
Abstract
Cardiac fibrosis is associated with non-ischemic dilated cardiomyopathy, increasing its morbidity and mortality. Cardiac fibroblast is the keystone of fibrogenesis, being activated by numerous cellular and humoral factors. Macrophages, CD4+ and CD8+ T cells, mast cells, and endothelial cells stimulate fibrogenesis directly by activating cardiac fibroblasts and indirectly by synthetizing various profibrotic molecules. The synthesis of type 1 and type 3 collagen, fibronectin, and α-smooth muscle actin is rendered by various mechanisms like transforming growth factor-beta/small mothers against decapentaplegic pathway, renin angiotensin system, and estrogens, which in turn alter the extracellular matrix. Investigating the underlying mechanisms will allow the development of diagnostic and prognostic tools and discover novel specific therapies. Serum biomarkers aid in the diagnosis and tracking of cardiac fibrosis progression. The diagnostic gold standard is cardiac magnetic resonance with gadolinium administration that allows quantification of cardiac fibrosis either by late gadolinium enhancement assessment or by T1 mapping. Therefore, the goal is to stop and even reverse cardiac fibrosis by developing specific therapies that directly target fibrogenesis, in addition to the drugs used to treat heart failure. Cardiac resynchronization therapy had shown to revert myocardial remodeling and to reduce cardiac fibrosis. The purpose of this review is to provide an overview of currently available data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonischemic dilated cardiomyopathy (NIDCM) is the most common cardiomyopathy having multiple causes and a clinical picture that includes heart failure (HF) and even sudden cardiac death (SCD) [1]. NIDCM is defined as a left ventricle (LV) enlargement and global systolic function impairment (LVEF < 45%) in the absence of coronary artery disease (CAD) or increased loading conditions (hypertension, valve disease) [2]. Due to late diagnosis and poor prognosis, the current research trend is the better understanding of its pathogenesis and the development of more efficient early diagnosis techniques [3].
Cardiac fibrosis occurs early in the progression of NIDCM, increasing cardiac rigidity, decreasing myocardial performance, and enhancing the risk of SCD and malignant arrhythmias [4, 5]. Fibrogenesis is based on myofibroblasts that come from the activation of cardiac fibroblasts (CFs) or from epithelial-to-mesenchymal trans-differentiation (EMTd). Macrophages, monocytes, T lymphocytes, mast cells, and endothelial cells are also involved in this process. Myofibroblasts enhance the synthesis of collagen fibers, fibronectin, and profibrotic mediators by various molecular pathways, disrupting the normal extracellular matrix (ECM) by increasing its myocardial percentage [6,7,8,9]. Transforming growth factor beta (TGF-β), the mothers against decapentaplegic homolog 2 and 3 (SMAD2 and SMAD3) complex, the renin angiotensin aldosterone system (RAAS), estrogens, tumoral necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) are the mediators that orchestrate the profibrotic process [10,11,12,13,14]. Serum biomarkers involved in cardiac fibrosis correlate with collagen volume fraction (CVF), and some of them could be used for prognosis and follow-up. Collagen metabolism products, galectin-3 (Gal3), soluble source of tumorigenicity 2 (sST2), and growth differentiation factor 15 (GDF-15) have also a role in dynamic tracking of fibrosis. MicroRNAs (miRs) are useful in the diagnosis, assessment, and even treatment of cardiac fibrosis [15, 16]. Nowadays, cardiac magnetic resonance imaging (cMRI) with gadolinium as contrast agent represents the gold standard diagnosis and is based on T1-weighted late gadolinium enhancement (LGE) or T1 mapping (T1M) [17,18,19]. Major current interests are the inhibition of fibrogenesis and the induction of its reversibility. Drugs used in HF have been shown to reduce cardiac fibrosis, but new substances are emerging that target cardiac fibrosis in various pathways are emerging; some modulate collagen metabolism, while others influence some pathogenetic pathways, most commonly the TGF-β/SMADs pathway, or miRs. In addition, cardiac resynchronization therapy (CRT) is an option that seems to positively influence fibrosis, but further studies are required to establish the large-scale impact [20,21,22,23,24]. This review describes the latest data on the mechanisms and biomarkers of cardiac fibrosis in NIDCM subjects, advanced cMRI imaging techniques, as well as prevention and treatment opportunities in this entity.
Pathogenesis
Cardiac fibrosis is characterized by fibroblastic activation and fibrogenesis. It increases the collagen fiber synthesis that will embed into the myocardium altering its architecture and potentiating HF [11]. The keystone of the fibrogenesis is the activation of CFs. Depending on the location and ECM characteristics, cardiac fibrosis can be classified as reactive interstitial, infiltrative interstitial, and replacement fibrosis [6].
Cardiac fibrogenesis is a multistep process initiated in response to pro-inflammatory stimuli and cytokines. In myocardial injury, fibroblasts are activated and fibrogenesis is initiated leading to lack of excitation-contraction coupling with cardiac dysfunction. Besides CFs, macrophages, mast cells, lymphocytes, cardiomyocytes and vascular cells are involved in fibrogenesis [11].
Within the ECM, fibrillar collagen accumulation increases, contributing to diastolic dysfunction and activation of CFs. Nonfibrillar type VI collagen is also synthesized, yet with uncertain role in cardiac fibrogenesis. Fibrin and fibronectin are embedded in the ECM during myocardial inflammation. They facilitate CF migration, activation, and proliferation through integrins and syndecans. In addition, the matricellular protein synthesis is stimulated resulting in a newly formed ECM. These proteins are presented in Table 1 [6, 25].
Cells in cardiac fibrogenesis
CFs synthesize connective tissue adapted to the heart, which, unlike in bone or tendon, it is dense, irregular, and has a different composition. Most CFs originate from the epicardium, but there are also endocardium-derived CFs originating from interventricular septum and right ventricle (RV). It has been shown that the resident CFs are mainly responsible for cardiac fibrogenesis as a response to various stimuli [7].They recruit inflammatory cells, stimulate type I and type III collagen synthesis, determine the ECM degradation mediated by matrix-metalloproteinases (MMPs), but also its proliferation [26]. CFs are difficult to identify, but recently biomarkers with role in detecting them have been described. α-Smooth muscle actin (αSMA) plays important roles in modulating CF activity and proliferation, and also in scar contraction and ventricular remodeling [27]. Collagen 1A1 (Col1a1) and discoidin domain receptor 2 are both markers of angiotensin II-induced CF activation and collagen fiber proliferation, being involved in tissue scarring and fibrogenesis [26, 28]. Fibroblast-specific protein 1 is a nonspecific marker of activated fibroblasts being useful in quantifying fibrosis when used with other specific markers [29]. Periostin and transcription factor 21 (TCF21) occur in CFs development, subsequently modulating ECM synthesis and activating CFs [25, 26]. Platelet-derived growth factor receptor-α (PDGFR-α) activates myocardial resident CFs by stimulating receptor tyrosine kinase inhibitor pathway [30].
Myofibroblasts originate from activated CFs and circulating progenitors, endothelial cells, pericytes, and epicardial endothelial cells [6]. Up to 17% of total cardiac myofibroblasts are bone-marrow derived cells [31]. Pathological stimuli enhance TGF-β synthesis which in turn activates CFs through the exposure of type VI collagen. Cell surface integrins and syndecans determine the transduction of cellular trans-differentiation signals [6] and mechanical stress induces the synthesis of αSMA through Rho/Rho kinase pathway [32].
The monocytes/macrophages (Mo/Mf) differentiate into myofibroblasts in response to diverse cytokines and produce various inflammatory mediators and profibrotic growth factors. Alternative activation of M2-type macrophages via IL-3 and IL-4 is associated with cardiac fibrosis. Conversely, due to their ability to phagocyte cell debris and apoptotic myofibroblasts, recent data suggest a possible antifibrotic effect of Mo/Mf [6]. Mast cells (MCs) are identified in all forms of myocardial injury, these being especially involved in fibrogenesis initiation. They can directly activate myofibroblasts and enhance fibrogenesis through tryptase, histamine, TGF-β1, and chymase stimulation [8]. Activated lymphocytes such as CD8+ and Th1, Th2, Th17, and T-reg CD4+ subsets promote cardiac fibrogenesis, myocardial remodeling, and cardiac dysfunction. Particularly, Th1 cells activate CFs by stimulating TGF-β via integrin-α4 [9, 33]. Lymphocyte T depletion has also been shown to reduce cardiac fibrosis [34]. Endothelial cells (ECs) promote cardiac fibrosis by enhancing angiotensin II (AG II) induced profibrotic factors and inflammatory cytokines, and also by EMTd. Conversely, ECs can produce antifibrotic mediators such as chemokine interferon-γ-inducible protein-10 (IP-10) that inhibit CFs and hypoxia-inducible factor-1 which in turn deplete TGF-β levels [6]. In elevated myocardial pressure, a cardiomyocyte-specific TGF-β pathway is activated, generating myocardial remodeling [35] (Fig. 1).

Cellular involvement in cardiac fibrogenesis. Lymphocytes activate cardiac fibroblast via integrin-α4 and stimulate the production of transforming growth factor-β (TGF-β), while periostin and TCF-21 directly activate them. Endothelial cells and pericytes undergo Epithelial-mesenchymal trans-differentiation (EMTd) in myofibroblast. Macrophages and monocytes among with angiotensin II stimulate myofibroblasts. Finally, activated cardiac fibroblast and myofibroblasts produces collagen and syndecans
Humoral factors and molecular pathways involved in cardiac fibrosis
TGF-β is a cytokine with major roles in cardiac fibrogenesis in NIDCM subjects. The profibrotic effect occurs in case of prolonged exposure to elevated levels of TGF-β [11]. It enhances myofibroblast activation, MC migration, and cardiac hypertrophy and also inhibits type I T helper lymphocytes [10]. Through its receptors (TGF-βR), type I and type II TGF-β receptors, TGF-β activates SMAD2/3 pathways, stimulating alternative pathogenetic pathways and regulating cell synthesis and differentiation promoting fibrogenesis. They will bind to SMAD4 forming a complex that will internalize into the cell nucleus and initiate the fibrotic response. Conversely, profibrotic conditions are able to activate the AMP-activated protein kinase α pathway which can antagonize SMAD3, counteracting its profibrotic effects [6, 10, 36, 37] [37]. TGF-β also activates protein kinase B (AKT) via the Ras homolog gene family-member A-dependent pathway which in turn activates the SMAD2 pathway while also determining EMTd; thus, it renders the production of ECM [36]. The activation of type II TGF-βR, transforming growth factor beta-activated kinase 1 (TAK1) is potentiated which in turn disrupts the normal myocardial cells and increases fibrogenesis [11]. Extracellular regulated protein kinases (ERKs) are also upregulated, playing major roles in load-induced cardiac fibrosis [6].
AG II is an important oligopeptide that renders cardiac fibrosis regardless of etiology. It potentiates cardiac fibrogenesis by interacting with its receptors, angiotensin II receptor type I and type II (AGTR1 and AGTR2) and by enhancing TGF-β production [12]. AG II stimulates myofibroblast cell adhesion and ECM production. Alternatively, by activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), it stimulates the synthesis of type I collagen and connective tissue growth factors (CTGF) [38]. Therefore, the blockade of RAAS could reverse cardiac fibrosis [6]. Endothelin 1 (ET-1) may induce cardiac fibrosis by stimulating the proliferation and activation of CFs, respectively, by enhancing collagen synthesis, but further studies are required in order to define the exact mechanisms [11].
TNF-α enhances collagen production and CF activation. TNF-α is associated with TGF-β and in murine models, with fibrogenesis and early onset of NIDCM [11, 39]. Interleukin-6 (IL-6) determines cardiac fibrosis along with AG II, but further studies are required to correctly establish the mechanisms [13]. IL-1β potentiates myocardial inflammation, apoptosis, fibrosis, and myocardial dysfunction. It decreases calcium channel functionality, reduces intracellular levels of phospholamban and sarcoplasmic ATPase, increases levels of nitrous oxide system which in turn induces mitochondrial toxicity, and stimulates TGF-β synthesis [40].
Recent studies have shown the role of the several molecular pathways involved in cardiac fibrosis. Wnt/β-catenin pathway inhibits the destruction complex formed of axin, adenomatosis polyposis coli (APC), protein phosphatase 2a (PP2a), glycogen synthase kinase 3 (GSK3), and casein kinase 1α leading to β-catenin accumulation. It also enhances myofibroblast infiltration by stimulating CF activation and epithelial to mesenchymal differentiation, hence determines myocardial dysfunction and NIDCM [41]. Moreover, in murine subjects, the CFs β-catenin knockout reduced cardiac fibrosis by downregulating Col1a1, collagen 3A1 (Col3a1), and periostin [42]. Lastly, the activating protein-1 (AP-1) stimulates the production of fibronectin, collagen, intracellular and vascular cell adhesion molecules, potentiating inflammation and cardiacfibrosis [11] (Fig. 2).

Molecular mechanisms in cardiac fibroblasts. Transforming growth factor-β (TGF-β) through its specific receptors activate the mothers against decapentaplegic homolog 2 and 3 pathways (SMAD2/3) that binds with SMAD4 forming a complex that will stimulate cardiac fibroblast (CFs) activation, proliferation, collagen synthesis, and cell apoptosis. TGF-β activates ERK inducing the same effects as SMADs. TGF-β also enhances protein kinase B (AKT) via Ras homolog gene family, member A (RHOA) that stimulates epithelial-mesenchymal trans-differentiation (EMTd) and activates SMAD2. AG II through its receptors (AGTRs) enhances collagen synthesis and stimulates CTGF. Interleukin-1β disrupts calcium channels and decreases the production of phospholamban and sarcoplasmic ATP-ase and increases nitrous oxide systems (NOS). The Wnt creates a complex formed of axin, adenomatosis polyposis coli (APC), protein phosphatase 2a (PP2a), glycogen synthase kinase 3 (GSK3), and casein kinase 1α (CK-1α) that will enter the CFs and will stimulate β-cathenin synthesis
The male-female paradigm in cardiac fibrosis
The male-female difference in cardiovascular pathology is widely recognized. It has been observed that in women over 50 years, there is an overexpression of ECM with increased cardiac fibrosis. Corroborated data emphasizes that female subjects, both murine and human, are more protected from cardiac fibrosis due to estrogens, predominantly 17β-estradiol (E2) [14]. E2 acts through its cytosolic receptors (E2R), ERα and ERβ, either at the nuclear level (genomic pathway) or through the G protein-coupled receptor 30 (GPR30) receptors at the membrane level (nongenomic pathway) [43].
In this matter, various experimental studies have been conducted and concluded that estrogens exert antifibrotic effects on CFs through various mechanisms. In ovariectomized mice, the E2 and GPR30 G1 agonist were able to inhibit the effects of isoproterenol and the fibrosis induced by TGF-β [44, 45]. E2 was also able to prevent AG II and arterial hypertension induced fibrosis via ERβ by reducing the levels of TGF-β, collagen, and fibronectin [14]. E2 is able to block ERK signalization by reducing the synthesis of reactive oxygen species (ROS) and by inhibiting NF-κB and mitogen-activated protein kinase (MAPK) lowering the expression of αSMA [14]. Moreover, by interacting with cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA), ERβ is able to lower the levels of TGF-β and brake the activation of SMAD3, decreasing the expression of fibronectin, type I and type III collagen [46]. Even though the protective effects of estrogens on female CFs are clearly established, on male CFs, they are contradictory. In male CFs, studies have shown that E2 renders fibrogenesis by increasing the synthesis of ECM proteinsdue to male-specific posttranslational modifications of ERs [47]. Interestingly, the activation of GPR30 expressed in male CFs exerted antifibrotic effects by reducing the levels of αSMA, type I and type III collagens and fibronectin, but the underlying mechanisms are not fully elucidated and further studies are required in order to correctly identify them [48].
Cardiac fibrogenesis in NIDCM
In both familial and nonfamilial NIDCMs, cardiac fibrosis is a progressive process that occurs from the onset of the disease with significant consequences on prognosis and mortality. NIDCM induced by sarcomere proteins mutations involve most commonly myosin and troponin, determining cardiomyocytes’ degeneration and interstitial fibrosis [4]. The truncating titin mutation, the most common cause of familial NIDCM, disturbs the mitochondrial energetic metabolism and alters cell cytoskeleton, potentiating cardiomyocyte dysfunction and inflammation, resulting in interstitial myocardial fibrosis and increased risk of arrhythmogenesis [49]. While in subjects with lamin A/C mutations there is an increased production of fibronectin, syndecans, nidogen and decorin, with enhanced activation of TGF-β and SMAD2/3 [50], in subjects with SCN5A mutations, the sodium channel homeostasis is disturbed and calcium pumps are imbalanced, damaging the myocardium and promoting fibrogenesis [51]. In toxic NIDCM, fibrogenesis varies depending on the toxin and often is being identified in the phase IV studies. Recently, it has been shown that doxorubicin (DXO) is able to modify the structure of ECM, to inhibit the activity of topoisomerase IIβ and to stimulate the synthesis of ROS, potentiating cardiomyocyte apoptosis and fibrogenesis [52]. It is presumed that apoptosis could be also determined by the interaction of DXO with substance P and neurokinin receptor 1 (SP/NK-1R). Moreover, its profibrotic effects are due to CF activation and in chronic administration, it enhances the production of TGF-β, MMP-2, MMP-9, fibronectin, and col1α1 [52,53,54]. Arsenic trioxide is able to upregulate profibrotic genes, to potentiate EMTd through AKT/GSK-3/Snail pathway and to stimulate TGF-β resulting in cardiac fibrogenesis and QT prolongation [55, 56]. In patients treated with anti-CTLA4 (cytotoxic T lymphocyte-associated protein-4), the presence of myocardial fibrosis was identified, while in those treated with anti-PDL-1 (programmed death-ligand 1) agents, it was shown to stimulate pulmonary fibroblastic proliferation, but the cardiac effects have not yet been studied [57,58,59].
Regarding the illicit drugs, alcohol is able to enhances the synthesis of TGF-β and myostatin and determines ECM disorganization, apoptosis and cardiomyocyte necrosis, both directly and through its metabolism products, acetaldehyde adducts and lipid peroxidation [60, 61]. It is known that cocaine and methamphetamine are associated with myocardial fibrosis, but further studies are needed to elucidate the underlying mechanisms [62, 63]. Several infectious agents are able to enhance cardiac fibrogenesis in NIDCM. Through its direct cytopathic effect, coxsackievirus B3 determines fibrosis secondary to myocarditis [64]. It also increases the production of type I collagen and TGF-β, the activation of CFs and inflammatory cells, potentiating cardiac fibrosis [65, 66]. In HIV-positive subjects, cardiac fibrosis develops due to excessive accumulation of collagen, increased synthesis of TGF-β and other cytokines, activated ROS and decreased ECM degradation [67]. In addition, antiretroviral therapy enhances TGF-β/SMADs pathway potentiating cardiac fibrogenesis in HIV patients [68].
Among the metabolic disorders, diabetes mellitus (DM) is best correlated with cardiac fibrosis and cardiomyopathy. DM determines cardiac impairment and diabetic cardiomyopathy both by potentiating ischemia and by stimulating alternative nonischemic pathological processes. The main nonischemic mechanisms of cardiac fibrosis are due to hyperglycemia, advanced glycation end-product (AGEs), ROS and neurohumoral activation, CF proliferation with high levels of αSMA [69], myofibroblast activation, and EMTd. Neurohumoral activation and hyperglycemia are able to activate the synthesis of TGF-β, the RAAS, and ROS. AG II activates the SMAD pathway directly by increasing TGF-β synthesis, stimulates TGF-β activity through endoglin, and induces ROS gene. Recent studies have shown that in subjects with DM, there are increased levels of profibrotic cytokines such as TNF-α and IL-1β. AGEs interact with their own receptors (RAGEs) and increases collagen and laminin synthesis, activates CFs and macrophages, and enhances TGF-β and AG II activity [70]. Moreover, recent data emphasize that hyperglycemia activates calcium-sensing receptors (CaSR) increasing type I and type III collagen synthesis, stimulating CF proliferation and enhancing TGF-β/SMADs pathway, potentiating cardiac fibrosis [71]. However, due to the complex mechanisms by which DM promotes myocardial ischemia, the mechanisms cannot be completely separated, and this pathological entity must be considered as a complex of ischemic and nonischemic factors. All these are summarized in Table 2.
Assessment of cardiac fibrosis in NIDCM
Endomyocardial biopsy and pathological aspects
Although cMRI examination can accurately evaluate and quantify the degree of myocardial fibrosis, pathological examination of endomyocardial biopsy (EMB) samples is still considered the gold standard diagnostic tool. Histopathological examination (HPE) in the complete assessment of NIDCM with cardiac fibrosis includes the description of the endocardium, interstitium, and myocardium, as well as the quantification of the fibrotic load. In the heart affected by NIDCM, the endocardium can be thickened, infiltrated with inflammatory cells and CFs, as well as murine thrombi and subendocardial fibrosis can also be detected. Within the myocardium, modified cardiomyocytes, myocardial disruption and interstitial fibrosis are frequently described. Cardiomyocytes present abnormalities in terms of number, size, and shape, and also within their intracellular compounds. These cells present large variations, hypertrophic, atrophic, and normal cardiomyocytes being identified. While their cytoplasm present vacuolar degeneration, myocytosis, reduction in myofibrils, and various pigment accumulation, their nuclei have variable dimensions, irregular forms, and variable pleomorphism [72].The interstitial structure is the main site of cardiac fibrosis. Initially, acute inflammation occurs with interstitial edema and activated inflammatory cells, activating the production of collagen fibers and therefore rendering the definitive cardiac fibrosis. Intramural blood vessels are characterized by perivascular inflammatory infiltrate leading to thickened vascular wall reduced blood flow [73].
The essential histopathological parameters that are widely used in the evaluation of heart’s fibrotic load of the heart tissue is CVF and the two specific forms, namely type I collagen volume fraction (C1VF) and type III collagen volume (C3VF). This parameters are able to quantitatively determine the extension of myocardial fibrosis, but their cutoff values for fibrosis grading have not yet been established [74, 75]. In a recent clinical study conducted on 172 subjects with nonischemic HF, Aoki et al. identified that CVF, as a marker of severe myocardial fibrosis, acted as an significant predictor for all cause mortality and for cardiac events in subjects with nonischemic HF with reduced ejection fraction (HFrEF) [76]. Still, the current usage of EMB is mainly prohibited due to the chance of missing out the fibrotic areas and the increased risk of major cardiovascular complications.
Serum biomarkers of cardiac fibrosis in NIDCM
According to WHO, a biomarker represents any measurement that is able to demonstrate the interaction between a biological system and a potential hazard, being characterized by its relevance, viability and reproducibility [77]. The current research trend is to early identify cardiac fibrosis by noninvasive serum biomarkers. These substances may raise suspicion of cardiac fibrosis and some of them may be correlated with disease progression. Due to the fact that NIDCM is commonly associated with cardiac fibrosis, the identification and usage of such biomarkers are required. To evaluate the ability of a biomarker to identify cardiac fibrosis, its levels need to be compared with the gold standard diagnosis tool for cardiac fibrosis which is represented by CVF. Subsequently, the biomarker’s dynamics are evaluated in relation to fibrosis progression or regression. Numerous potential biomarkers have been studied, but only a few have proved their utility, namely type 1 procollagen C-terminal pro-peptide (PICP), amino-terminal pro-peptide of type III procollagen (PIIINCP), Gal3, sST2, GDF-15, and circulating miRs [15, 16].
PICP and PIIICNP
Being part of the heterotrimeric structure of type I collagen, PICP is a peptide released in the blood when type 1 collagen is polymerized in extracellular fibrils. It is considered to be a marker of type 1 collagen synthesis, fibroblastic activity, bone formation, and various pathological processes [78]. Recent clinical studies have confirmed the use of PICP in the evaluation of cardiac fibrosis. Serum levels of PICP have been significantly correlated with CVF in hypertensive subjects both with and without HF [75] and, besides, Lopez et al. have shown that in those with HF, PICP is alsocorrelated with C1VF [79]. In addition, these serum values decreased simultaneously with CVF values in subjects treated with antifibrotic drugs such as losartan and torasemide [75]. It has also been shown that spironolactone was able to reduce the myocardial fibrotic load confirmed both by CVF and serum PICP, in patients with NIDCM [80]. Regarding PIIINCP, it is synthesized when procollagen type III converts into collagen type III [74]. Plasma PIIINCP levels are also associated with CVF and C3VF in patients with NIDCM [75]. Moreover, PIIINCP has been positively correlated with ventricular dilation and inversely associated with the improvement of systolic function in NIDCM patients [81].
Gal3
Gal3 is a β-galactoside-binding protein which is found in various organs and tissues having important roles in cell adhesion, macrophage activation, angiogenesis, chronic inflammation, apoptosis, and fibrosis [82].
Experimental studies
Nowadays, more and more studies confirm the primary role of Gal3 in cardiac fibrogenesis. Calvier et al. have shown that Gal3 is associated with cardiac fibrosis, while its inhibition diminishes the progression of it [83]. In a murine study, MacKinnon et al. identified the profibrotic effects of Gal3, being able to activate myofibroblasts, to control the immune-inflammatory modulation and to enhance the TGF-β signaling. Conversely, they have shown that the inhibition of Gal3 was correlated with decreased fibrosis due to TGF-β drop and lack of fibroblastic activation [84]. Furthermore, Wu et al. have found that in animal model, Gal3 was correlated with E/e’ as a marker of diastolic dysfunction induced by aortic banding and they also identified 32% higher values of Gal3 in stretched cardiomyocytes compared to normal ones [85]. Besides, in a recent experimental study conducted on mouse cardiomyocyte cell culture, it has been identified that mechanical stress, protein kinase C and AG II render the activation of Gal3 and its profibrotic effects. They have also shown that in animal models with HF, Gal3 levels were increased along with the αSMA, actin, and type I collagen [86]. Strong evidence emphasize that the administration of Gal3 in the pericardium renders cardiac fibrogenesis by interacting with syndecans and activating local macrophages [87]. Similarly, Liu et al. identified that the pericardial administration of Gal3 in rats has important profibrotic effects is by enhancing the TGF-beta expression and SMAD3 phosphorylation [88]. An interesting study conducted by Martinez et al. have shown that in human CF cell cultures, cardiothropin-1 (CT-1) upregulated Gal3 along with αSMA, vimentin, CTGF, TGF-β, osteopontin, collagen type I, as well as the proinflammatory markers IL-6, monocyte chemotactic protein 1 (CCL-2), and IL-1β. Conversely, in Gal3-knocked down cells, CT-1 did not render the expression of vimentin, CTGF, osteopontin, collagen type I, nor IL-6 and CCL-2. Furthermore, in the same published study, the authors showed that in animal models, CT-1 enhanced the expression of Gal3 which in turn was correlated with perivascular fibrosis [89].
Clinical studies
The role of Gal3 in cardiac fibrogenesis, pulmonary hypertension, and HF in human subjects have recently confirmed in clinical studies [90]. In a study conducted on 3353 patients from Framingham Offspring Cohort, Ho et al. identified that Gal3 was associated with increased LV mass and enhanced risk of HF and also with all-cause mortality in this population [91]. Moreover, de Boer et al. have shown that in patients with HF, Gal3 is an independent marker for rehospitalization for HF and all-cause mortality, especially in those with HFpEF, and it is also associated with inflammatory biomarkers [92]. In a study conducted on 150 patients with NIDCM, Vergaro et al. identified a significant correlation between serum Gal3 and myocardial replacement fibrosis determined by cMRI-LGE. In their research, Gal3 expressed a cutoff value of 14.6 ng/mL with a moderate sensibility and specificity for predicting the presence of LGE in cMRI [93]. Furthermore, Martinez et al. confirmed the role of CT-1 in potentiating Gal3 levels and its profibrotic effects even in human subjects with HF [89].
sST2
ST2 is part of the interleukin 1 receptor family having two isoforms, one transmembrane (ST2L) and another soluble circulating (sST2), and are synthetized mainly by CFs and cardiomyocytes. It is now recognized that ST2 specifically interacts with IL-33 providing a dual effect. By the interaction of IL-33 with ST2L, it provides cardioprotective effects, while its interaction with sST2 competes with ST2L acting as a receptor decoy and antagonizing its beneficial cardiac effects [94].
Recent data emphasize the diagnostic and prognostic value of sST2 in patients with acute and advanced chronic HF [95]. It has been shown that sST2 is correlated with increased LV end-diastolic filling pressure (E/e’ ratio > 15), NYHA classification and NT-proBNP levels [16]. In a study conducted on 876 patients with chronic HF, Bayes-Genis et al. confirmed that sST2 was superior to Gal3 in terms of risk stratification [96]. Furthermore, Santhanakrishnan et al. identified that patients with HF, sST2 serum levels were significantly higher in subjects with HFpEF compared to normal subjects, but after adjustment for age, sex and other comorbidities the correlation did not persist [97]. When compared between subjects with HFpEF, the sST2 serum levels are lower in the group with normal LV diastolic filling pressure (E/e’ ratio < 8) as opposed to those with undeterminable (E/e’ ratio = 8–15) or increased LV diastolic filling pressure (E/e’ ratio > 15) with a cut off value higher than 13.5 ng/mL [98]. Furthermore, in a study, it is identified that the levels of sST2 were associated with elevated secondary pulmonary hypertension in patients with HF and chronic obstructive pulmonary disease, but further studies are required in order to establish the correct association between sST2 and HF [99].
Regarding the prognostics, more and more data confirm the role of sST2 in prognostic evaluation in patients with HF and NIDCM. A study indicates a cut off value for sST2 of 17.53 ng/mL for mortality prediction [100]. sST2’s mortality prediction capacity is confirmed in another study conducted in pediatric patients with NIDCM and moreover, it confirms its usefulness in monitoring the outcome [101].
GDF-15
GDF-15 is a cytokine part of the TGF-β superfamily with important implications in inflammation, metabolism, and cancer. More and more evidence emphasized the primary roles of GDF-15 in the development and progression of cardiovascular disease and underline its diagnostic and prognostic importance, but studies are still few and incipient. It has been shown that in some clinical studies, GDF-15 was able to increase the prognostic value of other biomarkers used for predicting cardiac mortality in patients with HF and also in identifying asymptomatic HF [102]. A recent study conducted by Wang et al. emphasized a similar diagnostic ability of GDF-15 with NT-proBNP in subjects with HFpEF, while serum levels of GDF-15 were superior to NT-proNBP in identifying the degree of HF even after adjusting for confounders [103]. In a small but interesting study conducted on 32 patients with NIDCM, Nair et al. have shown that GDF-15 was significantly correlated with MMPs and sST2 levels as markers of myocardial dysfunction and fibrosis. They have also shown that GDF-15 was positively associated with the NYHA class, NT-proBNP levels, LVEF and LV diastolic function [104]. Furthermore, GDF-15 was significantly associated with markers of fibrogenesis such as type I collagen carboxy-terminal peptide and PIIINCP. It is also correlated with E/e’ ratio, left atrial pressure, and inversely with LVEF. Thus, GDF-15 appears to play roles in cardiac fibrosis, myocardial remodeling, and cardiac dysfunction, but further studies are needed to elucidate underlying mechanisms [105].
Circulating miRs
miRs are fragments of noncoding RNAs that play a primary role in controlling gene expression through gene silencing, either by blocking translation or inducing degradation of messenger RNAs. Their malfunction may induce aberrant gene expression with important pathogenetic consequences [106]. miRs are stable molecules that can be determined in all the body fluids and can be used in diagnostic and therapeutic purposes. Recent data identified an important number of miRs with implications in the whole spectrum of cardiovascular pathology, but the research is in the beginning [107]. Many miRs are described in cardiac fibrogenesis, but the most studied are miR-21, miR-29, and miR-133. miR-21 is associated with myocardial remodeling and fibrosis.
In an experimental study conducted on rat CFs, Cao et al. have shown that miR-21 was able to activate CF proliferation by enhancing signal transducer and activator of transcription 3 (STAT3) pathway and in turn decreasing the cell adhesion molecule 1 (CADM1), thus promoting cardiac fibrogenesis [108]. Furthermore, in the study of Yuan et al., TGF-β rendered the overexpression of miR-21 in a dose-dependent manner, while miR-21 blockage inhibited the CF proliferation. In addition, they have also demonstrated that by directly blocking the inhibitory SMAD7 pathway, miR-21 was able to activate TGF-β and SMAD2/3 pathway enhancing the synthesis of αSMA and collagen fibers, thus promoting cardiac fibrogenesis [109]. Nevertheless, Brønnum et al. have shown in an experimental research that miR-21 was capable to inhibit the protein sprouty homolog 1 (SPRY1) pathway, an inhibitor of ERK/MAPK pathway and in turn to render EMTd and fibroblastic phenotype expression [110]. The role of miR-21 in both in ischemic and NIDCM have been recently confirmed by Li et al. [111], but further studies are required in order to correctly establish the underlying mechanisms and utility.
The roles of miR-29 in modulating cardiac fibrogenesis have recently been demonstrated, yet some data emphasize its dual role in this process. It has been hypothesized that miR-29 could promote cardiomyocyte apoptosis by decreasing the expression of several antiapoptotic genes and, conversely, to hinder the synthesis of collagen fibers preventing cardiac fibrogenesis [112]. In a recent murine study, Sassi et al. have demonstrated that the lack of miR-29 expression was able to prevent myocardial remodeling and, even more than that, exogenous miR-29 blockade was able to impede the synthesis of Col1a1, Collagen 1 A2 (Col1a2), Col3a1 and lower the degree of cardiac hypertrophy and fibrosis. They have also identified that miR-29 stimulated glycogen synthase kinase 3 beta (GSK3B) and other proteins that were involved in the Wnt pathway, thus promoting cardiac fibrogenesis [113].On the contrary, in their studies, Yamada et al. and Drummond et al. highlighted the antifibrotic potential effects of miR-29, both on cardiac and extracardiac fibrosis [114, 115]. In addition to these findings, Liang et al. have found that miR-29b3p and miR-29c3p were able to block TGF-β and MMP2, and in turn TGF-β via the SMAD3 pathway was able to inactivate miR-29 molecules [116]. The adverse outcomes may be explained by the possible different roles of the miR-29 subtypes, but further studies are needed to accurately identify them.
miR-30 is significantly associated with myocardial hypertrophy, and it is inversely correlated with CTGF, collagen, and fibrosis [117]; thus, it could become a potential biomarker for cardiac fibrosis. Recently, in a murine study that evaluated the effects of miR-30 after myocardial infarction, Chen et al. demonstrated that miR-30 may be able to reduce the deposition of type I and type III collagen by directly blocking CTFC, exerting beneficial effects on cardiac fibrosis [118]. Yet, further studies must be conducted to evaluate its potential role in fibrosis in patients with NIDCM. Another miR that share similar effects of blocking CTGF is miR-133, which is also associated with cardiac fibrosis. Angelini et al. identified that miR-133 is able to reduce the expression of CTGF through the serum response factor (SRF)/miR-133/CTGF axis. Moreover, the hyperstimulation of SRF blocks miR-133 and potentiates CTGF, while normal levels of SRF do not exert any effect on miR-133 molecules [119]. Recently, it has been shown that miR-133a could be capable to hinder cardiac fibrosis by blocking the AKT pathway and by regulating β-adrenergic receptor transduction signalization [120]. The latter is achieved by downregulating β-arrestin and (cAMP)-dependent protein kinase A [121]. In the murine study of Chen et al. conducted on mice with NIDCM, it was identified that miR-133a inversely correlates with several markers of cardiac fibrogenesis namely with fibronectin, FGF1, TGF-β, AG II, and ET-1. Also, it has been shown that the overexpression of miR-133a inhibited TGF-β/SMAD2 pathway preventing diabetes induced fibrosis [122].
A recent study emphasized that the dysregulations in miRs expression in NIDCM models were able to differentiate between the compensated and the decompensated NIDCM. miR-454, miR-500, and miR-1423p were downregulated, while miR-1246 was increased and all three miRs significantly correlated with diastolic dysfunction. Moreover, miR-1246 was associated with the levels of NT-proBNP levels and with E/e’ as a marker of diastolic dysfunction [123]. The use of miRs patterns has also been shown to increase diagnostic accuracy. For instance, the pattern miR-125a5p, miR-190a, miR-550a5p, and miR-638 was able to differentiate between HFpEF and HFrEF, while the pattern miR-1833p, miR-190a, miR-1933p, miR-1935p, miR-5455p differentiated HFpEF subjects from a healthy group [124]. Other two miRs capable to distinguish between HFrEF and HFpEF are miR-375 and miR-328. Furthermore, miR-375 proved increased predictive power when it was associated with NT-proBNP [125]. Combining these miRs patterns with HF markers can enhance diagnostic accuracy in subjects with NIDCM.
cMRI
cMRI is the noninvasive gold standard investigation that uses advanced imaging techniques and contrast agents to accurately assess myocardial fibrosis [17]. Using cine-imaging, chambers volumes and cardiac function are determined, while for the assessment of myocardial fibrosis, specific cMRI protocols have been developed [18]. LGE increased signal intensity in the inversion-recovery sequence can detect irreversible replacement fibrosis [126] (Fig. 3), while post-contrast T1-weighted mapping identifies diffuse interstitial fibrosis [127]. Myocardial tissue stores gadolinium depending on its pathogenetic status, and in turn, it accelerates the longitudinal relaxation time T1, changing its intensity. Local infusion, extracellular distribution, internal water exchanges, and gadolinium wash-in/wash-out are parameters that influence the final aspect of the images [19]. Gadolinium persists longer in the fibrotic area because the extracellular distribution is increased and the wash-out time is prolonged. Image acquisition is based on T1-weighted gradient echo that can be achieved within a single heartbeat by the fast and highly reproductible modified look-locker inversion-recovery (MOLLI) sequence a single heartbeat technique [128]. There is also the double-heartbeat technique that gives an impeccable contrast being preferred in NIDCM’s characterization, but the long apnea and acquisition periods makes it sometimes prohibitive. These acquisitions evaluate the extent, magnitude and pattern of fibrosis with a global and segmental characterization. Specific LGE patterns can differentiate between various cardiomyopathies, inflammatory or storage myocardial diseases. LGE-identified fibrosis correlates with histological changes, fibrosis biomarkers and can assess myocardial viability [18].

Cardiac magnetic resonance imaging in a patient with clinical nonischemic dilated cardiomyopathy. Midwall late gadolinium enhancement in present in the anterior wall (arrows) in a short axis view. Focal midwall late gadolinium enhancement is visible on the infero-lateral wall (arrows) in a short axis views, b 4-chamber views, and c 2-chamber CMR views. Cine-imaging presenting mitral regurgitation (arrow) in d 4-chamber views
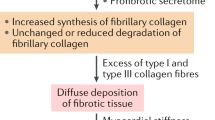
In NIDCM, three LGE patterns have been described, namely midwall, subendocardial, or transmural, which suggest a prior myocardial infarction, and the absence of LGE [129]. Linear or bandlike midwall LGE occur in 28–35% cases of NIDCM [129, 130] and corresponds to irreversible replacement fibrosis and fibro-fatty degeneration. In up to 59% of patients, histological fibrosis exists yet no LGE is being detected. This corresponds to diffuse interstitial fibrosis and can be unraveled when the null point method is used in LGE image acquisition. In previous reports, the presence of LGE was associated with major cardiac events and arrhythmias, but not with the cardiac function [131,132,133]. Particularly, myocardial LGE in NIDCM is associated with increased risk of hospitalization and mortality and low response rate to best medical therapy [126, 134]. Moreover, in LV midwall fibrosis (LVMWF), the global circumferential strain, strain rate, and torsion are reduced, and there is an increased risk of HFrEF [135]. In order to characterize diffuse cardiac fibrosis (DCF), the T1M technique was developed. DCF is caused by interstitial fibrosis which seizes the extracellular space, and the LGE technique is no longer able to identify it. T1M assesses the intrinsic T1 relaxation time of the myocardium, and combined with inversion pulses that suppress the tissue, it highlights the presence of gadolinium within the fibrotic areas by assessing the extracellular volume fraction (ECV) [136, 137]. In fact, T1M creates a myocardial map that is built from the intensity of the signals of each cardiac voxel. The MOLLI technique is the most used in T1M, but there are other procedural methods such as the short MOLLI (ShMOLLI) specifically designed to reduce the long apnea and acquisition time in patients with severe cardiac or respiratory disease [137]. T1M can accurately assess the degree of diffuse myocardial fibrosis, and in the future, typical fibrosis patterns can be created for various diseases. Studies have shown that the average precontrast T1 relaxation time is longer in the pathological myocardium. Also, in the presence of chronic scarring, it becomes significantly longer. Despite the extraordinary usefulness of the technique, it needs further studies to validate its clinical utility [126, 137]. There are other cMRI methods that evaluate myocardial fibrosis; some have not been validated for clinical usage, while others are under development. Equilibrium contrast cMRI is based on gadolinium bolus followed by continuous intravenous infusion in order to achieve humoral equilibrium, blood volume distribution determination, and T1-weighted measurements before and after equilibrium. This method was created for accurate assessment of diffuse myocardial fibrosis, but due to a complicated and complex acquisition protocol, it is not routinely used [138].
Current research trend aims to perfect the usage of cMRI-LGE in diagnosing and evaluating of cardiac fibrosis. The aforementioned LGE patterns have both diagnostic and prognostic value in patients with NIDCM. Besides the fact that LVMWF is able to exclude the ischemic etiology of DCM, in patients with NIDCM, it is associated with a 9-fold increased risk of SCD, ventricular tachycardia (VT), LV-assisted device requirement, cardiac transplantation, and 12-month mortality [5, 130, 134, 139]. Furthermore, a recent published study showed that subjects with LVEF > 30% and LV LGE > 5% are at similar risk as those with LVEF < 30%, and vice versa [140]. Recent data emphasize that cMRI-LGE could become useful in guiding CRT by identifying eligible patients more accurately [134]. A recent study has emphasized the ability of LGE-cMRI in assessing heterogeneous distributed fibrosis with important roles in predicting SCD and malignant arrhythmias in subjects with NIDCM, but further studies are needed in order to refine the technique [141]. Furthermore, a recently published meta-analysis that evaluated the prognostic value of cMRI-LGE on patients with NIDCM have shown that the presence of LGE seemed to be a strong predictor for cardiovascular events, particularly for malignant ventricular arrhythmias and HFrEF [17]. Current available data regarding cMRI-LGE and cardiac fibrosis in NIDCM are presented in Table 3 [139, 140, 142,143,144,145,146,147,148,149].
Fig. 3 cMRI in a patient with clinical NIDCM
Therapeutic approaches for cardiac fibrosis in NIDCM
Until recently, cardiovascular therapy in NIDCM targeted heart failure, arrhythmias, and support for systolic function. Current trend aims at reducing cardiac fibrosis, improving overall cardiac performance and cardiac resynchronization therapy, decreasing mortality, and increasing life quality.
Drugs
Pharmacological treatment represents the first line of therapy in NIDCM. ACCF/AHA and ESC recommend the use of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs), in association with β-blockers (βB) and aldosterone antagonists for improving LV dimension and function, reducing morbidity-mortality, but also reducing congestion symptoms [150, 151]. Furthermore, regarding cardiac fibrosis treatment, there are some studies that target some specific pathways and the results are promising, but further research is needed.
Recent studies have shown that ACEI and ARB could be able to inhibit AG II and TGF-β/SMADs pathway and to render the synthesis of N-acetyl seryl-aspartyl-lysyl-proline, a potent antifibrotic tetrapeptide, preventing myofibroblast activation and fibrogenesis [74, 152]. Moreover, βB, statins, and eplerenone have proven to exert antifibrotic effects [20]. Regarding specific antifibrotic therapy, direct inhibitors of TGF-β receptors such as GW 788388 and anti-TGF-β antibodies could block the activation of CFs; however, further studies are needed to strengthen such therapies [153]. TNF-α synthesis can also be blocked by p38 MAPK inhibitor [21]. Furthermore, being involved in CF adhesion and its inhibitors are able to mitigate renal fibrosis, periostin could become a potential cardiac antifibrotic target. Similarly, caveolin-1 could be a promising target, but studies are needed to evaluate this finding [154].
Modulating collagen synthesis is another way to influence cardiac fibrosis. Serelaxin, via relaxin-2 receptors, reduces inflammatory cells activation, apoptosis, ROS, and collagen synthesis, inhibiting fibrogenesis. It reduced the degree of dyspnea and mortality in those with acute HF [155]. In a murine study, Wu et al. identified that serelaxin could be able to inhibit TGF-β/SMADS pathway blocking the activation of CFs and to stimulate MMP2/TIMP2 ration enhancing ECM degradation [156]. Recently, it has been shown that polyphenols could block CTGF, TGF-β/SMADs pathway, ECM settlement, and NF-κB signaling; decrease TNF-α and ROS/ERK/TGF-β/periostin pathway; reduce ROS synthesis; and decrease Mo/Mf and MC infiltration. However, potency studies are needed to validate their clinical effects [157]. MMP inhibitors that stimulate collagen degradation could become promising therapeutic options, but the lack of their plasma stability and the insufficient data limit their use [74].
Certain miRs can be influenced either by miR mimicry and anti-miRs. The blockage of miR-21 could be able to reduce pressure-induced cardiac dysfunction and interstitial fibrosis, while antagonizing miR-208a could increase survival. The miR-29 mimicry could be capable ofTGF-β/SMADs pathway inhibition, reducing cardiac fibrogenesis [22, 74, 116]. Moreover, corroborated data have shown that decreased levels of miR-133 are correlated with increased levels of beclin-1, cardiac hypertrophy, HF severity, and inflammation, while miR-133 enhancement has beneficial effects. Interestingly, it has been shown that carvedilol could be able to upregulate miR-133, which could attenuate cardiomyocyte apoptosis [158]. In addition, low salt diet, adiponectin, and hydrogen sulfide could act as an exogenous and endogenous stimulator of miR-133. Targeting miRs is expanding, but significant studies are needed for the clinical validation of these therapies [120].
Other drugs have demonstrated their ability to reduce cardiac fibrosis. Rosiglitazone and pioglitazone reduce cardiac fibrosis through peroxisome proliferator-activated receptor-γ (PPARγ) and NF-κB, lowering CTGF levels. It also decreases myocardial remodeling by inhibiting RAAS [159]. Moreover, the combination of rosiglitazone and losartan attenuates interstitial fibrosis and collagen deposition by reducing TGF-β, TNFα, IL-1β, and IL-6 [160]. Imatinib, a tyrosine kinase inhibitor, blocks phosphorylation and activation of PDGFRs and thus inhibits profibrotic gene expression, type I and type III collagen deposition, decreases α-SMA expression, and reduces myocardial hypertrophy [161].These drug effects are summarized in Table 4.
Lastly, biomaterials can be used to obtain myocardial regeneration and ventricular stability, especially after acute myocardial injury. However, these methods could also be extrapolated to NIDCM. Thus, special polymers of myocardial support, implantation of special tissues containing active molecules are being tested. In a murine study conducted on canine models with ischemic HFrEF, Chaudhry et al. have shown that cardiac support device (CSD) developed by Acorn Cardiovascular Inc. that are wrapped around both LV and right ventricle (RV) could be able to normalize end-diastolic volume, reduce cardiac hypertrophy and cardiac fibrosis [162]. Even though the effects could be extrapolated to NIDCM, the results of clinical trials on CSD are questionable and yet the benefits are not fully proven. Hydrogels or cell-containing scaffolds may also be used in order to provide ventricular stability and render recovery [20].
Regarding the surgical treatment of NIDCM, several procedures such as Batista’s concept of reduction in LV wall tension by partial ventriculectomy have been tested, but with questionable results. In a clinical study conducted on five patients with NIDCM that underwent modified Dor’s procedure of endoventricular path plasty, Doenst et al. have shown that even though this procedure could be performed safely and could immediately improve cardiac function, the long-term results are questionable. Moreover, it cannot replace heart transplantation, but it could become an alternative to it [163]. In the study of Calafiore et al. conducted on 66 patients with ischemic DCM, NIDCM and post-valvular DCM, it has been shown that surgery performed in subjects with normal or moderately impaired RV function, pulmonary artery systolic pressure (PAPs) < 45 mmHg and elective surgery have better outcome than in those with severely impaired RV function, PAPs > 45 mmHg, necessity of intra-aortic balloon pump and/or inotropes [164]. Furthermore, in a study conducted on 56 subjects with NIDCM, Isomura et al. have demonstrated increased survival rate and better outcome due to intraoperative echocardiography in selecting operative procedure [165]. Recently, Suma et al. have found that in patients with NIDCM and HFrEF, NYHA class III/IV, septal akinesia and enlarged LV diastolic diameter, septal anterior ventricular exclusion with mitral reconstruction was associated with increased LVEF, decreased LV diastolic diameter, LV end-diastolic and end-systolic volumes, decrease in NTproBNP levels and in NYHA class [166]. Surgical treatment in NIDCM may be useful in certain situations, but it is grappling with important limitations due to the increased rate of periprocedural complications, high-risk interventions, reduced experience, and due to studies in small groups of patients.
CRT
CRT or biventricular pacing is a recently developed method of approaching patients with HFrEF and significant electrical and mechanical dyssynchrony [23]. NIDCM with HFrEF represent 43% of the patients with CRT [24]. This procedure is performed by placing two leads, one in RV at the apex and the other in LV through the coronary sinus at the lateral or posterolateral wall. Afterwards, the leads will synchronously stimulate both ventricles; thus, the electrical and mechanical dyssynchrony is solved and myocardial reverse remodeling is initiated. CRT is performed in patients with LVEF < 35%, electrical dyssynchrony with QRS complex > 120 ms with left bundle branch block and mechanical intraventricular, interventricular, and atrioventricular dyssynchrony [23].
Perspectives
The impact of CRT on cardiac fibrosis is being studied, but things are just at the beginning. In a recent murine study that evaluated the impact of CRT on myocardial fibrosis, Wang et al. have shown that CRT normalized QRS duration, reduced intraventricular dyssynchronism, and improved EF. Furthermore, they have shown that CRT was correlated with the reduction of myocardial fibrosis from LV quantified histologically by CVF, but insignificantly in RV, and was also associated with the reduction of TGF-β and SMAD2/3 pathway expression [167]. Thus, the early results are promising, but future studies are needed to accurately evaluate CRT on cardiac fibrosis in humans.
NIDCM is a severe form of primary myocardial disease characterized by progressive HF, with a poor prognosis, but still better than in ischemic HF. In patients with HFrEF, the 5-year-mortality rate exceeds 50%. Interestingly, in the study of Broch et al., patients with newly diagnosed NIDCM had better outcome when treated accordingly with the latest guidelines [168]. Furthermore, it is known that survival improvement in NIDCM patients are due to early diagnosis, heart transplantation, and HF management strategies [169]. Cardiac fibrosis is an important issue that should be evaluated promptly in each patient with NIDCM because it has a major impact on morbidity and mortality. Recent data certify that pathogenetic mechanisms in NIDCM stimulate various fibrogenesis pathways. However, some pathogenetic pathways are well explained, others require further studies to accurately identify them. Diagnosis by serum biomarkers begins to take shape and the use of miRs is a promising option for the future, yet validation studies are needed. Lately, EMB was replaced by cMRI with gadolinium administration having a high diagnostic accuracy. Given the consequences of cardiac fibrosis, the proper identification of therapeutic agents is another challenging aspect of the near future. There are drugs that exert antifibrotic effects and promising therapies targeting fibrosis are being developed, but things are still in the early beginning.
References
McKenna WJ, Maron BJ, Thiene G (2017) Classification, epidemiology, and global burden of cardiomyopathies. Circ Res 121:722–730. https://doi.org/10.1161/CIRCRESAHA.117.309711
Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, de Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio ALP, Charron P (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37:1850–1858. https://doi.org/10.1093/eurheartj/ehv727
Cecchi F, Tomberli B, Olivotto I (2012) Clinical and molecular classification of cardiomyopathies. Glob Cardiol Sci Pract 2012:4. https://doi.org/10.5339/gcsp.2012.4
Morita H, Seidman J, Seidman CE (2005) Genetic causes of human heart failure. J Clin Invest 115:518–526. https://doi.org/10.1172/JCI200524351
Venero JV, Doyle M, Shah M, Rathi VK, Yamrozik JA, Williams RB, Vido DA, Rayarao G, Benza R, Murali S, Glass J, Olson P, Sokos G, Biederman RWW (2015) Mid wall fibrosis on CMR with late gadolinium enhancement may predict prognosis for LVAD and transplantation risk in patients with newly diagnosed dilated cardiomyopathy-preliminary observations from a high-volume transplant centre. ESC Hear Fail 2:150–159. https://doi.org/10.1002/ehf2.12041
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71:549–574. https://doi.org/10.1007/s00018-013-1349-6
Ivey MJ, Tallquist MD (2016) Defining the cardiac fibroblast. Circ J 80:2269–2276. https://doi.org/10.1253/circj.CJ-16-1003
Legere SA, Haidl ID, Légaré J-F, Marshall JS (2019) Mast cells in cardiac fibrosis: new insights suggest opportunities for intervention. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.00580
Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P (2017) Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med 214:3311–3329. https://doi.org/10.1084/jem.20161791
Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee S-J, Karch J, Molkentin JD (2017) Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127:3770–3783. https://doi.org/10.1172/JCI94753
Ma Z-G, Yuan Y-P, Wu H-M, Zhang X, Tang Q-Z (2018) Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci 14:1645–1657. https://doi.org/10.7150/ijbs.28103
Jia G, Aroor AR, Hill MA, Sowers JR (2018) Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 72:537–548. https://doi.org/10.1161/HYPERTENSIONAHA.118.11065
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J (2012) Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One 7:e35144. https://doi.org/10.1371/journal.pone.0035144
Medzikovic L, Aryan L, Eghbali M (2019) Connecting sex differences, estrogen signaling, and microRNAs in cardiac fibrosis. J Mol Med. https://doi.org/10.1007/s00109-019-01833-6
Richards AM (2017) Circulating biomarkers of cardiac fibrosis. Circ Heart Fail 10. https://doi.org/10.1161/CIRCHEARTFAILURE.117.003936
Michalska-Kasiczak M, Bielecka-Dabrowa A, von Haehling S, Anker SD, Rysz J, Banach M (2018) Biomarkers, myocardial fibrosis and co-morbidities in heart failure with preserved ejection fraction: an overview. Arch Med Sci 14:890–909. https://doi.org/10.5114/aoms.2018.76279
Becker MAJ, Cornel JH, van de Ven PM, van Rossum AC, Allaart CP, Germans T (2018) The prognostic value of late gadolinium-enhanced cardiac magnetic resonance imaging in nonischemic dilated cardiomyopathy. JACC Cardiovasc Imaging 11:1274–1284. https://doi.org/10.1016/j.jcmg.2018.03.006
Valbuena-López S, Hinojar R, Puntmann VO (2016) Cardiovascular magnetic resonance in cardiology practice: a concise guide to image acquisition and clinical interpretation. Rev Española Cardiol English Ed 69:202–210. https://doi.org/10.1016/j.rec.2015.11.011
Croisille P, Revel D, Saeed M (2006) Contrast agents and cardiac MR imaging of myocardial ischemia: from bench to bedside. Eur Radiol 16:1951–1963. https://doi.org/10.1007/s00330-006-0244-z
Hinderer S, Schenke-Layland K (2019) Cardiac fibrosis – a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2019.05.011
Fan Z, Guan J (2016) Antifibrotic therapies to control cardiac fibrosis. Biomater Res 20:13. https://doi.org/10.1186/s40824-016-0060-8
Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E (2011) Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 124:1537–1547. https://doi.org/10.1161/CIRCULATIONAHA.111.030932
Jaffe LM, Morin DP (2014) Cardiac resynchronization therapy: history, present status, and future directions. Ochsner J 14:596–607
Zusterzeel R, Curtis JP, Caños DA, Sanders WE, Selzman KA, Piña IL, Spatz ES, Bao H, Ponirakis A, Varosy PD, Masoudi FA, Strauss DG (2014) Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 64:887–894. https://doi.org/10.1016/j.jacc.2014.06.1162
Landry NM, Cohen S, Dixon IMC (2018) Periostin in cardiovascular disease and development: a tale of two distinct roles. Basic Res Cardiol 113:1. https://doi.org/10.1007/s00395-017-0659-5
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC (2016) Cardiac fibrosis. Circ Res 118:1021–1040. https://doi.org/10.1161/CIRCRESAHA.115.306565
Shinde AV, Humeres C, Frangogiannis NG (2017) The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol basis Dis 1863:298–309. https://doi.org/10.1016/j.bbadis.2016.11.006
DeLeon-Pennell KY (2016) May the fibrosis be with you: is discoidin domain receptor 2 the receptor we have been looking for? J Mol Cell Cardiol 91:201–203. https://doi.org/10.1016/j.yjmcc.2016.01.006
Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG (2013) Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Circ Physiol 305:H1363–H1372. https://doi.org/10.1152/ajpheart.00395.2013
Wang L, Yue Y, Yang X, Fan T, Mei B, Hou J, Liang M, Chen G, Wu Z (2017) Platelet derived growth factor alpha (PDGFRα) induces the activation of cardiac fibroblasts by activating c-kit. Med Sci Monit 23:3808–3816. https://doi.org/10.12659/MSM.906038
Chu P-Y, Mariani J, Finch S, McMullen JR, Sadoshima J, Marshall T, Kaye DM (2010) Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. Am J Pathol 176:1735–1742. https://doi.org/10.2353/ajpath.2010.090574
Zhao X-H, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA (2007) Force activates smooth muscle -actin promoter activity through the Rho signaling pathway. J Cell Sci 120:1801–1809. https://doi.org/10.1242/jcs.001586
Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD (2017) Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Hear Fail:10. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003688
Abdullah CS, Li Z, Wang X, Jin Z-Q (2016) Depletion of T lymphocytes ameliorates cardiac fibrosis in streptozotocin-induced diabetic cardiomyopathy. Int Immunopharmacol 39:251–264. https://doi.org/10.1016/j.intimp.2016.07.027
Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA (2011) Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121:2301–2312. https://doi.org/10.1172/JCI44824
Yue Y, Meng K, Pu Y, Zhang X (2017) Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract 133:124–130. https://doi.org/10.1016/j.diabres.2017.08.018
Mishra R, Cool BL, Laderoute KR, Foretz M, Viollet B, Simonson MS (2008) AMP-activated protein kinase inhibits transforming growth factor-β-induced Smad3-dependent transcription and Myofibroblast Transdifferentiation. J Biol Chem 283:10461–10469. https://doi.org/10.1074/jbc.M800902200
Wei C, Kim I-K, Kumar S, Jayasinghe S, Hong N, Castoldi G, Catalucci D, Jones WK, Gupta S (2013) NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol 228:1433–1442. https://doi.org/10.1002/jcp.24296
Duerrschmid C, Trial J, Wang Y, Entman ML, Haudek SB (2015) Tumor necrosis factor. Circ Heart Fail 8:352–361. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001893
Szekely Y, Arbel Y (2018) A review of interleukin-1 in heart disease: where do we stand today? Cardiol Ther 7:25–44. https://doi.org/10.1007/s40119-018-0104-3
Lluri G, Deb A (2019) WNT signaling and cardiac fibrosis. Pp 319–334
Xiang F-L, Fang M, Yutzey KE (2017) Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun 8:712. https://doi.org/10.1038/s41467-017-00840-w
Menazza S, Murphy E (2016) The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res 118:994–1007. https://doi.org/10.1161/CIRCRESAHA.115.305376
Kang S, Liu Y, Sun D, Zhou C, Liu A, Xu C, Hao Y, Li D, Yan C, Sun H (2012) Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One 7:e48185. https://doi.org/10.1371/journal.pone.0048185
Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L (2012) Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res 94:96–104. https://doi.org/10.1093/cvr/cvs090
Pedram A, Razandi M, Narayanan R, Levin ER (2016) Estrogen receptor beta signals to inhibition of cardiac fibrosis. Mol Cell Endocrinol 434:57–68. https://doi.org/10.1016/j.mce.2016.06.018
Dworatzek E, Mahmoodzadeh S, Schriever C, Kusumoto K, Kramer L, Santos G, Fliegner D, Leung Y-K, Ho S-M, Zimmermann W-H, Lutz S, Regitz-Zagrosek V (2019) Sex-specific regulation of collagen I and III expression by 17β-estradiol in cardiac fibroblasts: role of estrogen receptors. Cardiovasc Res 115:315–327. https://doi.org/10.1093/cvr/cvy185
Wang H, Zhao Z, Lin M, Groban L (2015) Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem 405:135–148. https://doi.org/10.1007/s11010-015-2405-3
Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiarán Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca H-P, Brunner HG, Krapels IPC, Heymans SRB (2018) Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 39:864–873. https://doi.org/10.1093/eurheartj/ehx808
Chatzifrangkeskou M, Le Dour C, Wu W, Morrow JP, Joseph LC, Beuvin M, Sera F, Homma S, Vignier N, Mougenot N, Bonne G, Lipson KE, Worman HJ, Muchir A (2016) ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum Mol Genet 25:2220–2233. https://doi.org/10.1093/hmg/ddw090
Li W, Yin L, Shen C, Hu K, Ge J, Sun A (2018) SCN5A variants: association with cardiac disorders. Front Physiol 9. https://doi.org/10.3389/fphys.2018.01372
Levick SP, Soto-Pantoja DR, Bi J, Hundley WG, Widiapradja A, Manteufel EJ, Bradshaw TW, Meléndez GC (2018) Doxorubicin-induced myocardial fibrosis involves the neurokinin-1 receptor and direct effects on cardiac fibroblasts. Hear Lung Circ. https://doi.org/10.1016/j.hlc.2018.08.003
Robinson P, Kasembeli M, Bharadwaj U, Engineer N, Eckols KT, Tweardy DJ (2016) Substance P receptor signaling mediates doxorubicin-induced cardiomyocyte apoptosis and triple-negative breast cancer chemoresistance. Biomed Res Int 2016:1–9. https://doi.org/10.1155/2016/1959270
El-Agamy DS, El-Harbi KM, Khoshhal S, Ahmed N, Elkablawy MA, Shaaban AA, Abo-Haded HM (2018) Pristimerin protects against doxorubicin-induced cardiotoxicity and fibrosis through modulation of Nrf2 and MAPK/NF-kB signaling pathways. Cancer Manag Res Volume 11:47–61. https://doi.org/10.2147/CMAR.S186696
Chu W, Li C, Qu X, Zhao D, Wang X, Yu X, Cai F, Liang H, Zhang Y, Zhao X, Li B, Qiao G, Dong D, Lu Y, Du Z, Yang B (2012) Arsenic-induced interstitial myocardial fibrosis reveals a new insight into drug-induced long QT syndrome. Cardiovasc Res 96:90–98. https://doi.org/10.1093/cvr/cvs230
Zhang Y, Wu X, Li Y, Zhang H, Li Z, Zhang Y, Zhang L, Ju J, Liu X, Chen X, Glybochko PV, Nikolenko V, Kopylov P, Xu C, Yang B (2016) Endothelial to mesenchymal transition contributes to arsenic-trioxide-induced cardiac fibrosis. Sci Rep 6:33787. https://doi.org/10.1038/srep33787
Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, Pauschinger M, Gajewski TF, Lipson EJ, Luke JJ (2016) Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J Immunother Cancer 4:50. https://doi.org/10.1186/s40425-016-0152-y
Geng Y, Liu X, Liang J, Habiel DM, Vrishika K, Coelho AL, Deng N, Xie T, Wang Y, Liu N, Huang G, Kurkciyan A, Liu Z, Tang J, Hogaboam CM, Jiang D, Noble PW (2019) PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight. https://doi.org/10.1172/jci.insight.125326
Delgobo M, Frantz S (2018) Heart failure in cancer: role of checkpoint inhibitors. J Thorac Dis 10:S4323–S4334. https://doi.org/10.21037/jtd.2018.10.07
Maisch B (2016) Alcoholic cardiomyopathy. Herz 41:484–493. https://doi.org/10.1007/s00059-016-4469-6
Fernández-Solà J, Planavila Porta A (2016) New treatment strategies for alcohol-induced heart damage. Int J Mol Sci 17:1651. https://doi.org/10.3390/ijms17101651
Havakuk O, Rezkalla SH, Kloner RA (2017) The cardiovascular effects of cocaine. J Am Coll Cardiol 70:101–113. https://doi.org/10.1016/j.jacc.2017.05.014
Paratz ED, Cunningham NJ, MacIsaac AI (2016) The cardiac complications of methamphetamines. Hear Lung Circ 25:325–332. https://doi.org/10.1016/j.hlc.2015.10.019
Tschöpe C, Müller I, Xia Y, Savvatis K, Pappritz K, Pinkert S, Lassner D, Heimesaat MM, Spillmann F, Miteva K, Bereswill S, Schultheiss H-P, Fechner H, Pieske B, Kühl U, Van Linthout S (2017) NOD2 (nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circ hear fail 10. https://doi.org/10.1161/CIRCHEARTFAILURE.117.003870
Cao Y, Xu W, Xiong S (2013) Adoptive transfer of regulatory T cells protects against coxsackievirus B3-induced cardiac fibrosis. PLoS One 8:e74955. https://doi.org/10.1371/journal.pone.0074955
Chen P, Xie Y, Shen E, Li GG, Yu Y, Zhang CB, Yang Y, Zou Y, Ge J, Chen R, Chen H (2011) Astragaloside IV attenuates myocardial fibrosis by inhibiting TGF-β1 signaling in coxsackievirus B3-induced cardiomyopathy. Eur J Pharmacol 658:168–174. https://doi.org/10.1016/j.ejphar.2011.02.040
Hsue PY, Tawakol A (2016) Inflammation and fibrosis in HIV. Circ Cardiovasc Imaging 9. https://doi.org/10.1161/CIRCIMAGING.116.004427
Laurence J, Elhadad S, Robison T, Terry H, Varshney R, Woolington S, Ghafoory S, Choi ME, Ahamed J (2017) HIV protease inhibitor-induced cardiac dysfunction and fibrosis is mediated by platelet-derived TGF-β1 and can be suppressed by exogenous carbon monoxide. PLoS One 12:e0187185. https://doi.org/10.1371/journal.pone.0187185
Fowlkes V, Clark J, Fix C, Law BA, Morales MO, Qiao X, Ako-Asare K, Goldsmith JG, Carver W, Murray DB, Goldsmith EC (2013) Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci 92:669–676. https://doi.org/10.1016/j.lfs.2013.01.003
Russo I, Frangogiannis NG (2016) Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 90:84–93. https://doi.org/10.1016/j.yjmcc.2015.12.011
Yuan H, Fan Y, Wang Y, Gao T, Shao Y, Zhao B, Li H, Xu C, Wei C (2019) Calcium-sensing receptor promotes high glucose-induced myocardial fibrosis via upregulation of the TGF-β1/Smads pathway in cardiac fibroblasts. Mol Med Rep. https://doi.org/10.3892/mmr.2019.10330
Mitrut R, Stepan AE, Pirici D Histopathological aspects of the myocardium in dilated cardiomyopathy. Curr Heal Sci J 44:243–249. https://doi.org/10.12865/CHSJ.44.03.07
Cunningham KS (2006) An approach to endomyocardial biopsy interpretation. J Clin Pathol 59:121–129. https://doi.org/10.1136/jcp.2005.026443
Liu T, Song D, Dong J, Zhu P, Liu J, Liu W, Ma X, Zhao L, Ling S (2017) Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front Physiol 8. https://doi.org/10.3389/fphys.2017.00238
López B, González A, Ravassa S, Beaumont J, Moreno MU, San José G, Querejeta R, Díez J (2015) Circulating biomarkers of myocardial fibrosis. J Am Coll Cardiol 65:2449–2456. https://doi.org/10.1016/j.jacc.2015.04.026
Aoki T, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, Nakayama M, Shimokawa H (2011) Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. Circ J 75:2605–2613. https://doi.org/10.1253/circj.CJ-11-0568
Strimbu K, Tavel JA (2010) What are biomarkers? Curr Opin HIV AIDS 5:463–466. https://doi.org/10.1097/COH.0b013e32833ed177
Seo W-Y, Kim J-H, Baek D-S, Kim S-J, Kang S, Yang WS, Song J-A, Lee M-S, Kim S, Kim Y-S (2017) Production of recombinant human procollagen type I C-terminal propeptide and establishment of a sandwich ELISA for quantification. Sci Rep 7:15946. https://doi.org/10.1038/s41598-017-16290-9
López B, Querejeta R, González A, Larman M, Díez J (2012) Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure. Hypertension 60:677–683. https://doi.org/10.1161/HYPERTENSIONAHA.112.196113
Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, Ichihara S, Kato T, Ohshima S, Murase Y, Iino S, Obata K, Noda A, Okumura K, Yokota M (2005) Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy. Circulation 112:2940–2945. https://doi.org/10.1161/CIRCULATIONAHA.105.571653
Kaufman BD, Videon N, Zhang X, Harris MA, Shaddy RE, Goldmuntz E (2015) Procollagen type III amino-terminal propeptide: a serum biomarker of left ventricular remodelling in paediatric dilated cardiomyopathy. Cardiol Young 25:228–236. https://doi.org/10.1017/S1047951113001820
Sciacchitano S, Lavra L, Morgante A, Ulivieri A, Magi F, De Francesco G, Bellotti C, Salehi L, Ricci A (2018) Galectin-3: one molecule for an alphabet of diseases, from A to Z. Int J Mol Sci 19:379. https://doi.org/10.3390/ijms19020379
Calvier L, Martinez-Martinez E, Miana M, Cachofeiro V, Rousseau E, Sádaba JR, Zannad F, Rossignol P, López-Andrés N (2015) The impact of Galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Hear Fail 3:59–67. https://doi.org/10.1016/j.jchf.2014.08.002
MacKinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T (2012) Regulation of transforming growth factor-β1–driven lung fibrosis by galectin-3. Am J Respir Crit Care Med 185:537–546. https://doi.org/10.1164/rccm.201106-0965OC
Wu C-K, Su M-Y, Lee J-K, Chiang F-T, Hwang J-J, Lin J-L, Chen J-J, Liu F-T, Tsai C-T (2015) Galectin-3 level and the severity of cardiac diastolic dysfunction using cellular and animal models and clinical indices. Sci Rep 5:17007. https://doi.org/10.1038/srep17007
Song X, Qian X, Shen M, Jiang R, Wagner MB, Ding G, Chen G, Shen B (2015) Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochim Biophys Acta, Mol Cell Res 1853:513–521. https://doi.org/10.1016/j.bbamcr.2014.12.001
Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu F-T, de Boer RA (2018) Galectin-3 activation and inhibition in heart failure and cardiovascular disease: an update. Theranostics 8:593–609. https://doi.org/10.7150/thno.22196
Liu Y-H, D’Ambrosio M, Liao T, Peng H, Rhaleb N-E, Sharma U, André S, Gabius H-J, Carretero OA (2009) N -acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Circ Physiol 296:H404–H412. https://doi.org/10.1152/ajpheart.00747.2008
Martínez-Martínez E, Brugnolaro C, Ibarrola J, Ravassa S, Buonafine M, López B, Fernández-Celis A, Querejeta R, Santamaria E, Fernández-Irigoyen J, Rábago G, Moreno MU, Jaisser F, Díez J, González A, López-Andrés N (2019) CT-1 (cardiotrophin-1)-Gal-3 (galectin-3) axis in cardiac fibrosis and inflammation. Hypertension 73:602–611. https://doi.org/10.1161/HYPERTENSIONAHA.118.11874
Agoston-Coldea L, Bheecarry K, Petra C, Strambu L, Ober C, Revnic R, Lupu S, Mocan T, Fodor D (2018) The value of global longitudinal strain and galectin-3 for predicting cardiovascular events in patients with severe aortic stenosis. Med Ultrason 20:205. https://doi.org/10.11152/mu-1456
Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D (2012) Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 60:1249–1256. https://doi.org/10.1016/j.jacc.2012.04.053
de Boer RA, Lok DJA, Jaarsma T, van der Meer P, Voors AA, Hillege HL, van Veldhuisen DJ (2011) Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann Med 43:60–68. https://doi.org/10.3109/07853890.2010.538080
Vergaro G, Del Franco A, Giannoni A, Prontera C, Ripoli A, Barison A, Masci PG, Aquaro GD, Cohen Solal A, Padeletti L, Passino C, Emdin M (2015) Galectin-3 and myocardial fibrosis in nonischemic dilated cardiomyopathy. Int J Cardiol 184:96–100. https://doi.org/10.1016/j.ijcard.2015.02.008
Villacorta H, Maisel AS (2015) Soluble ST2 testing: a promising biomarker in the management of heart failure. Arq Bras Cardiol. https://doi.org/10.5935/abc.20150151
Lupu S, Agoston-Coldea L (2015) Soluble ST2 in ventricular dysfunction. Pp 139–159
Bayes-Genis A, de Antonio M, Vila J, Peñafiel J, Galán A, Barallat J, Zamora E, Urrutia A, Lupón J (2014) Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification. J Am Coll Cardiol 63:158–166. https://doi.org/10.1016/j.jacc.2013.07.087
Santhanakrishnan R, Chong JPC, Ng TP, Ling LH, Sim D, Leong KT, Yeo PS, Ong HY, Jaufeerally F, Wong R, Chai P, Low AF, Richards AM, Lam CSP (2012) Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail 14:1338–1347. https://doi.org/10.1093/eurjhf/hfs130
Wang Y-C, Yu C-C, Chiu F-C, Tsai C-T, Lai L-P, Hwang J-J, Lin J-L (2013) Soluble ST2 as a biomarker for detecting stable heart failure with a Normal ejection fraction in hypertensive patients. J Card Fail 19:163–168. https://doi.org/10.1016/j.cardfail.2013.01.010
Agoston-Coldea L, Lupu S, Hicea S, Paradis A, Mocan T (2014) Serum levels of the soluble IL-1 receptor family member ST2 and right ventricular dysfunction. Biomark Med 8:95–106. https://doi.org/10.2217/bmm.13.116
Wojciechowska C, Romuk E, Nowalany-Kozielska E, Jacheć W (2017) Serum galectin-3 and ST2 as predictors of unfavorable outcome in stable dilated cardiomyopathy patients. Hell J Cardiol 58:350–359. https://doi.org/10.1016/j.hjc.2017.03.006
You H, Jiang W, Jiao M, Wang X, Jia L, You S, Li Y, Wen H, Jiang H, Yuan H, Huang J, Qiao B, Yang Y, Jin M, Wang Y, Du J (2019) Association of soluble ST2 serum levels with outcomes in pediatric dilated cardiomyopathy. Can J Cardiol 35:727–735. https://doi.org/10.1016/j.cjca.2019.02.016
Desmedt S, Desmedt V, De Vos L, Delanghe JR, Speeckaert R, Speeckaert MM (2019) Growth differentiation factor 15: a novel biomarker with high clinical potential. Crit Rev Clin Lab Sci 56:333–350. https://doi.org/10.1080/10408363.2019.1615034
Wang F, Guo Y, Yu H, Zheng L, Mi L, Gao W (2010) Growth differentiation factor 15 in different stages of heart failure: potential screening implications. Biomarkers 15:671–676. https://doi.org/10.3109/1354750X.2010.510580
Nair N, Gongora E (2018) Correlations of GDF-15 with sST2, MMPs, and worsening functional capacity in idiopathic dilated cardiomyopathy. J Circ Biomarkers 7:184945441775173. https://doi.org/10.1177/1849454417751735
Wang F-F, Chen B-X, Yu H-Y, Mi L, Li Z-J, Gao W (2016) Correlation between growth differentiation factor-15 and collagen metabolism indicators in patients with myocardial infarction and heart failure. J Geriatr Cardiol 13:88–93. https://doi.org/10.11909/j.issn.1671-5411.2016.01.002
Cui J, Zhou B, Ross SA, Zempleni J (2017) Nutrition, microRNAs, and human health. Adv Nutr An Int Rev J 8:105–112. https://doi.org/10.3945/an.116.013839
Huang W (2017) MicroRNAs: biomarkers, diagnostics, and therapeutics. Pp 57–67
Cao W, Shi P, Ge J-J (2017) miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc Disord 17:88. https://doi.org/10.1186/s12872-017-0520-7
Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y, Gu M, Zhou Y, Zhu J, Ge T, Chen Q, Gao Y, Wang Y, Li X, Zhao Y (2017) Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem 42:2207–2219. https://doi.org/10.1159/000479995
Brønnum H, Andersen DC, Schneider M, Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R, Sheikh SP (2013) miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of epicardial mesothelial cells involving programmed cell death 4 and sprouty-1. PLoS one 8:e56280. https://doi.org/10.1371/journal.pone.0056280
Li X, Liu CY, Li YS, Xu J, Li DG, Han D (2016) Deep RNA sequencing elucidates microRNA-regulated molecular pathways in ischemic cardiomyopathy and nonischemic cardiomyopathy. Genet Mol Res:15. https://doi.org/10.4238/gmr.15027465
Dai Y, Dai D, Mehta JL (2014) MicroRNA-29, a mysterious regulator in myocardial fibrosis and circulating miR-29a as a biomarker. J Am Coll Cardiol 64:2181. https://doi.org/10.1016/j.jacc.2014.03.064
Sassi Y, Avramopoulos P, Ramanujam D, Grüter L, Werfel S, Giosele S, Brunner A-D, Esfandyari D, Papadopoulou AS, De Strooper B, Hübner N, Kumarswamy R, Thum T, Yin X, Mayr M, Laggerbauer B, Engelhardt S (2017) Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat Commun 8:1614. https://doi.org/10.1038/s41467-017-01737-4
Yamada Y, Takanashi M, Sudo K, Ueda S, Ohno S, Kuroda M (2017) Novel form of miR-29b suppresses bleomycin-induced pulmonary fibrosis. PLoS One 12:e0171957. https://doi.org/10.1371/journal.pone.0171957
Drummond CA, Fan X, Haller ST, Kennedy DJ, Liu J, Tian J (2018) Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PLoS One 13:e0197688. https://doi.org/10.1371/journal.pone.0197688
Liang J, Zou X, Fang X, Xu J, Xiao Z, Zhu J, Li H, Yang J, Zeng N, Yuan S, Pan R, Fu Y, Zhang M, Luo J, Wang S, Shan Z (2019) The Smad3-miR-29b/miR-29c axis mediates the protective effect of macrophage migration inhibitory factor against cardiac fibrosis. Biochim Biophys Acta Mol basis Dis 1865:2441–2450. https://doi.org/10.1016/j.bbadis.2019.06.004
Jiang X, Tsitsiou E, Herrick SE, Lindsay MA (2010) MicroRNAs and the regulation of fibrosis. FEBS J 277:2015–2021. https://doi.org/10.1111/j.1742-4658.2010.07632.x
Chen L, Ji Q, Zhu H, Ren Y, Fan Z, Tian N (2018) miR-30a attenuates cardiac fibrosis in rats with myocardial infarction by inhibiting CTGF. Exp Ther Med. https://doi.org/10.3892/etm.2018.5952
Angelini A, Li Z, Mericskay M, Decaux J-F (2015) Regulation of connective tissue growth factor and cardiac fibrosis by an SRF/MicroRNA-133a axis. PLoS One 10:e0139858. https://doi.org/10.1371/journal.pone.0139858
Li N, Zhou H, Tang Q (2018) miR-133: a suppressor of cardiac remodeling? Front Pharmacol 9. https://doi.org/10.3389/fphar.2018.00903
Wang DS, Zhang HQ, Zhang B, Yuan ZB, Yu ZK, Yang T, Zhang SQ, Liu Y, Jia XX (2016) miR-133 inhibits pituitary tumor cell migration and invasion via down-regulating FOXC1 expression. Genet Mol res 15. https://doi.org/10.4238/gmr.15017453
Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW, Chakrabarti S (2014) Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med 18:415–421. https://doi.org/10.1111/jcmm.12218
Nair N, Kumar S, Gongora E, Gupta S (2013) Circulating miRNA as novel markers for diastolic dysfunction. Mol Cell Biochem 376:33–40. https://doi.org/10.1007/s11010-012-1546-x
Wong LL, Armugam A, Sepramaniam S, Karolina DS, Lim KY, Lim JY, Chong JPC, Ng JYX, Chen Y-T, Chan MMY, Chen Z, Yeo PSD, Ng TP, Ling LH, Sim D, Leong KTG, Ong HY, Jaufeerally F, Wong R, Chai P, Low AF, Lam CSP, Jeyaseelan K, Richards AM (2015) Circulating microRNAs in heart failure with reduced and preserved left ventricular ejection fraction. Eur J Heart Fail 17:393–404. https://doi.org/10.1002/ejhf.223
Watson CJ, Gupta SK, O’Connell E, Thum S, Glezeva N, Fendrich J, Gallagher J, Ledwidge M, Grote-Levi L, McDonald K, Thum T (2015) MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur J Heart Fail 17:405–415. https://doi.org/10.1002/ejhf.244
Mewton N, Liu CY, Croisille P, Bluemke D, Lima JAC (2011) Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol 57:891–903. https://doi.org/10.1016/j.jacc.2010.11.013
Iles LM, Ellims AH, Llewellyn H, Hare JL, Kaye DM, McLean CA, Taylor AJ (2015) Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur Heart J Cardiovasc Imaging 16:14–22. https://doi.org/10.1093/ehjci/jeu182
Messroghli DR, Radjenovic A, Kozerke S, Higgins DM, Sivananthan MU, Ridgway JP (2004) Modified Look-Locker inversion recovery (MOLLI) for high-resolutionT1 mapping of the heart. Magn Reson Med 52:141–146. https://doi.org/10.1002/mrm.20110
McCrohon JA, Moon JCC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJS, Pennell DJ (2003) Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation 108:54–59. https://doi.org/10.1161/01.CIR.0000078641.19365.4C
Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ (2006) Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol 48:1977–1985. https://doi.org/10.1016/j.jacc.2006.07.049
Duan X, Li J, Zhang Q, Zeng Z, Luo Y, Jiang J, Chen Y (2015) Prognostic value of late gadolinium enhancement in dilated cardiomyopathy patients: a meta-analysis. Clin Radiol 70:999–1008. https://doi.org/10.1016/j.crad.2015.05.007
Machii M, Satoh H, Shiraki K, Saotome M, Urushida T, Katoh H, Takehara Y, Sakahara H, Ohtani H, Wakabayashi Y, Ukigai H, Tawarahara K, Hayashi H (2014) Distribution of late gadolinium enhancement in end-stage hypertrophic cardiomyopathy and dilated cardiomyopathy: differential diagnosis and prediction of cardiac outcome. Magn Reson Imaging 32:118–124. https://doi.org/10.1016/j.mri.2013.10.011
Tachi M, Amano Y, Inui K, Takeda M, Yamada F, Asai K, Kumita S (2016) Relationship of postcontrast myocardial T1 value and delayed enhancement to reduced cardiac function and serious arrhythmia in dilated cardiomyopathy with left ventricular ejection fraction less than 35%. Acta Radiol 57:430–436. https://doi.org/10.1177/0284185115580840
Patel AR, Kramer CM (2017) Role of cardiac magnetic resonance in the diagnosis and prognosis of nonischemic cardiomyopathy. JACC Cardiovasc Imaging 10:1180–1193. https://doi.org/10.1016/j.jcmg.2017.08.005
Taylor RJ, Umar F, Lin ELS, Ahmed A, Moody WE, Mazur W, Stegemann B, Townend JN, Steeds RP, Leyva F (2015) Mechanical effects of left ventricular midwall fibrosis in non-ischemic cardiomyopathy. J Cardiovasc Magn Reson 18:1. https://doi.org/10.1186/s12968-015-0221-2
Jellis CL, Kwon DH (2014) Myocardial T1 mapping: modalities and clinical applications. Cardiovasc Diagn Ther 4:126–137. https://doi.org/10.3978/j.issn.2223-3652.2013.09.03
Burt JR, Zimmerman SL, Kamel IR, Halushka M, Bluemke DA (2014) Myocardial T1 mapping: techniques and potential applications. RadioGraphics 34:377–395. https://doi.org/10.1148/rg.342125121
Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, McGregor C, Moon JC (2010) Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis. Circulation 122:138–144. https://doi.org/10.1161/CIRCULATIONAHA.109.930636
Halliday BP, Gulati A, Ali A, Guha K, Newsome S, Arzanauskaite M, Vassiliou VS, Lota A, Izgi C, Tayal U, Khalique Z, Stirrat C, Auger D, Pareek N, Ismail TF, Rosen SD, Vazir A, Alpendurada F, Gregson J, Frenneaux MP, Cowie MR, Cleland JGF, Cook SA, Pennell DJ, Prasad SK (2017) Association between midwall late gadolinium enhancement and sudden cardiac death in patients with dilated cardiomyopathy and mild and moderate left ventricular systolic dysfunction. Circulation 135:2106–2115. https://doi.org/10.1161/CIRCULATIONAHA.116.026910
Klem I, Weinsaft JW, Bahnson TD, Hegland D, Kim HW, Hayes B, Parker MA, Judd RM, Kim RJ (2012) Assessment of myocardial scarring improves risk stratification in patients evaluated for cardiac defibrillator implantation. J Am Coll Cardiol 60:408–420. https://doi.org/10.1016/j.jacc.2012.02.070
Ota S (2019) The pattern of myocardial fibrosis detected by cardiovascular magnetic resonance imaging provides prognostic information in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 73:1551. https://doi.org/10.1016/S0735-1097(19)32157-6
Kono AK, Ishii K, Kumagai H, Taniguchi Y, Kajiya T, Sugimura K (2010) Late gadolinium enhancement on cardiac magnetic resonance imaging: is it associated with a higher incidence of nonsustained ventricular tachycardia in patients with idiopathic dilated cardiomyopathy? Jpn J Radiol 28:355–361. https://doi.org/10.1007/s11604-010-0433-1
Leyva F, Foley PW, Chalil S, Ratib K, Smith RE, Prinzen F, Auricchio A (2011) Cardiac resynchronization therapy guided by late gadolinium-enhancement cardiovascular magnetic resonance. J Cardiovasc Magn Reson 13:29. https://doi.org/10.1186/1532-429X-13-29
Leong DP, Chakrabarty A, Shipp N, Molaee P, Madsen PL, Joerg L, Sullivan T, Worthley SG, De Pasquale CG, Sanders P, Selvanayagam JB (2012) Effects of myocardial fibrosis and ventricular dyssynchrony on response to therapy in new-presentation idiopathic dilated cardiomyopathy: insights from cardiovascular magnetic resonance and echocardiography. Eur Heart J 33:640–648. https://doi.org/10.1093/eurheartj/ehr391
Leyva F, Taylor RJ, Foley PWX, Umar F, Mulligan LJ, Patel K, Stegemann B, Haddad T, Smith REA, Prasad SK (2012) Left ventricular midwall fibrosis as a predictor of mortality and morbidity after cardiac resynchronization therapy in patients with nonischemic cardiomyopathy. J Am Coll Cardiol 60:1659–1667. https://doi.org/10.1016/j.jacc.2012.05.054
Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TDH, Ismail NA, Dweck MR, Di Pietro E, Roughton M, Wage R, Daryani Y, O’Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK (2013) Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 309:896. https://doi.org/10.1001/jama.2013.1363
Puntmann VO, Carr-White G, Jabbour A, Yu C-Y, Gebker R, Kelle S, Hinojar R, Doltra A, Varma N, Child N, Rogers T, Suna G, Arroyo Ucar E, Goodman B, Khan S, Dabir D, Herrmann E, Zeiher AM, Nagel E (2016) T1-mapping and outcome in nonischemic cardiomyopathy. JACC Cardiovasc Imaging 9:40–50. https://doi.org/10.1016/j.jcmg.2015.12.001
Pi S-H, Kim SM, Choi J-O, Kim EK, Chang S-A, Choe YH, Lee S-C, Jeon E-S (2018) Prognostic value of myocardial strain and late gadolinium enhancement on cardiovascular magnetic resonance imaging in patients with idiopathic dilated cardiomyopathy with moderate to severely reduced ejection fraction. J Cardiovasc Magn Reson 20:36. https://doi.org/10.1186/s12968-018-0466-7
Oh J, Hong YJ, Ha J, Chun KH, Kim H, Lee CJ, Kim YJ, Choi BW, Kang SM (2019) P3555Lower native T1, extracellular volume and T2 on cardiac magnetic resonance imaging is related to more left ventricular reverse remodeling in nonischemic dilated cardiomyopathy. Eur heart J 40. https://doi.org/10.1093/eurheartj/ehz745.0418
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL (2013) 2013 ACCF/AHA guideline for the management of heart failure. J Am Coll Cardiol 62:e147–e239. https://doi.org/10.1016/j.jacc.2013.05.019
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González-Juanatey JR, Harjola V-P, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P (2016) 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 37:2129–2200. https://doi.org/10.1093/eurheartj/ehw128
Russo V, Papa AA, Williams EA, Rago A, Palladino A, Politano L, Nigro G (2018) ACE inhibition to slow progression of myocardial fibrosis in muscular dystrophies. Trends Cardiovasc Med 28:330–337. https://doi.org/10.1016/j.tcm.2017.12.006
Leask A (2015) Getting to the heart of the matter. Circ Res 116:1269–1276. https://doi.org/10.1161/CIRCRESAHA.116.305381
Rog-Zielinska EA, Norris RA, Kohl P, Markwald R (2016) The living scar – cardiac fibroblasts and the injured heart. Trends Mol Med 22:99–114. https://doi.org/10.1016/j.molmed.2015.12.006
Tietjens J, Teerlink JR (2016) Serelaxin and acute heart failure. Heart 102:95–99. https://doi.org/10.1136/heartjnl-2014-306786
Wu X, Wang H, Wang Y, Shen H, Tan Y (2018) Serelaxin inhibits differentiation and fibrotic behaviors of cardiac fibroblasts by suppressing ALK-5/Smad2/3 signaling pathway. Exp Cell Res 362:17–27. https://doi.org/10.1016/j.yexcr.2017.10.004
Zhang N, Wei W-Y, Li L-L, Hu C, Tang Q-Z (2018) Therapeutic potential of polyphenols in cardiac fibrosis. Front Pharmacol 9. https://doi.org/10.3389/fphar.2018.00122
Xu C, Hu Y, Hou L, Ju J, Li X, Du N, Guan X, Liu Z, Zhang T, Qin W, Shen N, Bilal MU, Lu Y, Zhang Y, Shan H (2014) β-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J Mol Cell Cardiol 75:111–121. https://doi.org/10.1016/j.yjmcc.2014.07.009
Ihm S-H, Chang K, Kim H-Y, Baek SH, Youn H-J, Seung K-B, Kim J-H (2010) Peroxisome proliferator-activated receptor-γ activation attenuates cardiac fibrosis in type 2 diabetic rats: the effect of rosiglitazone on myocardial expression of receptor for advanced glycation end products and of connective tissue growth factor. Basic Res Cardiol 105:399–407. https://doi.org/10.1007/s00395-009-0071-x
Shim CY, Song B-W, Cha M-J, Hwang K-C, Park S, Hong G-R, Kang S-M, Lee JE, Ha J-W, Chung N (2014) Combination of a peroxisome proliferator-activated receptor-gamma agonist and an angiotensin II receptor blocker attenuates myocardial fibrosis and dysfunction in type 2 diabetic rats. J Diabetes Investig 5:362–371. https://doi.org/10.1111/jdi.12153
Wang L-X, Yang X, Yue Y, Fan T, Hou J, Chen G-X, Liang M-Y, Wu Z-K (2017) Imatinib attenuates cardiac fibrosis by inhibiting platelet-derived growth factor receptors activation in isoproterenol induced model. PLoS One 12:e0178619. https://doi.org/10.1371/journal.pone.0178619
Chaudhry PA, Mishima T, Sharov VG, Hawkins J, Alferness C, Paone G, Sabbah HN (2000) Passive epicardial containment prevents ventricular remodeling in heart failure. Ann Thorac Surg 70:1275–1280. https://doi.org/10.1016/S0003-4975(00)01755-0
Doenst T, Ahn-Veelken L, Schlensak C, Berchtold-Herz M, Sarai K, Schaefer M, van de Loo A, Beyersdorf F (2001) Left ventricular reduction for idiopathic dilated cardiomyopathy as alternative to transplant - truth or dare?*. Thorac Cardiovasc Surg 49:70–74. https://doi.org/10.1055/s-2001-11709
Calafiore A (1999) Surgical treatment of dilated cardiomyopathy with conventional techniques*1. Eur J Cardio-Thoracic Surg 16:S73–S78. https://doi.org/10.1016/S1010-7940(99)00193-1
Isomura T, Suma H, Horii T, Sato T, Kikuchi N (2000) Partial left ventriculectomy, ventriculoplasty or valvular surgery for idiopathic dilated cardiomyopathy – the role of intra-operative echocardiography. Eur J Cardio-Thoracic Surg 17:239–245. https://doi.org/10.1016/S1010-7940(00)00322-5
Suma H, Isomura T, Horii T, Nomura F (2006) Septal anterior ventricular exclusion procedure for idiopathic dilated cardiomyopathy. Ann Thorac Surg 82:1344–1348. https://doi.org/10.1016/j.athoracsur.2006.04.096
WANG J, GONG X, CHEN H, QIN S, ZHOU N, SU Y, GE J (2017) Effect of cardiac resynchronization therapy on myocardial fibrosis and relevant cytokines in a canine model with experimental heart failure. J Cardiovasc Electrophysiol 28:438–445. https://doi.org/10.1111/jce.13171
Broch K, Murbræch K, Andreassen AK, Hopp E, Aakhus S, Gullestad L (2015) Contemporary outcome in patients with idiopathic dilated cardiomyopathy. Am J Cardiol 116:952–959. https://doi.org/10.1016/j.amjcard.2015.06.022
Keeling PJ, Goldman JH, Slade AKB, Elliott PM, Caforio ALP, Poloniecki J, McKenna WJ (1995) Prognosis of idiopathic dilated cardiomyopathy. J Card Fail 1:337–345. https://doi.org/10.1016/S1071-9164(05)80002-8
Author information
Authors and Affiliations
Contributions
B.C.M., A.Z., and L.A.C. researched data for the article. B.C.M. and A.Z. wrote the article. L.A.C. discussed the content of the article, and B.C.M., A.Z., and L.A.C. reviewed and edited before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cojan-Minzat, B.O., Zlibut, A. & Agoston-Coldea, L. Non-ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail Rev 26, 1081–1101 (2021). https://doi.org/10.1007/s10741-020-09940-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-020-09940-0