
Abstract
Heart failure (HF) represents a global public health and economic problem associated with unacceptable rates of death, hospitalization, and healthcare expenditure. Despite available therapy, HF carries a prognosis comparable to many forms of cancer with a 5-year survival rate of ~50 %. The current treatment paradigm for HF with reduced ejection fraction (EF) centers on blocking maladaptive neurohormonal activation and decreasing cardiac workload with therapies that concurrently lower blood pressure and heart rate. Continued development of hemodynamically active medications for stepwise addition to existing therapies carries the risk of limited tolerability and safety. Moreover, this treatment paradigm has thus far failed for HF with preserved EF. Accordingly, development of hemodynamically neutral HF therapies targeting primary cardiac pathologies must be considered. In this context, a partial adenosine A1 receptor (A1R) agonist holds promise as a potentially hemodynamically neutral therapy for HF that could simultaneous improve cardiomyocyte energetics, calcium homeostasis, cardiac structure and function, and long-term clinical outcomes when added to background therapies. In this review, we describe the physiology and pathophysiology of HF as it relates to adenosine agonism, examine the existing body of evidence and biologic rationale for modulation of adenosine A1R activity, and review the current state of drug development of a partial A1R agonist for the treatment of HF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The global prevalence of heart failure (HF) continues to rise and carry significant mortality, morbidity, and healthcare costs. Despite provision of guideline-recommended therapies, survival for patients with HF is comparable or worse than many cancers [1]. Although multiple early therapeutic breakthroughs have substantially improved outcomes for patient with chronic HF and reduced ejection fraction (EF), with few notable exceptions [2, 3], there have been few successes in the HF clinical trial arena over the past decade [4]. Built on the success of early trials, the current treatment paradigm for patients with HF with reduced EF (HFrEF) centers on systemic blockade of maladaptive neurohormones in efforts to attenuate, halt, or reverse adverse cardiac remodeling and improve clinical outcomes. However, these therapies may carry significant concurrent hemodynamic effects (i.e., reducing heart rate, preload, and/or afterload) and repeated stepwise addition of such hemodynamically active medications raises tolerability and safety concerns (e.g., hypotension and bradyarrhythmias) [5]. Likewise, the high failure rate of phase III HFrEF trials in recent years suggests that incremental benefit with the addition of such agents may be difficult or unattainable [6], and hemodynamic compromise represents a frequent reason for failed HF drug development [5]. This difficult landscape for HF drug development is particularly troubling considering the complete absence of effective therapy for HF with preserved EF (HFpEF), a condition that accounts for approximately half of all HF diagnoses. Attempts to reproduce benefits of neurohormonal antagonism seen in HFrEF have thus far been unsuccessful in HFpEF populations.
The difficulties encountered in the discovery of novel effective drugs for the treatment of HFrEF and HFpEF have fostered research efforts to develop so-called hemodynamically neutral treatments that directly target intrinsic myocardial pathophysiology, while manipulating central hemodynamics to a much lesser extent [5, 7–11]. Along these lines, partial adenosine A1 receptor (A1R) agonism represents a promising novel approach. Preclinical data using these partial agonists suggest an ability to attenuate or reverse progressive left ventricular (LV) remodeling and favorably influence myocardial energetics without eliciting hemodynamic effects such as hypotension or bradycardia [12]. In this review, we examine the physiology and pathophysiology of HF as it relates to adenosine agonism, review the existing body of evidence and biologic rationale for modulation of adenosine A1R activity in HF, and describe the current state of drug development of a partial A1R agonist for HF.
Adenosine A1 receptor physiology
Adenosine is a purine nucleoside that exerts a variety of physiological actions by binding to adenosine cell surface receptor subtypes, namely A1, A2a, A2b, and A3. Adenosine acts as a cytoprotective modulator linking cardiac function to metabolic demand under physiological and pathophysiologic conditions. The primary physiological undertaking of adenosine is to preclude tissue injury and promote repair in response to stress by different mechanisms [13–15]. The cardio-protective effects of adenosine have been extensively studied and are primarily mediated by activation of the A1R subtype. The adenosine A1R is a G-protein-coupled receptor expressed in multiple body tissues including the heart, brain, adipose tissue, and kidney. In the heart, the highest densities of receptors are found in smooth muscle cells and cardiomyocytes within the atria, with lower expression in the ventricles [16]. Activation of adenosine A1R results in downstream inhibition of adenyl cyclase and reduction in intracellular levels of cyclic adenosine monophosphate (cAMP) [15, 17]. Such adenyl cyclase modulation combats sympathetic over-activation and augments release of atrial natriuretic peptide, thus favoring a cardioprotective state [18–20]. Adenosine A1Rs also influence phospholipase C-mediated processes. The modulation of mitochondrial KATP channels via A1 receptor activation of protein kinase C results in reduction in mitochondrial protein transition pore opening and thereby improvement of mitochondrial function under hypoxic conditions [21]. Intracardiac calcium currents are also modulated by the A1R, primarily at the atrioventricular node via nitric oxide-dependent mechanisms [22]. Furthermore, the A1R is coupled to pertussis-toxin-sensitive potassium channels and KATP channels [23].
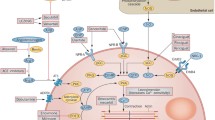
Collectively, via these processes, A1R activation favors cardiac protection. However, activation of the A1R using full agonists, while offering potential therapeutic benefits, is limited by side effects including bradycardia, atrioventricular blocks, vasoconstriction, negative inotropy and dromotropy, sedation, and anti-diuretic effects [15]. These potentially undesirable physiological effects may be overcome with use of a partial A1R agonist (Fig. 1) [12, 24].

Adenosine A1 receptor activation by a full or partial agonist or blockade by an antagonist in the presence of adenosine
Adenosine modulation in heart failure
To date, there has been only one published preclinical study exploring the impact of partial adenosine A1R agonism in HF [12]. In contrast, extensive work has explored adenosine A1R antagonism, driven primarily by perceived benefits of adenosine blockade on renal function, including prevention of afferent arteriole constriction and proximal tubule sodium reabsorption [25]. This potential for renal benefit with adenosine antagonism led to large-scale drug development programs with adenosine A1R antagonists, such as rolofylline, that included multiple phase II and phase III clinical trials. Phase II experiences were generally “positive,” suggesting rolofylline could augment urine output while preserving or improving glomerular filtration rate among patients with HFrEF and moderate renal insufficiency [4, 26, 27]. However, the subsequent large phase III study was neutral, demonstrating no benefit on symptoms, renal function, mortality, or rehospitalization [28]. Also, higher rates of persistent renal impairment, seizure, and stroke were noted in the rolofylline group [28, 29]. Indeed, ~1 % of rolofylline patients experienced seizures, a well-recognized risk of adenosine A1R antagonism, as compared to none with placebo [28, 30]. Failure of the rolofylline program provided support, albeit indirect, to continue development of a partial adenosine agonist program for the treatment of HF.
Rationale for partial adenosine A1 receptor agonism for heart failure
The goal of HF therapy is to restore cardiac structure and function, or at least, slow the rate of progression toward a worsening HF state. Ideally, such improvements in intrinsic cardiac structure would be durable and persist for a reasonable period of time even after withdrawal of the therapy (i.e., not related directly to the immediate ingestion of the medication). Therapies proven to improve survival in HFrEF (e.g., beta-blockers, angiotensin-converting enzyme inhibitors) have shown the ability to reverse remodel the failing LV. This benefit, however, may come at a risk of significant hemodynamic effects that can limit tolerability or prevent achievement of target dosing (e.g., in one long-term registry, only 30 % of patients received target dosages of these drugs) [31]. In contrast, positive inotropic agents can improve hemodynamics acutely but at the risk of increased mortality [32].
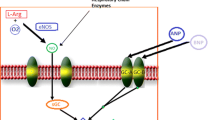
Accordingly, alternative therapeutic targets with hemodynamically neutral mechanisms (i.e., minimal hypotension and bradycardia) are being increasingly explored. Such agents include those that improve mitochondrial function, energetics, intracellular calcium handling, cardioprotection, and reversal of interstitial fibrosis. Despite potential therapeutic benefits with full A1R agonism, undesired effects of this strategy have limited further drug development. Accordingly, efforts have been directed toward development of partial A1R agonists that elicit beneficial effects (e.g., cardioprotection, reverse LV remodeling) while maximizing safety by avoiding off target side effects. Based on pharmacologic and preclinical data, as compared to full agonists, partial agonists exert minimal effects on blood pressure, heart rate, AV conduction, central nervous system, and renal function [12, 24]. Furthermore, no relevant negative inotropy is observed in animal models [12]. Thus, a partial A1R agonist may fulfill many of the characteristics of an ideal hemodynamically neutral HF therapy and exert benefits via multiple mechanisms (Table 1; Fig. 2).

Adenosine A1 receptor signaling pathways in the failing heart. AC adenylate cyclase; K ATP ATP-dependent potassium channel, MPTP mitochondrial permeability transition pore, PKA phosphokinase A, PLC phospholipase C, PLD phospholipase D, SR sarcoplasmic reticulum
Mitochondrial function
The failing heart is characterized by abnormal mitochondrial structure and function including hyperplasia and reduced organelle size, poor organelle respiration, reduced mitochondrial membrane potential, opening of membrane permeability pores, and reduced rates of adenosine triphosphate (ATP) synthesis [8, 12, 33, 34]. In addition, mitochondrial dysfunction is associated with derangements in the electron transport chain and excessive production of reactive oxygen species (ROS) [8, 34, 35]. Excessive generation of ROS by mitochondria can exert widespread adverse effects on the heart through damage to cellular and subcellular components that include key signaling pathways, membrane lipid sublayers, and the extracellular matrix [8, 36]. Mitochondrial uncoupling proteins, particularly UCP-2 and UCP-3, are necessary for preserving appropriate mitochondrial membrane potential and regulating production of ATP and ROS [37, 38]. Thus, restoring the integrity of mitochondrial membranes and optimizing their number and function represent emerging therapeutic targets.
Canine models receiving the partial A1R agonist capadenoson offer strong evidence that modulation of this pathway is a viable strategy for treating mitochondrial dysfunction in HF [12]. In the untreated control HF arm, expression of UCP-2 and UCP-3 was reduced compared to normal, whereas in HF animals treated with capadenoson, a near normalization of UCP-2 and UCP-3 protein levels was observed [12]. Likewise, treatment with capadenoson improved sarcoplasmic reticulum calcium-ATPase (SERCA-2a) activity, leading to decreased intracellular calcium overload (Fig. 3) [12]. Lastly, capadenoson appeared to regulate mitochondrial permeability transition pores (mPTP), a key regulator of cellular apoptosis [12]. In HF, these pores may demonstrate increased opening, facilitating increased rates of apoptosis [39]. Preclinical evidence suggests adenosine agonists are capable of preventing this increased rate of mPTP opening and maintaining mitochondrial and cell viability [21]. These results are consistent with findings from the capadenoson canine study, where adenosine normalized expression of citrate synthase, an established marker of intact mitochondria [12]. In aggregate, these effects of partial adenosine A1R agonism support improvement in the efficiency of mitochondrial electron transport chain and, therefore, a potential for an improvement in the rate of ATP synthesis and attenuation of excessive ROS formation.

Left panel: Bar graphs depicting changes in Ca2+-ATPase activity (Vmax) in left ventricular (LV) myocardium. Right panel: Bar graphs depicting changes in affinity for calcium (Ka) in LV myocardium. Changes are for normal (NL) dogs, untreated heart failure control dogs (CON), and dogs with heart failure treatment with capadenoson (CAP). Data are shown as mean ± SEM. *P < 0.05 versus NL; **P < 0.05 versus CON. Reprinted with permission from Sabbah et al. [12]
Myocardial energy substrate utilization
Besides mitochondrial dysfunction, the failing heart may undergo other separate maladaptive changes in energetics. HF is characterized by an added reliance on fatty acid oxidation, with downregulation of myocardial glucose transporters [40, 41]. These changes characterize the transition of the failing heart to a fetal metabolic phenotype and gene profile, an adaptation that can further promote HF progression [40–42]. Animal studies suggest that partial A1R agonists can augment expression of the GLUT-1 and GLUT-4 glucose transporters to near-normal levels [12]. Moreover, therapy with a partial A1R agonist has also been associated with normalization of protein levels that mediate fatty acid oxidation and reduction in free fatty acid plasma levels [12, 43]. Thus, A1R agonism appears capable of partially correcting derangements in cardiac substrate utilization and restoring a physiological metabolic profile in HF.
Reverse remodeling
Decreases in ventricular filling pressures and augmentation of cardiac output can be seen acutely with multiple medications and are not uniformly associated with improved outcome [32]. Structural recovery of the failing heart with global reverse ventricular remodeling represents a more elusive goal but one that is more often associated with long-term clinical benefit. Overall, the biology of cardiac structural recovery in HF is not fully understood but has been described as at least partial reversal of cardiomyocyte structure and function and extracellular matrix abnormalities facilitating improved ventricular geometry and contractile performance [44]. The relative proportion of dysfunctional viable myocardium and the nature of the myocardial interstitium are recognized as key factors in this process [7, 9].
Beyond beneficial effects on cardiac myocytes themselves, adenosine A1R agonism may exhibit favorable remodeling by altering the extracellular matrix with a reduction in interstitial fibrosis. Such changes would favor improvements in ventricular compliance and filling, as well as preservation of acceptable cardiac capillary density and oxygen diffusion distances. Twelve-week treatment with capadenoson was associated with a significant increase in EF and decreases in end-diastolic and systolic volumes and natriuretic peptides (Fig. 4) [12]. Moreover, capadenoson treatment mediated improvements in volume fraction of interstitial fibrosis, capillary density, oxygen diffusion distance, and myocyte hypertrophy [12].

Left panel: Change (treatment effect) between pre-treatment and 12 weeks post-treatment of left ventricular (LV) end-diastolic volume (EDV), end-systolic volume (ESV), ejection fraction (EF), and stroke volume (SV) in untreated control dogs and dogs treated for 3 months with capadenoson. Right panel: Change (treatment effect) between pre-treatment and 12 weeks post-treatment in plasma levels of n-terminal pro-brain natriuretic peptide (NT-proBNP). Data are shown as mean ± SEM. Probability values are comparisons between untreated control and capadenoson. Reprinted with permission from Sabbah et al. [12]
Cardioprotection
Adenosine has been shown in animal and human studies to be cardioprotective [45, 46]. The major underlying mechanism appears to be maintenance of intracellular homeostasis and ischemia prevention, potentially mediated by activation of downstream effectors (e.g., protein kinase C, KATP channels, mitogen-activated protein kinase) and inhibition of adenylate cyclase activation and reduction in cyclic adenosine monophosphate [47, 48]. Among patients with angina pectoris, treatment with capadenoson prolonged total exercise time and time to ischemia [49]. Adenosine carries anti-adrenergic properties that can protect the heart from adverse mechanical and metabolic activation from excessive catecholamine stimulation, thereby limiting ischemia. Activation of the A1R may also inhibit norepinephrine release [50]. Conceivably, these effects may be important for preventing disease progression and further adverse remodeling, particularly in those ongoing or latent ischemia.
Development of a partial A1R agonist for human heart failure
HF is not a disease but a final common syndrome stemming from various admixtures of cardiac and noncardiac abnormalities. As we increasingly recognize the heterogeneity in cardiac substrates, comorbidities, biomarkers, and reasons for HF progression, it has been proposed that future development of novel therapeutics begins with investigation in homogenous HF subsets deemed most likely to benefit from the study drug [51]. Generally speaking, it is felt that patients with large segments of dysfunctional but viable myocardium constitute an ideal platform for testing novel therapies, but other traditional parameters such as EF, natriuretic peptides, and comorbidities (e.g., atrial fibrillation, coronary artery disease, diabetes, renal disease) should continue to be considered in conjunction [4, 7, 11].
Identifying hurdles to successful development of these agents is difficult at this early stage. Obvious attention must be given to monitoring for side effects typical of full adenosine agonists to ensure that the sound preclinical safety profile of partial agonists remains applicable to human subjects. New opportunities are emerging in the area of “eHealth” that offer potentially useful tools for ensuring patient safety. For example, remote telemonitoring devices can continuously monitor and automatically transmit electrocardiogram data to centers for real-time monitoring (e.g., auto-trigger electrocardiogram data upon event onset with notification episode reporting). However, although such novel methods of patient monitoring and selection may be helpful in homogenizing study cohorts in smaller phase II/proof-of-concept studies (e.g., monitoring devices, biomarkers, genomics, cardiac magnetic resonance), the feasibility of these instruments in subsequent large trials remains to be seen.
Conclusions
Heart failure remains a growing public health problem with unacceptable long-term outcomes despite the use of guideline-directed medical and device therapies. Continued development of more potent hemodynamically active medications for stepwise addition or replacement of existing therapy carries an inherent risk of poor tolerability and safety [5]. Moreover, application of this approach in HFpEF has been unsuccessful. It is under this framework where a partial A1R agonist for HF holds promise—a potentially hemodynamically neutral therapy that could simultaneously improve cardiomyocyte energetics, cardiac structure and function, and prevent further tissue injury while being well tolerated when added to background medical therapy. Although drug development of a partial A1R agonist remains at an early stage, compelling biologic rationale and encouraging preclinical evidence support continued attention be given to ongoing and future trials with these agents in HF.
References
Butler J, Fonarow GC, Gheorghiade M (2013) Need for increased awareness and evidence-based therapies for patients hospitalized for heart failure. JAMA 310:2035–2036
Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost-Brama A, Lerebours G, Tavazzi L (2010) Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 376:875–885
McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR (2014) Angiotensin–neprilysin inhibition versus enalapril in heart failure. N Engl J Med 371:993–1004
Vaduganathan M, Greene SJ, Ambrosy AP, Gheorghiade M, Butler J (2013) The disconnect between phase II and phase III trials of drugs for heart failure. Nat Rev Cardiol 10:85–97
Vaduganathan M, Butler J, Pitt B, Gheorghiade M (2015) Contemporary drug development in heart failure: call for hemodynamically neutral therapies. Circ Heart Fail 8:826–831
Gheorghiade M, Bohm M, Greene SJ, Fonarow GC, Lewis EF, Zannad F, Solomon SD, Baschiera F, Botha J, Hua TA, Gimpelewicz CR, Jaumont X, Lesogor A, Maggioni AP (2013) Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: the ASTRONAUT randomized trial. JAMA 309:1125–1135
Bayeva M, Sawicki KT, Butler J, Gheorghiade M, Ardehali H (2014) Molecular and cellular basis of viable dysfunctional myocardium. Circ Heart Fail 7:680–691
Bayeva M, Gheorghiade M, Ardehali H (2013) Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61:599–610
Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M (2014) Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J Am Coll Cardiol 63:2188–2198
Greene SJ, Gheorghiade M, Borlaug BA, Pieske B, Vaduganathan M, Burnett JC Jr, Roessig L, Stasch JP, Solomon SD, Paulus WJ, Butler J (2013) The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc 2:e000536
Wilcox JE, Fonarow GC, Ardehali H, Bonow RO, Butler J, Sauer AJ, Epstein SE, Khan SS, Kim RJ, Sabbah HN, Diez J, Gheorghiade M (2015) “Targeting the Heart” in heart failure: myocardial recovery in heart failure with reduced ejection fraction. JACC Heart Fail 3:661–669
Sabbah HN, Gupta RC, Kohli S, Wang M, Rastogi S, Zhang K, Zimmermann K, Diedrichs N, Albrecht-Kupper BE (2013) Chronic therapy with a partial adenosine A1-receptor agonist improves left ventricular function and remodeling in dogs with advanced heart failure. Circ Heart Fail 6:563–571
Linden J (2005) Adenosine in tissue protection and tissue regeneration. Mol Pharmacol 67:1385–1387
Brodde OE, Michel MC (1999) Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 51:651–690
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552
Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118:1461–1468
Akbar M, Okajima F, Tomura H, Shimegi S, Kondo Y (1994) A single species of A1 adenosine receptor expressed in Chinese hamster ovary cells not only inhibits cAMP accumulation but also stimulates phospholipase C and arachidonate release. Mol Pharmacol 45:1036–1042
Wang D, Belardinelli L (1994) Mechanism of the negative inotropic effect of adenosine in guinea pig atrial myocytes. Am J Physiol 267:H2420–2429
Yuan K, Cao C, Han JH, Kim SZ, Kim SH (2005) Adenosine-stimulated atrial natriuretic peptide release through A1 receptor subtype. Hypertension 46:1381–1387
Schutte F, Burgdorf C, Richardt G, Kurz T (2006) Adenosine A1 receptor-mediated inhibition of myocardial norepinephrine release involves neither phospholipase C nor protein kinase C but does involve adenylyl cyclase. Can J Physiol Pharmacol 84:573–577
Xiang F, Huang YS, Zhang DX, Chu ZG, Zhang JP, Zhang Q (2010) Adenosine A1 receptor activation reduces opening of mitochondrial permeability transition pores in hypoxic cardiomyocytes. Clin Exp Pharmacol Physiol 37:343–349
Martynyuk AE, Kane KA, Cobbe SM, Rankin AC (1996) Nitric oxide mediates the anti-adrenergic effect of adenosine on calcium current in isolated rabbit atrioventricular nodal cells. Pflugers Arch 431:452–457
Kirsch GE, Codina J, Birnbaumer L, Brown AM (1990) Coupling of ATP-sensitive K+ channels to A1 receptors by G proteins in rat ventricular myocytes. Am J Physiol 259:H820–826
Albrecht-Kupper BE, Leineweber K, Nell PG (2012) Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal 8:91–99
Vallon V, Muhlbauer B, Osswald H (2006) Adenosine and kidney function. Physiol Rev 86:901–940
Givertz MM, Massie BM, Fields TK, Pearson LL, Dittrich HC (2007) The effects of KW-3902, an adenosine A1-receptor antagonist, on diuresis and renal function in patients with acute decompensated heart failure and renal impairment or diuretic resistance. J Am Coll Cardiol 50:1551–1560
Dittrich HC, Gupta DK, Hack TC, Dowling T, Callahan J, Thomson S (2007) The effect of KW-3902, an adenosine A1 receptor antagonist, on renal function and renal plasma flow in ambulatory patients with heart failure and renal impairment. J Card Fail 13:609–617
Massie BM, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Weatherley BD, Cleland JG, Givertz MM, Voors A, DeLucca P, Mansoor GA, Salerno CM, Bloomfield DM, Dittrich HC (2010) Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med 363:1419–1428
Teerlink JR, Iragui VJ, Mohr JP, Carson PE, Hauptman PJ, Lovett DH, Miller AB, Pina IL, Thomson S, Varosy PD, Zile MR, Cleland JG, Givertz MM, Metra M, Ponikowski P, Voors AA, Davison BA, Cotter G, Wolko D, Delucca P, Salerno CM, Mansoor GA, Dittrich H, O’Connor CM, Massie BM (2012) The safety of an adenosine A(1)-receptor antagonist, rolofylline, in patients with acute heart failure and renal impairment: findings from PROTECT. Drug Saf 35:233–244
Dunwiddie TV, Worth T (1982) Sedative and anticonvulsant effects of adenosine analogs in mouse and rat. J Pharmacol Exp Ther 220:70–76
Maggioni AP, Anker SD, Dahlstrom U, Filippatos G, Ponikowski P, Zannad F, Amir O, Chioncel O, Leiro MC, Drozdz J, Erglis A, Fazlibegovic E, Fonseca C, Fruhwald F, Gatzov P, Goncalvesova E, Hassanein M, Hradec J, Kavoliuniene A, Lainscak M, Logeart D, Merkely B, Metra M, Persson H, Seferovic P, Temizhan A, Tousoulis D, Tavazzi L (2013) Are hospitalized or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12,440 patients of the ESC Heart Failure Long-Term Registry. Eur J Heart Fail 15:1173–1184
Francis GS, Bartos JA, Adatya S (2014) Inotropes. J Am Coll Cardiol 63:2069–2078
Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN (2000) Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32:2361–2367
Sharov VG, Todor A, Khanal S, Imai M, Sabbah HN (2007) Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol 42:150–158
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A (1999) Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 85:357–363
Siwik DA, Colucci WS (2004) Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev 9:43–51
Laskowski KR, Russell RR 3rd (2008) Uncoupling proteins in heart failure. Curr Heart Fail Rep 5:75–79
Bugger H, Guzman C, Zechner C, Palmeri M, Russell KS, Russell RR 3rd (2011) Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother Pharmacol 67:1381–1388
Sabbah HN (2000) Apoptotic cell death in heart failure. Cardiovasc Res 45:704–712
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104:2923–2931
Sack MN, Kelly DP (1998) The energy substrate switch during development of heart failure: gene regulatory mechanisms (review). Int J Mol Med 1:17–24
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J (2007) MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116:258–267
Staehr PM, Dhalla AK, Zack J, Wang X, Ho YL, Bingham J, Belardinelli L (2013) Reduction of free fatty acids, safety, and pharmacokinetics of oral GS-9667, an A(1) adenosine receptor partial agonist. J Clin Pharmacol 53:385–392
Mann DL, Barger PM, Burkhoff D (2012) Myocardial recovery and the failing heart: Myth, magic, or molecular target? J Am Coll Cardiol 60:2465–2472
Urmaliya VB, Pouton CW, Devine SM, Haynes JM, Warfe L, Scammells PJ, White PJ (2010) A novel highly selective adenosine A1 receptor agonist VCP28 reduces ischemia injury in a cardiac cell line and ischemia-reperfusion injury in isolated rat hearts at concentrations that do not affect heart rate. J Cardiovasc Pharmacol 56:282–292
Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB (1999) Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol 34:1711–1720
Shneyvays V, Mamedova LK, Leshem D, Korkus A, Shainberg A (2002) Insights into the cardioprotective function of adenosine A(1) and A(3) receptors. Exp Clin Cardiol 7:138–145
Kitakaze M, Hori M, Takashima S, Sato H, Inoue M, Kamada T (1993) Ischemic preconditioning increases adenosine release and 5′-nucleotidase activity during myocardial ischemia and reperfusion in dogs. Implications for myocardial salvage. Circulation 87:208–215
Tendera M, Gaszewska-Zurek E, Parma Z, Ponikowski P, Jankowska E, Kawecka-Jaszcz K, Czarnecka D, Krzeminska-Pakula M, Bednarkiewicz Z, Sosnowski M, Ochan Kilama M, Agrawal R (2012) The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin Res Cardiol 101:585–591
Bott-Flugel L, Bernshausen A, Schneider H, Luppa P, Zimmermann K, Albrecht-Kupper B, Kast R, Laugwitz KL, Ehmke H, Knorr A, Seyfarth M (2011) Selective attenuation of norepinephrine release and stress-induced heart rate increase by partial adenosine A1 agonism. PLoS ONE 6:e18048
Greene SJ, Gheorghiade M (2014) Matching mechanism of death with mechanism of action: considerations for drug development for hospitalized heart failure. J Am Coll Cardiol 64:1599–1601
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Greene reports no conflicts. Dr. Sabbah has received research Grants and/or is consultant to Bayer Healthcare AG, Stealth Peptides, Inc., Amgen Corp., Johnson & Johnson, Inc., Novartis Corp., Merck, and Takeda Pharmaceuticals. Dr. Butler reports research support from the National Institutes of Health, European Union, and Health Resources Service Administration and is a consultant to Amgen, Bayer, BG Medicine, Cardiocell, Celladon, Gambro, GE Healthcare, Medtronic, Novartis, Ono Pharma, Takeda, Trevena, and Zensun. Dr Voors received consultancy fees and/or research Grants from: Alere, Amgen, Bayer, Boehringer, Cardio3Biosciences, Celladon, GSK, Merck/MSD, Novartis, Servier, Singulex, Sphingotec, Trevena, Vifor, ZS Pharma, and is supported by a Grant from the European Commission: FP7-242209-BIOSTAT-CHF. Drs. Albrecht-Küpper and Dinh are employees of Bayer Healthcare. Dr. Düngen has received research grants and/or is consultant to Bayer Healthcare AG, Amgen Corp., Novartis, Trevena, Celladon, AstraZeneca and reports research support from German Ministry of Education and Research. Dr. Gheorghiade has been a consultant for Abbott Laboratories, AstraZeneca, Bayer Schering Pharma AG, Cardiocell LLC, Cardiorentis Ltd, GlaxoSmithKline, Johnson & Johnson, Medtronic, Merck, Novartis Pharma AG, Ono Pharmaceuticals USA, Otsuka Pharmaceuticals, Sanofi-Aventis, Sigma Tau, Solvay Pharmaceuticals, Stealth BioTherapeutics, Sticares InterACT, Takeda Pharmaceuticals North America, Inc and Trevena Therapeutics.
Rights and permissions
About this article

Cite this article
Greene, S.J., Sabbah, H.N., Butler, J. et al. Partial adenosine A1 receptor agonism: a potential new therapeutic strategy for heart failure. Heart Fail Rev 21, 95–102 (2016). https://doi.org/10.1007/s10741-015-9522-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-015-9522-7