
Abstract
Heart failure represents the end result of different pathophysiologic processes, which culminate in functional impairment. Regardless of its aetiology, the presentation of heart failure usually involves symptoms of pump failure and congestion, which forms the basis for clinical diagnosis. Pathophysiologic descriptions of heart failure with reduced ejection fraction (HFrEF) are being established. Most commonly, HFrEF is centred on a reactive model where a significant initial insult leads to reduced cardiac output, further triggering a cascade of maladaptive processes. Predisposing factors include myocardial injury of any cause, chronically abnormal loading due to hypertension, valvular disease, or tachyarrhythmias. The pathophysiologic processes behind remodelling in heart failure are complex and reflect systemic neurohormonal activation, peripheral vascular effects and localised changes affecting the cardiac substrate. These abnormalities have been the subject of intense research. Much of the translational successes in HFrEF have come from targeting neurohormonal responses to reduced cardiac output, with blockade of the renin-angiotensin-aldosterone system (RAAS) and beta-adrenergic blockade being particularly fruitful. However, mortality and morbidity associated with heart failure remains high. Although systemic neurohormonal blockade slows disease progression, localised ventricular remodelling still adversely affects contractile function. Novel therapy targeted at improving cardiac contractile mechanics in HFrEF hold the promise of alleviating heart failure at its source, yet so far none has found success. Nevertheless, there are increasing calls for a proximal, ‘cardiocentric’ approach to therapy. In this review, we examine HFrEF therapy aimed at improving cardiac function with a focus on recent trials and emerging targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure represents the end result of different pathophysiologic processes, which culminate in functional impairment. Regardless of its aetiology, the presentation of heart failure usually involves symptoms of pump failure and congestion, which forms the basis for clinical diagnosis [1]. Contemporary practice has categorised heart failure into several distinct groups: acute vs. chronic, left sided vs. right sided and preserved vs. reduced ejection fraction. During acute presentations, the schema of ‘wet-dry’ and ‘warm-cold’ often aids in appropriate management. However, unlike the dichotomous nature of classification systems, clinical manifestations of heart failure can be subtle and lie on a spectrum between asymptomatic dysfunction to severe end-stage disease. Nevertheless, the distinction between different classes of heart failure is important as they represent divergent pathophysiology and treatment. Heart failure with preserved ejection fraction (HFpEF) remains a poorly understood entity, with a lack of effective therapy, reflecting our lack of insight into the disease process. Increasingly, HFpEF is being viewed not as a primary cardiac disease but as a disease driven by complex and heterogeneous comorbidities [2].
On the other hand, pathophysiologic descriptions of heart failure with reduced ejection fraction (HFrEF) are better established [3]. Most commonly, HFrEF is centred on a reactive model where a significant initial insult leads to reduced cardiac output, further triggering a cascade of maladaptive processes [4, 5]. Predisposing factors include myocardial injury of any cause, chronically abnormal loading due to hypertension, valvular disease or tachyarrhythmias. The pathophysiologic processes behind remodelling in heart failure are complex and reflect systemic neurohormonal activation, peripheral vascular effects and localised changes affecting the cardiac substrate. These abnormalities have been the subject of intense research.
Much of the translational successes in HFrEF have come from targeting neurohormonal responses to reduced cardiac output, with blockade of the renin-angiotensin-aldosterone system (RAAS) and beta-adrenergic blockade being particularly fruitful. However, mortality and morbidity associated with heart failure remains high [6]. Although systemic neurohormonal blockade slows disease progression, localised ventricular remodelling still adversely affects contractile function.
Historically, a direct approach to stimulating cardiac contraction through the use of inotropic agents was employed. However, an association with poor clinical outcomes lead to re-evaluation in their routine use. Mechanistically, multiple aspects of the cardiac contractile apparatus are dysregulated in HFrEF and subject to adverse re-modelling. Bluntly increasing contractile force via influencing cardiac calcium levels did not address the underlying derangement in electrochemical coupling and energetic inefficiencies that are now well established. Novel therapy targeted at improving cardiac contractile mechanics and metabolic efficiency in HFrEF hold the promise of alleviating heart failure at its source, yet so far none has found success, i.e. been incorporated into current heart failure management guidelines [7, 8]. Nevertheless, there are increasing calls for a proximal, ‘cardiocentric’ approach to therapy [5, 9, 10]. In this review, we summarise translational progress aimed at improving cardiac pump function, focusing particularly on excitation-contraction coupling, novel inotropes and cardiac energetics.
Excitation contraction coupling
The mechanical action of the heart depends on the coordinated action of individual cardiomyocytes, which are composed mainly of contractile proteins and mitochondria. Myofibrils mainly contain actin and myosin and span the entire length of the cardiomyocyte. They are subdivided into individual sarcomeric elements identifiable by electron microscopy. The actin filaments are anchored at Z-lines found on either end of each sarcomere, whilst myosin filaments in the A band are interconnected at the central M line and to the Z-disc by the titin filaments. Invaginations of sarcolemma in the form a complex network of transverse tubules (T-tubules) are also anchored to the Z-discs. The T-tubules are rich in ion channels and serve to regulate excitation contraction coupling within the myocyte.
Contraction begins with an action potential that causes Ca2+ release from voltage gated L type Ca2+ channels (LTCCs) in the sarcolemma and within the T-tubules. The resulting increase in Ca2+ concentration triggers Ca2+-induced Ca2+ release (CICR) from ryanodine receptors (RyR) located on the closely apposed sarcoplasmic reticulum (SR). The propagating Ca2+ binds to cardiac troponin C and induces conformational changes between tropomyosin and actin on the thin filament. This exposes the myosin-binding sites on actin, enabling myosin to bind to it, thus activating the cross-bridge cycle (systole). Cardiac relaxation (diastole) requires the active uptake of Ca2+ into the SR through sarcoplasmic reticulum Ca2+ ATPase 2a (SERCA-2a) and also active Ca2+ efflux through the sodium-calcium exchanger (NCX) [Fig. 1]. Significant derangements in excitation-contraction (EC) coupling are found in HFrEF and correspond with abnormalities in the systems involved in Ca2+ handling. These include remodelling of the T-tubules, decoupling of RyRs and reduced activity of SERCA. The outcomes correspond to reduced amplitudes of the Ca2+ transients and reduced SR Ca2+ concentration, which negatively impact on both systolic contractility and diastolic relaxation [11]. Importantly, elevation of intracellular Ca2+ and Ca2+ leak from the SR predisposes to malignant arrhythmias and myocyte death [12].

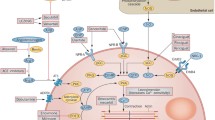
Excitation contraction coupling in the cardiomyocyte. The cardiac action potential [1] is initiated by influx of Na+ ions via Na+ channels which brings the net cellular potential from negative to positive. This triggers Ca2+ release from LTCC [2] found within the invagination of the T-tubule. The localised elevation of Ca2+ concentration induces CICR from the closely opposed SR via RyR receptors [3]. Contraction [4] and relaxation [5] of myofilaments are dependent on binding and dissociation of Ca2+ from troponin. Diastole is therefore dependent on efflux of Ca2+ to either the SR via SERCA or extracellularly via NCX. Return to net negative cellular potential occurs during repolarisation and involves efflux of K+ ions via K+ channels and NKX. LTCC, L-type Ca2+ channels; CICR, calcium induced calcium release; SR, sarcoplasmic reticulum; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase; NCX, sodium-calcium exchanger; NKX, sodium-potassium exchanger
T-tubule and ryanodine receptor dyad
In animal and (some) human studies, T-tubule disruption is a feature of established heart failure and hampers cardiac contractility and synchrony [13,14,15,16,17]. Impaired intracellular Ca2+ cycling is evident long before the development of clinical heart failure, coinciding with T-tubule disorganisation [18]. Migration of the T-tubules away from the Z-line alters LTCC distribution and leaves behind ‘orphaned RyRs’ [19], which exhibit ‘leaky’ SR Ca2+ release. This leads to asynchronous and regionally heterogeneous CICR, which correlates strongly with poor contractility in human HFrEF [20] and is postulated to be arrhythmogenic [21]. There is probably a linear reduction in ejection fraction with increasing disorganisation [22].
The underlying cause of T-tubule remodelling has not been defined, but in small animal studies, it has been linked to reduced expression and altered distribution of the anchor protein junctophilin-2 (JPH-2), possibly in response to increased wall stress [23, 24]. The T-tubule/SR triad is held together by JPH-2, which is essential for stabilisation of local ion channels including RyR and LTCC [25]. Cardiac-specific knock-down of JPH-2 in a mouse model led to the development of acute heart failure, with loss of the junctional membrane complexes and reduced CICR [26]. However, contradictory studies exist which show no correlation of JPH-2 expression with T-tubule distribution in both rats and sheep [27]. Whilst JPH-2 mutations has been found in individuals with hypertrophic cardiomyopathy (HCM), they remain at a case study level without a definitive link to HFrEF [28].
Recovery of tubule morphology has been reported in animal models with cardiac resynchronisation [29], sildenafil [30] and beta-blockade [31]; however, the efficacy of these treatments is based on unloading of the myocardium. Novel therapeutic approaches include gene therapy and miRNA inhibition. Overexpression of JPH-2 via viral-mediated gene therapy halted the progression of heart failure in a mouse model, with preservation of T-tubule structures [32]. Suppression of miRNA-24, which is known to target JPH-2, rescued LTCC signalling and stabilised JPH-2 expression in a mouse model of heart failure secondary to aortic constriction. It has been recently shown that JPH-2 fragments found in stressed hearts localise to the nucleus and activate cardioprotective transcriptional programming, attenuating hypertrophic remodelling in mice [33]. However, given the uncertain role of JPH-2 in human HFrEF, preclinical studies using human heart failure samples will be necessary prior to clinical trials.
SERCA
The Ca2+ re-uptake channel SERCA-2a has long been implicated in the pathogenesis of heart failure and has been the target of several clinical trials in HFrEF [34]. Normally, SERCA is the principal determinant of the rate of Ca2+ efflux, determining SR levels. Hence, SERCA activity greatly impacts on both the rate of diastole and the subsequent force of systolic contraction induced by Ca2+ release from the SR. SERCA is required for complete diastolic relaxation and minimises delayed after-depolarisations. The primary regulator of SERCA is phospholamban (PLB), which reduces the activity of SERCA by reducing its affinity for Ca2+. Phosphorylation of PLB by protein kinase A (PKA) or Ca-dependent protein kinase II (CaMKII) leads to disinhibition of SERCA resulting in both increased lusitropy and inotropy.
Animal and human studies in heart failure have variously confirmed the reduced expression of SERCA-2a at mRNA and protein levels [34,35,36]. SERCA activity is reduced by a relative decrease in SERCA/PLB levels and by reduced phosphorylation of PLB [37, 38]. SERCA KO mice exhibit inefficient Ca2+ handling, reduced contractile efficiency and eventual heart failure [39]. These and other experiments highlight the causal relationship between SERCA loss and heart failure, identifying it as a target for therapy.
PLB ablation improves EC coupling and Ca2+ handling in small and large animal models of heart failure, a result which has been replicated in humans [40,41,42]. A recent murine model of heart failure demonstrated reduced mortality following PLB ablation [43]. Modulation of PLB function via increased phosphorylation (inhibition of protein phosphatase 1 or enhancing PKA activity) has also been studied with some success in animals [44]. However, there are significant concerns with downstream effects on other signalling pathways and non-specific induction or inhibition of phosphorylation may have unforeseen outcomes. Importantly, mutations in PLB have been linked to hereditary dilated cardiomyopathy [45] and arrhythmogenic cardiomyopathy [46]. Multiple binding partners underlie the complexity of PLB signalling, and more preclinical work on human models is needed to translate these findings [47].
The direct SERCA activator, istaroxime, was investigated in the HORIZON-HF trial for acute heart failure (AHF). Istaroxime is a novel non-glycoside inhibitor of Na+/K+ ATPase, which also directly stimulates SERCA-2a activity, likely by disrupting the SERCA-PLB complex [48]. It exerts a dual inotropic/lusitropic effect by increasing SR Ca2+ sequestration in diastole and increasing cytosolic Ca2+ in systole. Increased SERCA activity had been demonstrated in guinea pigs and human cardiomyocyte preparations [49]. Istaroxime improved systolic and diastolic functions in a canine chronic heart failure model without increased myocardial oxygen consumption or an increase in heart rate [50]. In the HORIZON-HF trial randomising 120 AHF patients to istaroxime infusion or placebo, istaroxime decreased pulmonary capillary wedge pressure and diastolic stiffness whilst improving contractility without an increase in adverse events. Compared with other commonly used inotropes, istaroxime has a better safety profile than digoxin and does not increase energy consumption as is the case with dobutamine [51]. Further phases I and II trials of istaroxime in AHF are underway (ClinicalTrials.gov Identifier NCT02617446 and NCT02477449).
SERCA gene therapy has garnered much attention in preclinical research with extensive modelling both in vitro and in vivo. Transgenic murine models overexpressing SERCA demonstrate improved cardiac function and protection against developing heart failure [52]. Human cardiomyocytes from HFrEF patients [53] transfected with SERCA-2a carrying adenovirus have improved contractile characteristics in vitro [53]. Multiple small animal models provide proof of concept for gene therapy, with reports of restoration of contractile function in heart failure [54], reversal of negative remodelling [55], protection against malignant arrhythmias [56] and improved survival [57]. Large animal studies of canine, ovine and porcine models followed suit, with promising outcomes despite heterogeneity in the choice of the heart failure model [58]. Proposed mechanisms to explain the beneficial effect of SERCA gene therapy include improvement in mechano-energetic efficiency [59], modulation of apoptotic signalling and more recently altered miRNA expression [60]. Evidence for SERCA gene therapy has been described as ‘overwhelming’, years prior to the initiation of its first human trial [61].
The phase 2 CUPID trial enrolled 39 patients with advanced heart failure and utilised intracoronary delivery of SERCA-2a via adeno-associated virus (AAV) vector. The authors reported promising results across multiple domains to a prespecified p value < 0.2, assessing the likelihood of a false-positive result to be 2.7% [62]. A 3-year follow-up of CUPID participants reported an 82% reduction in recurrent cardiovascular events and a favourable safety profile [63]. Nevertheless, as is often the case for promising heart failure therapies, the phase 2b CUPID II trial in 250 patients failed to meet its primary endpoint of time to recurrent cardiovascular events, nor its secondary outcome of time to all-cause of death [64]. Whilst no substantial differences in patient characteristics exist between the two trials to explain these results, the study authors point to a possible reduction in viral transduction efficacy in the CUPID II trial. However, significant weaknesses exist in the design of the CUPID trial, including less stringent p values, subdivision of an already small sample size and the lack of dose response to treatment, all of which bias to a false-positive result. Following the disappointing result of the CUPID II trial, enthusiasm for SERCA gene therapy waned, with withdrawal of the companion phase II AGENT-HF trial examining its effect on ventricular remodelling [65]. Preliminary data favoured placebo, although the study at termination was too underpowered to detect a difference between the two arms. Despite the generally favourable safety profile of gene therapy for HFrEF, challenges remain in optimising its delivery and establishing its efficacy [66].
Post-translational regulation of SERCA2a is now recognised. Glutathionylation increases SERCA activity, whereas glycosylation decreases it [67]. Specifically, O-GlcNAcylation refers to the addition of O-linked N-acetylglucosamine and has been shown to reduce SERCA expression [68]. SUMOylation of SERCA2a has gained the lion’s share of attention in heart failure research and has been identified as essential for normal cardiac development [69]. SUMOylation refers to attachment of small ubiquitin-like modifier proteins which modify the function of the targeted protein [70]. SUMO1 gene transfer rescues decreased levels of SUMO1, increases SERCA2a expression and potentiates contractile function in mice with heart failure [71]. This result was replicated in a swine model of ischaemic heart failure [72]. A small molecule activator of SUMOylation has been discovered, which acts as a dose-dependent inotrope and improves left ventricular function in vivo [73]. Better elucidation of Ca2+ handling and its effect on cardiac function continues to supplement an ever-expanding menu of treatment options. Given its essential role not only in EC coupling but also its critical function in gene transcription [74], mechano-energetics, growth and apoptosis [11], it is likely that Ca2+ signalling will remain an active area of translational research.
Inotropes in heart failure
Inotropes were first introduced into clinical practice over 200 years ago with William Withering’s treatise ‘An Account of the Foxglove’, where digitalis was used to treat the ‘most hopeless and deplorable’ cases of heart failure [75]. Due to its significant symptomatic benefit in acute heart failure, digoxin use gained widespread acceptance but was plagued by significant toxicity. The search for a ‘digitalis replacement’ in heart failure took centre stage in the 1980s, with the development of adrenergic agonists and phosphodiesterase inhibitors as novel inotropes [76]. It soon became apparent, however, that their clinical benefit over digoxin was overstated.
Theoretically, augmenting systolic function should blunt maladaptive hormonal responses that are associated with remodelling. However, inotrope use was soon linked to increased arrhythmias, myocardial ischaemia and mortality. Current international clinical guidelines do not endorse the use of inotropes in chronic stable heart failure but advocate for its limited application to acute decompensated heart failure or as a bridge to destination therapy [1]. However, novel inotropes continue to be developed, which may improve our understanding of the pathophysiology of heart failure.
Calcium mobilisers
Medications that increase inotropy by elevating cytosolic Ca2+ as ‘calcium mobilisers’ include digoxin, milrinone and dobutamine [77]. Dobutamine is an adrenergic agonist and milrinone is a phosphodiesterase inhibitor. Both increase contractility by increasing cAMP whereas digoxin acts by blocking the Na+/K+ ATPase pump, thereby reducing the chemical gradient for Ca2+ efflux through NCX.
Neurohormonal hyperactivity in heart failure leads to desensitisation and down-regulation of adrenoceptors, reduction in cAMP levels and ultimately depleted intracellular Ca2+, all proportional to the severity of heart failure [78]. Medications that enhance cAMP rationally target the reduced cAMP levels found in heart failure. Whilst calcium mobilisers provide modest short-term haemodynamic benefit [79, 80], chronic administration leads to further loss of contractile reserve [81]. There was a 28% increase in all-cause mortality in the PROMISE trial of milrinone in severe chronic heart failure [82], a result echoed in studies involving dobutamine [83]. Undesirable pathophysiologic effects of increased cytosolic Ca2+ include increased myocardial oxygen demand, ventricular arrhythmias, reduced diastolic relaxation and acceleration of myocyte death [84]. Methods to circumvent these adverse effects such as reduced or intermittent dosing and the use of partial agonists have all failed to demonstrate a clinical benefit in HFrEF [85]. Digoxin itself did not improve all-cause mortality in heart failure as reported in the Digitalis Investigator Group Study [86]. Many began questioning the wisdom of stimulating rather than resting the failing heart, and long-term use of inotropes in stable HFrEF was abandoned soon after.
Intravenous inotropes are still used for acute heart failure (AHF) refractory to vasodilators and diuretics and/or accompanied by hypotension. Calcium mobilisers improve haemodynamics in the short-term, although evidence suggests that even brief use may lead to increased mortality. The prospective OPTIME-CHF trial of milrinone failed to show a clinical benefit in AHF, with higher rates of hypotension and ventricular arrhythmias [87]. Retrospective analysis of the FIRST trial identified dobutamine infusions as a strong independent risk factor for mortality [88]. Recent systematic reviews of dobutamine [89] and milrinone [90] continue to point to possible harm, although the reported trials tended to be small and of poor methodological quality, often utilising surrogate outcomes with significant clinical heterogeneity. Contrary with the mortality neutral results of the DIG trial, systematic review of nine large trials of digoxin in heart failure yielded a hazard ratio of 1.14 for all-cause mortality [91]. This has placed doubt on the role of the most established calcium mobilising agent. Given that large observational studies such as the ADHERE registry [92] echo the concern in controlled trials regarding the use of calcium mobilisers in acute heart failure, larger, better designed trials are needed to conclusively settle this question.
Calcium sensitisers
Calcium sensitisers enhance myocyte contractility by increasing myofilament Ca2+ sensitivity. They are thought to be superior to calcium mobilisers as they do not elevate cytosolic Ca2+ levels, in addition to avoiding down-regulation of the adrenergic signalling pathway in heart failure. The best studied calcium sensitisers are pimobendan and levosimendan, both of which augment troponin C binding to Ca2+.
In vitro animal studies of pimobendan using muscle preparations, demonstrated superior mechano-energetic efficiency compared with dobutamine [93]. In vivo, pimobendan exerts positive inotropic, lusitropic and vasodilatory effects [94]. Double-blinded RCTs in canine-dilated cardiomyopathy models suggested a strong mortality benefit, similar to those previously been observed with angiotensin converting enzyme (ACE) inhibitors [95]. However, this benefit never translated in the human trials. Like other inotropic agents of its time, pimobendan improved exercise tolerance at the expense of increased mortality [96]. Use of pimobendan in humans was subsequently abandoned, despite subsequent contradictory studies [97].
Levosimendan is routinely used in clinical practice, demonstrating several pharmacologic effects that act in concert to improve myocardial function [98]. Inotropy is sustained via stabilisation of the Ca2+-troponin C complex [99], with possible contribution from highly selective PDE III inhibition. Levosimendan has prominent vasodilatory effects brought about through activation of ATP-dependent potassium channels (KATP), which leads to relaxation of smooth muscles and after-load reduction. Cardioprotection is mediated by improvement of coronary flow and opening of mitochondrial KATP, which may reduce ischaemia-related cell damage [100]. Advantages of levosimendan over other agents include its potency despite β-blockade, lack of propensity for tachyphlaxis and sustained duration of action due to active metabolites [77].
Preclinical and clinical models of levosimendan indicate a beneficial effect in heart failure. Preservation of myocardial contractile efficiency despite inotropy has been demonstrated in guinea pig hearts [101] and human volunteers [102]. In vivo, levosimendan reduces infarct size in rat and pig models of LAD ligation [103, 104]. Open-label human studies of levosimendan in acute heart failure secondary to myocardial infarction suggest preservation of contractile function, improvements in coronary perfusion and mortality [105]. Double-blinded RCTs of levosimendan have been performed with inconsistent results reflecting heterogeneous study designs. The LIDO study of 203 patients with severe heart failure found levosimendan to be superior to dobutamine with haemodynamic improvement and lower 6-month mortality [106]. The RUSSLAN study randomised 504 patients with acute heart failure post-myocardial infarction to levosimendan or placebo, with safety being the primary outcome. Again, levosimendan correlated with lower mortality without an increase in hypotension or ischaemia [107].
However, larger follow-up trials SURVIVE [108] and REVIVE-II failed to demonstrate superiority of levosimendan in terms of safety and mortality. A meta-analysis of 45 trials involving levosimendan found a significant reduction in mortality despite the negative mortality benefit in the two largest studies [109]. However, the authors excluded singular studies at a time in their sensitivity analysis, which is not as rigorous as the exclusion of all low quality or unblinded studies [110]. Only smaller studies exist for the intermittent use of levosimendan in the chronic heart failure setting, such as in the LevoREP study [111]. No difference was found in the primary outcome of functional capacity or quality of life [112].
The jury is still out on whether calcium sensitisers are beneficial in both acute and chronic heart failure. However, there seems to be increasing clinical equipoise for larger trials of levosimendan therapy [113]. It remains controversial whether the inotropic effects of levosimendan can be partially or fully explained by phosphodiesterase inhibition [99, 114, 115]. Pleotropic effects of levosimendan extend to multiple organ systems, improving circulation in pulmonary, hepatic and renal vasculature, whilst protecting against reperfusion injury [116]. More research is needed to further elucidate the role of calcium sensitisers at both preclinical and clinical levels in heart failure.
Direct myosin activators
The actin-myosin cycle is where chemical energy is converted to mechanical energy in myocytes. Improvements in contractility can be gained by pharmacologically targeting actin-myosin kinetics. The steps of the cycle are well described. Hydrolysis of ATP bound to the myosin head allows weak interaction between actin and myosin, which is strengthened by the release of phosphate. Conformational change of the myosin head follows the release of phosphate and leads to a power stroke of roughly 10 nm on the actin fibres. The conversion from a weak to a strong bond between actin and myosin, also termed cross-bridge formation, is the rate-limiting step of this cycle. If the release of ADP and phosphate occurs before cross-bridge formation, no power stroke occurs. Direct myosin activators were first discovered via high-throughput screening of agents catalysing cross-bridge formation without increasing cytosolic Ca2+ concentrations [117]. Of the many myosin and actin modulators currently being investigated [118], the standout agent is omecamtiv mecarbyl (OM), a selective activator of cardiac myosin [119].
Cardiomyocyte preparations exposed to OM exhibit an increased duration of contraction without an acceleration of contraction velocity. This inotropic effect has been confirmed in a rat model of heart failure and also canine models with both tachycardia and ischaemia induced heart failure [117]. The investigators report a 20–30% increase in contractile efficiency. Early human studies were consistent with animal modelling, whereby measures of left ventricular function improved with administration of OM in a dose-dependent manner in healthy volunteers and patients with chronic heart failure. [120, 121]. Increases in systolic duration and stroke volume were noted without an increase in heart rate.
The ATOMIC-HF and COSMIC-HF trials were phase 2b trials published in 2016 that investigated OM in acute and chronic heart failure, respectively. The former did not meet the primary endpoint of relieving dyspnoea nor any of the secondary endpoints including length of hospitalisation and short term mortality [122]. Nevertheless, the authors point to a possible benefit in a subgroup using the highest OM concentration (425 ± 173 ng/mL).
Oral administration of OM over 20 weeks produced the more encouraging result of increased stroke volume and a reduction of NT-proBNP [123]. The main safety concern with OM is ischaemia due to a reduction in the duration of diastole in healthy individuals, which emerge at serum concentrations greater than 1200 ng/mL [120]. However, in patients with ischaemic cardiomyopathy, administration of OM at standard doses did not result in signs and symptoms of ischaemia with exercise induced stress [124]. Despite being generally well-tolerated with no increase in adverse event rates, both phase 2b studies associated OM administration with a small, concentration independent increase in troponin levels. It is likely that dosing of OM will need to be within a narrow therapeutic range.
The underlying mechanisms of action of OM may be more complex than first thought. The specificity of OM for cardiac myosin is under question due to a study reporting an increase in slow twitch skeletal muscle fibre contractility with OM [125]. More recently, the idea that OM increases contractile efficiency has been challenged, with reports of impaired contractile efficiency and increased myocardial demand in porcine models, attributed to increased resting myosin ATPase activity [126]. In the same paper, it was suggested that OM may also have calcium sensitising effects due to improved interaction between the thick and thin filaments. Regardless of these lingering questions, enthusiasm for myosin activators remains high. GALACTIC-HF is a large phase III trial of OM in chronic HFrEF currently under way, which seeks to answer the question of whether supporting cardiac contractility with direct myosin activators will lead to reduced cardiovascular mortality (ClinicalTrials.gov Identifier: NCT02929329) (Table 1).
Energetics in heart failure
Consideration of myocardial energetics is essential for any therapy aimed at improving contractile function. Under normal circumstances, oxidative metabolism in the mitochondria supplies 95% of cardiac energy with 5% derived from anaerobic glycolysis [133]. In terms of substrate utilisation, fatty acid oxidation (FAO) accounts for 70% of cardiac metabolic requirements, with the remainder being derived from oxidation of glucose, ketone bodies, lactate and other amino acids [134]. Substrate metabolism in the heart responds to the physiologic environment, hence fatty acid oxidation is suppressed in presence of excess glucose and vice versa [135]. The majority of energy consumption is for maintenance of EC coupling, including cross-bridge cycling and powering ion fluxes. The efficient turnover of metabolic substrates is therefore a prerequisite for normal contractile function and energetic deficits are strongly linked to both cellular oxidative stress and contractile failure.
Heart failure represents impairment of energetic reserve and substrate utilisation. This is corroborated by declining stores of both ATP and phosphocreatine (PCr) with progressive left ventricular dysfunction in failing human hearts [136, 137]. Even in the absence of lower concentrations of adenosine nucleotides, reduced shuttling of the creatine kinase system contributes to failure to meet energy demands [138]. In particular, a reduction in PCr/ATP ratio is predictive of adverse outcomes in heart failure [139]. Modification of substrate use is seen in both cardiac hypertrophy and heart failure, with increasing dependence on glucose metabolism and reduced FAO. The physiological impetus for this change is not a reduction in availability of fatty acids but from alterations in transcription signalling [140]. Peroxisome proliferator-activated receptor-⍺ (PPAR-⍺) and its co-activator peroxisome proliferator-activated receptor gamma .co-activator-1 (PGC-1) induce fatty acid metabolism and mitochondrial biogenesis. They are both down-regulated in human heart failure [141, 142]. Although oxidative metabolism of glucose is more efficient than FAO, there is uncoupling of glucose oxidation from glycolysis in heart failure [134, 143]. Products of glycolysis such as pyruvate and lactate can be channelled to the Krebs cycle via accessory or ‘anaplerotic’ pathways. However, efflux of substrate and loss to other non-productive pathways such as protein glycosylation and polyol formation results in reduced oxidative efficiency, as observed in animal models of heart failure [143, 144]. Given the paradigm of energetic exhaustion in the failing heart and the trepidation for the current use of inotropes (‘flog a dead horse’), it is plausible that enhancing cardiac energetics may improve contractile function.
Metabolic substrate modulation
An approach to improve energetic balance in the failing heart is to focus on inducing glucose oxidation. Amongst the investigated agents, anti-anginals target the imbalance in myocardial oxygen supply and demand, with perhexiline and trimetazidine demonstrating promising results. Trimetazidine promotes glucose oxidation by competitively inhibiting long chain 3-ketoacyl-coenzyme A thiolase (3-KAT), the enzyme responsible for the last step in beta-oxidation of FA. Administration of trimetazidine to heart failure patients positively impacted on cardiac PCr/ATP ratio with additional beneficial effects including improved endothelial function, restoration of cytosolic Ca2+ levels and protection against free radical injury and fibrosis [145]. Recent interest in extending the use of trimetazidine to heart failure led to a series of small trials of variable methodological quality, which demonstrated an improvement in LVEF and NYHA class. A systematic review and meta-analysis performed by Gao et al. in 2011 was significant for an improvement in cardiovascular events, hospitalisation and overall mortality [146]. The results are echoed by a more recent retrospective cohort study [147]. However, larger double-blinded RCTs are needed to clarify the benefits of trimetazidine in HFrEF.
Perhexiline is an anti-anginal agent, which inhibits carnitine palmitoyl transferase-1 (CPT-1) responsible for the transport of FA into mitochondria. Evidence for use of perhexiline in heart failure lies mainly in one trial conducted by Lee et al., which demonstrated improvements in contractile function and heart failure symptoms in patients with advanced heart failure [148]. A more recent study of perhexiline in patients with non-ischaemic heart failure demonstrated improvements in PCr/ATP ratio and patient symptoms, without evidence for change in LVEF or substrate utilisation [149]. This is in agreement with an earlier murine study by the same group [150]. At present, multiple mechanisms have been proposed for perhexiline without consensus [151]. Concerns with hepatotoxicity and neurotoxicity places an increased burden of proof for benefit in heart failure before perhexiline can be used for this indication [152].
Ranolazine is an inhibitor of the late Na+ channel current with expanding roles in management of angina, cardiac arrhythmias and diastolic dysfunction [153]. Perhaps reflective of the complexities in extrapolating animal studies to human heart failure, earlier suggestions of enhanced glucose oxidation by ranolazine in murine models did not translate to humans [154]. Agents that directly increase glucose oxidation have also been explored. Dichloroacetate (DCA) inactivates mitochondrial pyruvate dehydrogenase kinase to release inhibition of pyruvate dehydrogenase, the rate-limiting enzyme, which converts pyruvate into acetylCoA for entry into the Krebs cycle. Early clinical experience with DCA suggested an improvement in cardiac function and efficiency [155], but this was not always replicated [156]. Poor pharmacokinetic properties of DCA also make it unsuitable for chronic use, despite benefits found in animal models [157].
The benefit of repressing FAO to induce glucose oxidation has been questioned. FA remains the most important metabolic substrate in heart failure, and acute depletion of FA levels actually impairs contractile function [158]. Inhibition of FAO may also lead to accumulation of lipid metabolites, which are a reversible cause of contractile dysfunction and leads to structural damage in a process termed cardiac lipotoxicity [159]. A newer approach to managing heart failure energetics has been to induce FAO, which in addition to boosting supply of ATP, may clear FA derivatives that accumulate in heart failure. Induction of PPAR-⍺, the main regulator of FAO, can have positive effects on the contractility of the failing heart. Overexpression of PPAR-⍺ under normal circumstances can repress glucose utilisation, resulting in a diabetic cardiomyopathy in murine models [160]. However, in the context of heart failure, PPAR-⍺ activation preserves the level of high-energy phosphates and in particular, exerts a cardioprotective effect in vivo [161, 162]. Subgroup analyses of the VA-HIT trial of fibrates, which act via PPAR-⍺ agonism, suggest a benefit in heart failure [163]. However, this may reflect enhanced endothelial function and anti-inflammatory effects of PPAR-⍺ agonists rather than the promotion of FA metabolism [164]. In fact, fibrates may reduce cardiac FAO by inducing FA consumption in the periphery [165]. It remains to be demonstrated that enhancement of cardiac FAO occurs with fibrates in humans as no RCTs have directly proven their benefit in heart failure [166].
Restoring mitochondrial activity
Defects in multiple domains of mitochondrial function occur in heart failure and result in abnormal ion handling, oxidative stress and programmed cell death amongst other pathophysiologic effects [134]. Directly relevant to the issue of energetics is the disruption of the mitochondria electron transport chain (ETC), noted in both animal and human studies of heart failure [167]. The ETC facilitates transformation of free energy released from oxidative reactions within the mitochondria into ATP through the action of a series of enzyme complexes. Decreased expression of mitochondrial complexes has been described in failing human hearts of multiple aetiologies [168]. On the other hand, mitochondrial myopathies are also associated with contractile dysfunction and defective oxidation [169]. Inefficient ETC function additionally increases reactive oxygen species (ROS), which results in myocyte apoptosis, pathological remodelling and progressive cardiac contractile dysfunction [170]. Modulation of ETC function is seen with proven therapies in HFrEF, including cardiac resynchronisation and neprilysin inhibition [171]. Direct modulation of mitochondrial ETC activity may be the target for future treatments in heart failure.
Coenzyme Q10 (CoQ10) is an over-the-counter supplement with limited but promising data for HFrEF treatment. CoQ10 primarily participates in the ETC as an electron shuttle, facilitating the production of ATP. However, it also has anti-oxidant and endothelial protective effects [172]. Reduced levels of CoQ10 are found in HFrEF and correlate with lower LVEF and increased mortality [173]. In agreement with animal studies, clinical trials of CoQ10 supplementation have shown improvements in LVEF and the recent Q-SYMBIO study found a 50% relative reduction in major cardiovascular events and all-cause mortality. Nevertheless, significant weaknesses exist with the existing trials, including small patient numbers, heterogeneous study populations and protocols and large margins of error for outcome measures.
Other agents that target respiratory chain function include flavonoids and melatonin. Flavonoids refer to naturally occurring pigments found in a variety of plants whose consumption has been associated with reduced incidence of heart failure. Members of the flavonoid family have been shown to improve ETC activity and it has been proposed for long term preventative therapy for heart failure [174]. In animal models, melatonin stabilises the inner mitochondrial membrane leading to improved ETC function, whilst also exerting anti-oxidant effects [175]. Both classes of compounds have limited evidence in human heart failure and require further study.
Although there is consensus that alterations to ETC expression and function occur in HFrEF, the reported defects are highly variable between different heart failure models and human heart failure of differing aetiologies [167]. ETC phosphorylation status, respiratory complex assembly and regional distribution of mitochondrial defects are also increasingly recognised as important aspects of a complex metabolic network. The fundamental question of whether heart failure progression is a direct result of metabolic dysfunction or whether energetic remodelling is a consequence of adapting to a cardiac insult remains unanswered. A recent study of failing vs. non-failing human hearts found no difference in oxidative capacity for fatty acid or glucose [176] compared with controls; whilst a separate study on fresh human heart failure samples found preserved in vitro oxidative capacity of cardiac mitochondria [177]. The complex physiology of cardiac metabolism has proven difficult to capture with current experimental models, especially those based on animals.
Towards human models of heart failure
Cardiac contractility relies on complex and multifaceted physiology, which is modulated at the level of ion fluxes, myofibrillar interaction and energetics. As can be seen by our brief exploration of current therapies (summarised in Fig. 2), many gaps remain in our understanding of the pathophysiology of human heart failure. In adopting a cardiocentric approach to heart failure, we continue to rely on fragmented approaches, which are often incompletely characterised. Manipulation of one aspect of the system may simultaneously be counteracted by another or have unintended consequences, and the increased risk of arrhythmias with calcium mobilising inotropes is a case in point. Even in the case of OM, which was rationally screened and designed as a cardiac selective inotrope, it also has pleotropic effects.

Summary diagram of pharmacologic agents modulating cardiomyocyte function and their molecular targets. Contractile function can be enhanced via optimisation of excitation contraction coupling through influencing the activity of SERCA and Junctophilin-2. Calcium-mobilising inotropes increase contractility by increasing cytosolic calcium either via action of mediators such as cAMP or directly acting on ion channels and exchangers. Calcium-sensitising inotropes increase sensitivity to the action of calcium whereas myofilament activators directly increase cross-bridge cycling. Contractile efficiency may be appropriately targeted via modulating the metabolic substrate as in the case of the anti-anginal agents or through manipulation of the electron transport chain within mitochondria
There is a great discrepancy between successful animal studies and lacklustre performance in larger human trials. Although this can be partly attributed to small sample sizes and biases in publication and study design [178], even in methodologically robust animal trials, results lack external validity when applied to humans. Preclinical animal studies often fail to replicate the complex patient phenotype. On a macroscopic level, animal models generally do not capture the progressive and degenerative nature of human disease nor are they able to replicate the multiple comorbidities commonly found in the HFrEF population. Significant differences on a genetic level are further confounded by heterogeneity in the results of inter- and even intraspecies studies [179].
Research utilising human tissue can help shed light on the intertwining factors contributing to a poorly contractile heart without needing to rely on rough disease models. A vogue area of research that may prove insightful is that of cardiac recovery, specifically recovery of failing human hearts requiring mechanical unloading via left ventricular assist devices (LVADs). It is now well established that following a period of support and pharmacological therapy, significant structural reverse remodelling occurs which allow successful explant of the supporting device in a minority (1.3%) of patients [180]. Importantly, success has been reported not just for acute cardiomyopathy but also for chronic heart failure. Whilst clinical research has emphasised on establishing robust clinical criteria to identify those most suitable for device explantation, availability of human cardiac tissue at inplantation, explantation and transplantation has proven a boon for understanding underlying pathophysiology and its reversibility. With LVAD support, remodelling occurs across multiple domains, with improvement in Ca2+ homeostasis, T-tubular structure, mitochondrial function and sarcomeric contraction [181]. Nevertheless, there remains a tendency for patient phenotype to relapse, and current mechanical and pharmacologic support may be better described to provide remission rather than full recovery [182, 183]. Currently, supportive pharmacotherapy to enable weaning of mechanical support relies heavily on systemic neurohormonal approach such as RAAS blockade and beta-blockers—although some localised beneficial effects on the myocardium may also exist [184]. To achieve durable recovery, targeting aetiology specific findings within the cardiac substrate will likely be necessary.
In an era of increasingly personalised medicine, specific, human relevant molecular targets are more important than ever. Although fresh human cardiac tissue samples such as those utilised in the cardiac recovery studies represent the gold standard for in vitro research, there is generally very limited availability. The key to improving access will be the development of reliable, collaborative tissue banking [185]. Whether at the scale of elucidating transcriptional modification in heart failure or in large clinical trials of therapeutics, ultimately, all animal studies require validation with human studies.
References
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ et al (2016) 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the heart failure association (HFA) of the ESC. Eur J Heart Fail 18(8):891–975
van Heerebeek L, Paulus WJ (2016) Understanding heart failure with preserved ejection fraction: where are we today? Neth Hear J 24(4):227–236
Reed BN, Sueta CA (2015) A practical guide for the treatment of symptomatic heart failure with reduced ejection fraction (HFrEF). Curr Cardiol Rev 11(1):23–32
Kemp CD, Conte JV (2012) The pathophysiology of heart failure. Cardiovasc Pathol 21(5):365–371
MacIver DH, Dayer MJ (2012) An alternative approach to understanding the pathophysiological mechanisms of chronic heart failure. Int J Cardiol 154(2):102–110
Maggioni AP, Dahlstrom U, Filippatos G, Chioncel O, Leiro MC, Drozdz J et al (2013) EURObservational research programme: regional differences and 1-year follow-up results of the heart failure pilot survey (ESC-HF pilot). Eur J Heart Fail 15(7):808–817
Yancy CW, Jessup M, Masoudi FA, McBride PE, Peterson PN, Stevenson LW et al (2016) 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure. J Am Coll Cardiol 68(13):1476–1488
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS et al (2016) 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 37(27):2129–U130
Gheorghiade M, Larson CJ, Shah SJ, Greene SJ, Cleland JGF, Colucci WS et al (2016) Developing new treatments for heart failure focus on the heart. Circ Heart Fail 9(5):1–8
Vaduganathan M, Butler J, Pitt B, Gheorghiade M (2015) Contemporary drug development in heart failure: call for hemodynamically neutral therapies. Circ Heart Fail 8(4):826–831
Lipskaia L, Hulot JS, Lompre AM (2009) Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflügers Archiv: Eur J Physiol 457(3):673–685
Marks AR (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Investig 123(1):46–52
Lyon AR, MacLeod KT, Zhang YJ, Garcia E, Kanda GK, Lab MJ et al (2009) Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A 106(16):6854–6859
Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W et al (2004) Reduced synchrony of Ca2+ release with loss of T-tubules—a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 62(1):63–73
Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I et al (2006) T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 574(2):519–533
Crossman DJ, Ruygrok PR, Soeller C, Cannell MB (2011) Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One 6(3):1–10
Crossman DJ, Shen X, Jullig M, Munro M, Hou Y, Middleditch M et al (2017) Increased collagen within the transverse tubules in human heart failure. Cardiovasc Res 113(8):879–891
Shah SJ, Aistrup GL, Gupta DK, O'Toole MJ, Nahhas AF, Schuster D et al (2014) Ultrastructural and cellular basis for the development of abnormal myocardial mechanics during the transition from hypertension to heart failure. Am J Phys Heart Circ Phys 306(1):H88–H100
Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng HP (2006) Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103(11):4305–4310
Crossman DJ, Young AA, Ruygrok PN, Nason GP, Baddelely D, Soeller C et al (2015) T-tubule disease: relationship between T-tubule organization and regional contractile performance in human dilated cardiomyopathy. J Mol Cell Cardiol 84:170–178
Crocini C, Coppini R, Ferrantini C, Yan P, Loew LM, Tesi C et al (2014) Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure. Proc Natl Acad Sci U S A 111(42):15196–15201
Wei S, Guo A, Chen BY, Kutschke W, Xie YP, Zimmerman K et al (2010) T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107(4):520–U163
Frisk M, Ruud M, Espe EKS, Aronsen JM, Roe AT, Zhang LL et al (2016) Elevated ventricular wall stress disrupts cardiomyocyte T-tubule structure and calcium homeostasis. Cardiovasc Res 112(1):443–451
Zhang CM, Chen BY, Guo A, Zhu YQ, Miller JD, Gao S et al (2014) Microtubule-mediated defects in Junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 129(17):1742–1750
Beavers DL, Landstrom AP, Chiang DY, Wehrens XHT (2014) Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res 103(2):198–205
van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N et al (2011) Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 123(9):979–988
Caldwell JL, Smith CER, Taylor RF, Kitmitto A, Eisner DA, Dibb KM et al (2014) Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ Res 115(12):986–U157
Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR et al (2007) Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol 42(6):1026–1035
Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF et al (2012) Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res 110(4):588–U197
Huang CK, Chen BY, Guo A, Chen R, Zhu YQ, Kutschke W et al (2016) Sildenafil ameliorates left ventricular T-tubule remodeling in a pressure overload-induced murine heart failure model. Acta Pharmacol Sin 37(4):473–482
Chen B, Li Y, Jiang S, Xie YP, Guo A, Kutschke W et al (2012) Beta-adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J 26(6):2531–2537
Reynolds JO, Quick AP, Wang QL, Beavers DL, Philippen LE, Showell J et al (2016) Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int J Cardiol 225:371–380
Guo A, Wang Y, Chen B, Wang Y, Yuan J, Zhang L et al (2018) E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 362(6421)
Mercadier JJ, Lompré AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C et al (1990) Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Investig 85(1):305–309
Arai M, Alpert NR, Maclennan DH, Barton P, Periasamy M (1993) Alterations in sarcoplasmic-reticulum gene-expression in human heart failure—a possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res 72(2):463–469
Kiss E, Ball NA, Kranias EG, Walsh RA (1995) Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)-ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ Res 77(4):759–764
Schwinger RHG, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M et al (1995) Unchanged protein levels of SERCA-II and phospholamban but reduced Ca2+ uptake and Ca2+ ATPase activity of cardiac sarcoplasmic-reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 92(11):3220–3228
Sande JB, Sjaastad I, Hoen IB, Bokenes J, Tonnessen T, Holt E et al (2002) Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res 53(2):382–391
Boardman NT, Aronsen JM, Louch WE, Sjaastad I, Willoch F, Christensen G et al (2014) Impaired left ventricular mechanical and energetic function in mice after cardiomyocyte-specific excision of SERCA2. Am J Physiol-Heart Circ Physiol 306(7):H1018–H1024
Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y et al (2002) Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med 8(8):864–871
del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ (2002) Targeting phospholamban by gene transfer in human heart failure. Circulation. 105(8):904
Kaye DM, Preovolos A, Marshall T, Byrne M, Hoshijima M, Hajjar R et al (2007) Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J Am Coll Cardiol 50(3):253
Kaneko M, Hashikami K, Yamamoto S, Matsumoto H, Nishimoto T (2016) Phospholamban ablation using CRISPR/Cas9 system improves mortality in a murine heart failure model. PLoS One 11(12):1–16
Yamada M, Ikeda Y, Yano M, Yoshimura K, Nishino S, Aoyama H et al (2006) Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J 20(8):1197–1199
Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO et al (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Investig 111(6):869–876
van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC et al (2012) Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail 14(11):1199–1207
Kranias EG, Hajjar RJ (2017) The phospholamban journey 4 decades after setting out for Ithaka. Circ Res 120(5):781–783
Huang CLH (2013) SERCA2a stimulation by istaroxime: a novel mechanism of action with translational implications. Br J Pharmacol 170(3):486–488
Micheletti R, Palazzo F, Barassi P, Glacalone G, Ferrandi M, Schiavone A et al (2007) Istaroxime, a stimulator of sarcoplasmic, reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am J Cardiol 99(2A):24A–32A
Sabbah HN, Imai M, Cowart D, Amato A, Carminati P, Gheorghiade M (2007) Hemodynamic properties of a new-generation positive luso-inotropic agent for the acute treatment of advanced heart failure. Am J Cardiol 99(2A):41A–46A
Aditya S, Rattan A (2012) Istaroxime: a rising star in acute heart failure. J Pharmacol Pharmacother 3(4):353–355
Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B et al (2004) Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 109(15):1898–1903
Federica del M, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW et al (1999) Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 100(23):2308
Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T et al (2000) Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A 97(2):793–798
Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang LF et al (2007) Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Phys Heart Circ Phys 292(2):H1204–H12H7
Lyon AR, Bannister ML, Collins T, Pearce E, Sepehripour AH, Dubb SS et al (2011) SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol 4(3):362–372
del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK et al (2001) Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation. 104(12):1424–1429
Ishikawa K, Tilemann L, Ladage D, Aguero J, Leonardson L, Fish K et al (2012) Cardiac gene therapy in large animals: bridge from bench to bedside. Gene Ther 19(6):670–677
Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF et al (2007) Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol 42(4):852–861
Kumarswamy R, Lyon AR, Volkmann I, Mills AM, Bretthauer J, Pahuja A et al (2012) SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur Heart J 33(9):1067–1075
del Monte F, Hajjar RJ, Harding SE (2001) Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circulation Research 88(11):E66-E
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B et al (2011) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 124(3):304–313
Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M et al (2014) Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 114(1):101–108
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS et al (2016) Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 387(10024):1178–1186
Hulot JS, Salem JE, Redheuil A, Collet JP, Varnous S, Jourdain P et al (2017) Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT-HF randomized phase 2 trial. Eur J Heart Fail 19(11):1534–1541
Penny WF, Hammond HK (2017) Randomized clinical trials of gene transfer for heart failure with reduced ejection fraction. Hum Gene Ther 28(5):378–384
Stammers AN, Susser SE, Hamm NC, Hlynsky MW, Kimber DE, Kehler DS et al (2015) The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can J Physiol Pharmacol 93(10):843–854
Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M et al (2003) Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 278(45):44230–44237
Lee A, Oh JG, Gorski PA, Hajjar RJ, Kho C (2016) Post-translational modifications in heart failure: small changes, big impact. Heart Lung Circ 25(4):319–324
Hay RT (2005) SUMO: a history of modification. Mol Cell 18(1):1–12
Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E et al (2011) SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 477(7366):601–605
Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C et al (2013) SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med 5(211):211ra159
Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K et al (2015) Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun 6:7229
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Withering W (1785) An account of the foxglove, and some of its medical uses: with practical remarks on dropsy, and other diseases. Birmingham, England
Scholz H (1984) Inotropic drugs and their mechanisms of action. J Am Coll Cardiol 4(2):389–397
Pollesello P, Papp Z, Papp JG (2016) Calcium sensitizers: what have we learned over the last 25 years? Int J Cardiol 203:543–548
Bohm M, La Rosee K, Schwinger RH, Erdmann E (1995) Evidence for reduction of norepinephrine uptake sites in the failing human heart. J Am Coll Cardiol 25(1):146–153
Unverferth DA, Blanford M, Kates RE, Leier CV (1980) Tolerance to dobutamine after a 72 hour continuous infusion. Am J Med 69(2):262–266
Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K et al (1982) Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med 307(4):205–211
Packer M, Medina N, Yushak M (1984) Hemodynamic and clinical limitations of long-term inotropic therapy with amrinone in patients with severe chronic heart failure. Circulation. 70(6):1038
Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM et al (1991) Effect of oral milrinone on mortality in severe chronic heart failure. N Engl J Med 325(21):1468–1475
Krell MJ, Kline EM, Bates ER, Hodgson JM, Dilworth LR, Laufer N et al (1986) Intermittent, ambulatory dobutamine infusions in patients with severe congestive heart failure. Am Heart J 112(4):787–791
Francis GS, Bartos JA, Adatya S (2014) Inotropes. J Am Coll Cardiol 63(20):2069–2078
Packer M (1993) The development of positive inotropic agents for chronic heart failure: how have we gone astray. J Am Coll Cardiol 22(4 Suppl A):119A–126A
Perry G, Brown E, Thornton R, Shiva T, Hubbard J, Reddy KR et al (1997) The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 336(8):525–533
Felker GM, Benza RL, Chandler AB, Leimberger JD, Cuffe MS, Califf RM et al (2003) Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol 41(6):997–1003
O'Connor CM, Gattis WA, Uretsky BF, Adams KF Jr, McNulty SE, Grossman SH et al (1999) Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (FIRST). Am Heart J 138(1 Pt 1):78–86
Tacon CL, McCaffrey J, Delaney A (2012) Dobutamine for patients with severe heart failure: a systematic review and meta-analysis of randomised controlled trials. Intensive Care Med 38(3):359–367
Koster G, Bekema HJ, Wetterslev J, Gluud C, Keus F, van der Horst ICC (2016) Milrinone for cardiac dysfunction in critically ill adult patients: a systematic review of randomised clinical trials with meta-analysis and trial sequential analysis. Intensive Care Med 42(9):1322–1335
Ziff OJ, Lane DA, Samra M, Griffith M, Kirchhof P, Lip GYH et al (2015) Safety and efficacy of digoxin: systematic review and meta-analysis of observational and controlled trial data. Br Med J 351:1–9
Adams KF Jr, Fonarow GC, Emerman CL, LeJemtel TH, Costanzo MR, Abraham WT et al (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 149(2):209–216
Goto Y, Hata K (1997) Mechanoenergetic effect of pimobendan in failing dog hearts. Heart Vessel 12(Suppl):103–105
Asanoi H, Ishizaka S, Kameyama T, Ishise H, Sasayama S (1994) Disparate inotropic and lusitropic responses to pimobendan in conscious dogs with tachycardia-induced heart failure. J Cardiovasc Pharmacol 23(2):268–274
Boswood A (2010) Current use of pimobendan in canine patients with heart disease. Vet Clin N Am Small Anim Pract 40(4):571–580
Lubsen J, Just H, Hjalmarsson AC, La Framboise D, Remme WJ, Heinrich-Nols J et al (1996) Effect of pimobendan on exercise capacity in patients with heart failure: main results from the Pimobendan in Congestive Heart Failure (PICO) trial. Heart. 76(3):223–231
Kato K, Iizuka M, Yazaki Y, Sasayama S, Nakashima M, Ohashi Y et al (2002) Effects of pimobendan on adverse cardiac events and physical activities in patients with mild to moderate chronic heart failure—the effects of pimobendan on chronic heart failure study (EPOCH study). Circulation 66(2):149–157
Papp Z, Edes I, Fruhwald S, De Hert SG, Salmenpera M, Leppikangas H et al (2012) Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol 159(2):82–87
Robertson IM, Pineda-Sanabria SE, Yan Z, Kampourakis T, Sun YB, Sykes BD et al (2016) Reversible covalent binding to cardiac troponin C by the Ca2+-sensitizer Levosimendan. Biochemistry. 55(43):6032–6045
Facundo HTF, Fornazari M, Kowaltowski AJ (2006) Tissue protection mediated by mitochondrial K+ channels. Biochim Biophys Acta (BBA)-Mol Basis Dis 1762(2):202–212
Kaheinen P, Pollesello P, Levijoki J, Haikala H (2004) Effects of levosimendan and milrinone on oxygen consumption in isolated guinea-pig heart. J Cardiovasc Pharmacol 43(4):555–561
Ukkonen H, Saraste M, Akkila J, Knuuti MJ, Lehikoinen P, Nagren K et al (1997) Myocardial efficiency during calcium sensitization with levosimendan: a noninvasive study with positron emission tomography and echocardiography in healthy volunteers. Clin Pharmacol Ther 61(5):596–607
Tarkia M, Stark C, Haavisto M, Kentala R, Vähäsilta T, Savunen T et al (2016) Effect of levosimendan therapy on myocardial infarct size and left ventricular function after acute coronary occlusion. Heart. 102(6):465
Honisch A, Theuring N, Ebner B, Wagner C, Strasser RH, Weinbrenner C (2010) Postconditioning with levosimendan reduces the infarct size involving the PI3K pathway and KATP-channel activation but is independent of PDE-III inhibition. Basic Res Cardiol 105(2):155–167
Nieminen MS, Buerke M, Cohen-Solal A, Costa S, Edes I, Erlikh A et al (2016) The role of levosimendan in acute heart failure complicating acute coronary syndrome: a review and expert consensus opinion. Int J Cardiol 218:150–157
Follath F, Cleland JG, Just H, Papp JG, Scholz H, Peuhkurinen K et al (2002) Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomised double-blind trial. Lancet. 360(9328):196–202
Moiseyev VS, Poder P, Andrejevs N, Ruda MY, Golikov AP, Lazebnik LB et al (2002) Safety and efficacy of a novel calcium sensitizer, levosimendan, in patients with left ventricular failure due to an acute myocardial infarction. A randomized, placebo-controlled, double-blind study (RUSSLAN). Eur Heart J 23(18):1422–1432
Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ et al (2007) Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE randomized trial. J Am Med Assoc 297(17):1883–1891
Landoni G, Biondi-Zoccai G, Greco M, Greco T, Bignami E, Morelli A et al (2012) Effects of levosimendan on mortality and hospitalization. A meta-analysis of randomized controlled studies. Crit Care Med 40(2):634–646
Jüni P, Witschi A, Bloch R, Egger M (1999) The hazards of scoring the quality of clinical trials for meta-analysis. J Am Med Assoc 282(11):1054–1060
Nieminen MS, Fruhwald S, Heunks LMA, Suominen PK, Gordon AC, Kivikko M et al (2013) Levosimendan: current data, clinical use and future development. Heart Lung Vessels 5(4):227–245
Altenberger J, Parissis JT, Costard-Jaeckle A, Winter A, Ebner C, Karavidas A et al (2014) Efficacy and safety of the pulsed infusions of levosimendan in outpatients with advanced heart failure (LevoRep) study: a multicentre randomized trial. Eur J Heart Fail 16(8):898–906
Nieminen MS, Altenberger J, Ben-Gal T, Bohmer A, Comin-Colet J, Dickstein K et al (2014) Repetitive use of levosimendan for treatment of chronic advanced heart failure: clinical evidence, practical considerations, and perspectives: an expert panel consensus. Int J Cardiol 174(2):360–367
Orstavik O, Ata SH, Riise J, Dahl CP, Andersen GO, Levy FO et al (2014) Inhibition of phosphodiesterase-3 by levosimendan is sufficient to account for its inotropic effect in failing human heart. Br J Pharmacol 171(23):5169–5181
Hasenfuss G, Pieske B, Castell M, Kretschmann B, Maier LS, Just H (1998) Influence of the novel inotropic agent levosimendan on isometric tension and calcium cycling in failing human myocardium. Circulation. 98(20):2141–2147
Farmakis D, Alvarez J, Ben Gal T, Brito D, Fedele F, Fonseca C et al (2016) Levosimendan beyond inotropy and acute heart failure: evidence of pleiotropic effects on the heart and other organs: an expert panel position paper. Int J Cardiol 222:303–312
Teerlink JR (2009) A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev 14(4):289–298
Hwang PM, Sykes BD (2015) Targeting the sarcomere to correct muscle function. Nat Rev Drug Discov 14(5):313–328
Mamidi R, Gresham KS, Li A, dos Remedios CG, Stelzer JE (2015) Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein-C. J Mol Cell Cardiol 85:262–272
Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD et al (2011) Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 378(9792):667–675
Cleland JGF, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJV, Lang CC et al (2011) The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet. 378(9792):676–683
Teerlink JR, Felker GM, McMurray JJV, Ponikowski P, Metra M, Filippatos GS et al (2016) Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure the ATOMIC-AHF study. J Am Coll Cardiol 67(12):1444–1455
Teerlink JR, Felker GM, McMurray JJV, Solomon SD, Adams KF, Cleland JGF et al (2016) Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC-HF): a phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet. 388(10062):2895–2903
Greenberg BH, Chou W, Saikali KG, Escandón R, Lee JH, Chen MM et al (2015) Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail 3(1):22–29
Nagy L, Kovacs A, Bodi B, Pasztor ET, Fulop GA, Toth A et al (2015) The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol 172(18):4506–4518
Bakkehaug JP, Kildal AB, Engstad ET, Boardman N, Naesheim T, Ronning L et al (2015) Myosin activator omecamtiv mecarbil increases myocardial oxygen consumption and impairs cardiac efficiency mediated by resting myosin ATPase activity. Circ-Heart Fail 8(4):766–775
Oliva F, Latini R, Politi A, Staszewsky L, Maggioni AP, Nicolis E et al (1999) Intermittent 6-month low-dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am Heart J 138(2 Pt 1):247–253
Cohn JN, Goldstein SO, Greenberg BH, Lorell BH, Bourge RC, Jaski BE et al (1998) A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. N Engl J Med 339(25):1810–1816
Cuffe MS, Califf RM, Adams KF Jr, Benza R, Bourge R, Colucci WS et al (2002) Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 287(12):1541–1547
De Luca L, Colucci WS, Nieminen MS, Massie BM, Gheorghiade M (2006) Evidence-based use of levosimendan in different clinical settings. Eur Heart J 27(16):1908–1920
Packer M (2005) Revive II trial investigators: REVIVE II: multicenter placebo-controlled trial of levosimendan on clinical status in acutely decompensated heart failure. Circulation. 112:3363
Gheorghiade M, Blair JEA, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J et al (2008) Hemodynamic, echocardiographic, and neurohormonal effects of istaroxime, a novel intravenous inotropic and lusitropic agent—a randomized controlled trial in patients hospitalized with heart failure. J Am Coll Cardiol 51(23):2276–2285
Doenst T, Nguyen TD, Abel ED (2013) Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113(6):709–724
Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR et al (2017) Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14(4):238–250
Depre C, Vanoverschelde J-LJ, Taegtmeyer H (1999) Glucose for the heart. Circulation. 99(4):578–588
Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W et al (1985) The creatine kinase system in normal and diseased human myocardium. N Engl J Med 313(17):1050–1054
Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S et al (2003) Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol 42(9):1587–1593
Weiss RG, Gerstenblith G, Bottomley PA (2005) ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A 102(3):808–813
Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W et al (1997) Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 96(7):2190–2196
Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD (2011) Targeting fatty acid and carbohydrate oxidation—a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 1813(7):1333–1350
Karbowska J, Kochan Z, Smolenski RT (2003) Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett 8(1):49–53
Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ (2009) PGC-1 alpha and ERR alpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol 46(2):201–212
Kolwicz SC, Tian R (2011) Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90(2):194–201
Sorokina N, O’Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF et al (2007) Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation. 115(15):2033–2041
Lopatin YM, Rosano GM, Fragasso G, Lopaschuk GD, Seferovic PM, Gowdak LH et al (2016) Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int J Cardiol 203:909–915
Gao D, Ning N, Niu X, Hao G, Meng Z (2011) Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart. 97(4):278–286
Fragasso G, Rosano G, Baek SH, Sisakian H, Di Napoli P, Alberti L et al (2013) Effect of partial fatty acid oxidation inhibition with trimetazidine on mortality and morbidity in heart failure: results from an international multicentre retrospective cohort study. Int J Cardiol 163(3):320–325
Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L et al (2005) Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 112(21):3280–3288
Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F et al (2015) Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. J American C C: Heart Fail 3(3):202–211
Unger SA, Kennedy JA, McFadden-Lewis K, Minerds K, Murphy GA, Horowitz JD (2005) Dissociation between metabolic and efficiency effects of perhexiline in normoxic rat myocardium. J Cardiovasc Pharmacol 46(6):849–855
Chong CR, Sallustio B, Horowitz JD (2016) Drugs that affect cardiac metabolism: focus on Perhexiline. Cardiovasc Drugs Ther 30(4):399–405
Barclay ML, Sawyers SM, Begg EJ, Zhang M, Roberts RL, Kennedy MA et al (2003) Correlation of CYP2D6 genotype with perhexiline phenotypic metabolizer status. Pharmacogenetics 13(10):627–632
Banerjee K, Ghosh RK, Kamatam S, Banerjee A, Gupta A (2017) Role of Ranolazine in cardiovascular disease and diabetes: exploring beyond angina. Int J Cardiol 227:556–564
Belardinelli L, Shryock JC, Fraser H (2006) Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 92(suppl 4):iv6
Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K et al (1994) Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 23(7):1617–1624
Lewis JF, DaCosta M, Wargowich T, Stacpoole P (1998) Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol 21(12):888–892
Lopaschuk GD (2016) Metabolic modulators in heart disease: past, present, and future. Can J Cardiol 33(7):838–849
Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE et al (2006) Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation. 114(20):2130–2137
Schulze PC, Drosatos K, Goldberg IJ (2016) Lipid use and misuse by the heart. Circ Res 118(11):1736–1751
Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A et al (2002) The cardiac phenotype induced by PPAR-alpha overexpression mimics that caused by diabetes mellitus. J Clin Investig 109(1):121–130
Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K et al (2017) Activation of PPAR-alpha in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am J Phys Heart Circ Phys 312(2):H305–HH13
Pol CJ, Lieu M, Drosatos K (2015) PPARs: protectors or opponents of myocardial function? PPAR Res 2015:1–19
Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P et al (1987) Helsinki heart study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 317(20):1237–1245
Sarma S, Ardehali H, Gheorghiade M (2012) Enhancing the metabolic substrate: PPAR-alpha agonists in heart failure. Heart Fail Rev 17(1):35–43
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90(1):207–258
Cleland JG, Hutchinson K, Pellicori P, Clark A (2014) Lipid-modifying treatments for heart failure: is their use justified? Heart Fail Clin 10(4):621–634
Rosca MG, Tandler B, Hoppel CL (2013) Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol 55:31–41
Schwarz K, Siddiqi N, Singh S, Neil CJ, Dawson DK, Frenneaux MP (2014) The breathing heart—mitochondrial respiratory chain dysfunction in cardiac disease. Int J Cardiol 171(2):134–143
Brunel-Guitton C, Levtova A, Sasarman F (2015) Mitochondrial diseases and cardiomyopathies. Can J Cardiol 31(11):1360–1376
Arcaro A, Pirozzi F, Angelini A, Chimenti C, Crotti L, Giordano C et al (2016) Novel perspectives in redox biology and pathophysiology of failing myocytes: modulation of the intramyocardial redox milieu for therapeutic interventions—a review article from the working group of cardiac cell biology, Italian Society of Cardiology. Oxidative Med Cell Longev 2016:6353469
Grois L, Hupf J, Reinders J, Schroder J, Dietl A, Schmid PM et al (2017) Combined inhibition of the renin-angiotensin system and neprilysin positively influences complex mitochondrial adaptations in progressive experimental heart failure. PLoS One 12(1):1–21
Sharma A, Fonarow GC, Butler J, Ezekowitz JA, Felker GM (2016) Coenzyme Q10 and heart failure: a state-of-the-art review. Circ-Heart Fail 9(4):1–8
Molyneux SL, Florkowski CM, George PM, Pilbrow AP, Frampton CM, Lever M et al (2008) Coenzyme Q10: an independent predictor of mortality in chronic heart failure. J Am Coll Cardiol 52(18):1435–1441
Parihar P, Parihar MS (2017) Metabolic enzymes dysregulation in heart failure: the prospective therapy. Heart Fail Rev 22(1):109–121
Paradies G, Petrosillo G, Paradies V, Reiter RJ, Ruggiero FM (2010) Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J Pineal Res 48(4):297–310
Holzem KM, Vinnakota KC, Ravikumar VK, Madden EJ, Ewald GA, Dikranian K et al (2016) Mitochondrial structure and function are not different between nonfailing donor and end-stage failing human hearts. FASEB J 30(8):2698–2707
Cordero-Reyes AM, Gupte AA, Youker KA, Loebe M, Hsueh WA, Torre-Amione G et al (2014) Freshly isolated mitochondria from failing human hearts exhibit preserved respiratory function. J Mol Cell Cardiol 68:98–105
Hackam DG (2007) Translating animal research into clinical benefit—poor methodological standards in animal studies mean that positive results may not translate to the clinical domain. Br Med J 334(7586):163–164
Chandrasekera PC, Pippin JJ (2015) The human subject: an integrative animal model for 21(st) century heart failure research. Am J Transl Res 7(9):1636–1647
Wever-Pinzon O, Drakos SG, McKellar SH, Horne BD, Caine WT, Kfoury AG et al (2016) Cardiac recovery during long-term left ventricular assist device support. J Am Coll Cardiol 68(14):1540–1553
Dandel M, Hetzer R (2018) Recovery of failing hearts by mechanical unloading: pathophysiologic insights and clinical relevance. Am Heart J 206:30–50
Halliday BP, Wassall R, Lota AS, Khalique Z, Gregson J, Newsome S et al (2019) Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED-HF): an open-label, pilot, randomised trial. Lancet 393(10166):61–73
Uriel N, Kim G, Burkhoff D (2017) Myocardial recovery after LVAD implantation a vision or simply an illusion? J Am Coll Cardiol 70(3):355–357
Frigerio M, Roubina E (2005) Drugs for left ventricular remodeling in heart failure. Am J Cardiol 96(12A):10L–18L
Jweied E, de Tombe P, Buttrick PM (2007) The use of human cardiac tissue in biophysical research: the risks of translation. J Mol Cell Cardiol 42(4):722–726
Acknowledgements
For Figs. 1 and 2, the authors acknowledge the use of Motifolio© PowerPoint SmartArt Objects that were modified and re-edited to suite content.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ge, Z., Li, A., McNamara, J. et al. Pathogenesis and pathophysiology of heart failure with reduced ejection fraction: translation to human studies. Heart Fail Rev 24, 743–758 (2019). https://doi.org/10.1007/s10741-019-09806-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-019-09806-0