Lung Transplant 35(1):1–23
Raichlin E, Daly RC, Rosen CB, McGregor CG, Charlton MR, Frantz RP, Clavell AL, Rodeheffer RJ, Pereira NL, Kremers WK, Kushwaha SS, Edwards BS (2009) Combined heart and liver transplantation: a single-center experience. Transplantation 88(2):219–225
Rosenbaum AN, AbouEzzeddine OF, Grogan M, Dispenzieri A, Kushwaha S, Clavell A, Daly RC, Edwards BS (2018) Outcomes after cardiac transplant for wild type transthyretin amyloidosis. Transplantation 102(11):1909–1913
Ridouani F, Damy T, Tacher V, Derbel H, Legou F, Sifaoui I, Audureau E, Bodez D, Rahmouni A, Deux JF (2018) Myocardial native T2 measurement to differentiate light-chain and transthyretin cardiac amyloidosis and assess prognosis. J Cardiovasc Magn Reson 20(1):58
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Bistola reports honoraria for lectures from Novartis, Pfizer, and Servier. Dr. Parissis reports horonaria for lectures and advisory boards from Novartis, Pfizer, Servier, Orion Pharma, and Roche Diagnostics. Dr. Kastritis has received honoraria for lectures and advisory boards from Amgen, Genesis Pharma, Janssen, Takeda, Pfizer, and research funding from Amgen and Janssen. Dr. Foukarakis reports honoraria for lectures and advisory boards from Pfizer, Menarini and Bayer. G. Koutsis has received research grants from Genesis Pharma and Teva, consultation fees, advisory boards, and honoraria from Genzyme, Genesis Pharma, Teva, and Novartis. Drs. Valsamaki, Anastasakis, and Efthimiadis report honoraria from Pfizer.
Consent for publication
All authors agree to publish the present manuscript in its current form.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bistola, V., Parissis, J., Foukarakis, E. et al. Practical recommendations for the diagnosis and management of transthyretin cardiac amyloidosis. Heart Fail Rev 26, 861–879 (2021). https://doi.org/10.1007/s10741-020-10062-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-020-10062-w