
Abstract
Myocardial infarction (MI) is a great threat to patients all over the word. MicroRNAs (miRNAs) are a group of non-coding RNAs and can regulate initiation and progression of MI. The current research aimed to investigate the role of miR-467a-5p in MI. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was conducted to detective relative expression of miR-467a-5p in cardiac tissues and mouse cardiomyocytes (MCMs). Hematoxylin and eosin staining was used to reveal the histology of the myocardium. Echocardiography was utilized to reveal cardiac function of mice. Flow cytometer analysis was used to reveal cell apoptosis. Luciferase reporter assay was applied for determining the binding capacity between molecules. We discovered that the level of miR-467a-5p was up-regulated in MI mice and in MCMs induced by H2O2 or hypoxia. Functionally, an elevation of left ventricular end-diastolic diameter and left ventricular end-systolic diameter, as well as a decrease of left ventricular ejection fraction and left ventricular fractional shortening were observed in MI mice. In addition, deficiency of miR-467a-5p improved MI in mice by increasing the contents of lactate dehydrogenase, creatine kinase and malondialdehyde and reducing the activity of superoxide dismutase in serum. Moreover, silencing of miR-467a-5p reversed hypoxia-induced apoptosis of MCMs. Mechanistically, zinc finger E-box binding homeobox 1 (ZEB1) was confirmed as the target of miR-467a-5p. Moreover, miR-467a-5p negatively regulated ZEB1 level in MI mice and MCMs. Finally, the promotive effect of miR-467a-5p inhibition on cell apoptosis was reversed by knockdown of ZEB1. All the experimental results demonstrate that miR-467a-5p aggravates MI by modulating ZEB1 expression in mice, which may provide a novel therapeutic strategy for MI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction (MI) refers to myocardial necrosis mainly caused by persistent ischemia and hypoxia in the coronary arteries (Lim et al. 2018; Venetsanos et al. 2017). Clinically, the MI patients are often diagnosed with severe and continuous post-sternal pain (Rozado et al. 2017). Unfortunately, MI might be complicated by arrhythmia, shock or heart failure, which has caused tremendous mortalities worldwide (Liang et al. 2016; Wincewicz and Sulkowski 2017). In China, approximately 24 million individuals will experience MI each year by 2030 based on the prediction of World Health Organization (Lee et al. 2016). Known factors like smoking, drinking, metabolic syndrome and hypertension are closely associated with the progression of MI (Mathiew-Quiros et al. 2017; Pedrinelli et al. 2012; Yusuf et al. 2004). However, the molecular regulatory mechanisms in MI are required to be further investigated.
MicroRNA (miRNA) is a member of non-coding RNAs (ncRNAs) with less than 24 nucleotides in length (Dong et al. 2013). Numerous evidence has showed that the dysregulated miRNA may contribute to the initiation or progression of diseases (Ameres and Zamore 2013). Particularly, miRNAs were supposed to be implicated in several biological processes. For instance, miR-377 inhibitor was reported to inhibit renal ischemia–reperfusion injury by suppressing inflammation and oxidative stress through a VEGF-dependent mechanism (Liu et al. 2019). MiR-150 inhibits cell apoptosis and autophagy in LPS-induced acute lung injury by modulating AKT3 level (Li et al. 2019). Most recently, miR-467a-5p has been identified to be upregulated in MI mice model (Zhang et al. 2019a, b). However, the specific function of miR-467a-5p in MI remains to be explored.
Mechanistically, miRNAs are able to bind with complementary sequences of downstream target genes to inhibit their expression in the posttranscriptional level (Sotillo and Thomas-Tikhonenko 2011). MiR-26a targets ataxia-telangiectasia mutated to attenuate cardiac apoptosis in MI (Chiang et al. 2020). MiR-132 inhibits myocardial remodeling in MI through inhibition on IL-1β (Zhao et al. 2020). MiR-494 induces proliferation and migration of vascular endothelial cells in rats following Myocardial Infarction through targeting LRG1 Expression (Su et al. 2019). The miR-let-7a/TGFBR3 pathway regulates apoptosis of cardiomyocyte after infarction (Chen et al. 2019).
The current project aimed to explore the specific function and molecular mechanism of miR-467a-5p in MI, which may provide a new insight into the potential therapeutic target for MI.
Materials and methods
Myocardial infarction mouse model
C57BL/6 male mice (10 weeks old, 20–25 g) were obtained from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and were randomly divided into 5 groups (n = 8 in each group). The MI mouse model was established by coronary artery ligation as described before (Zhang et al. 2019b). For heart rate analysis, once body temperature reached 37 °C, heart rate was allowed to stabilize for 10 min to determine the baseline rate using subcutaneous electrodes in lead II configuration. Animals underwent 30 min ischemia and 60 min reperfusions, and then injected with adeno-associated virus (AAV). After 4 weeks, mice were sacrificed via cervical dislocation, and the hearts were collected for follow-up analysis. The animal experimental protocols were approved by The Third Xiangya Hospital of Central South University (Hunan, China).
Cell culture and cell treatments
Mouse cardiomyocytes (MCMs) were acquired from Cell Bank of Chinese Academy of Science, (Shanghai, China) and grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Hyclone, Logan, UT, USA). Then, 10% (v/v) fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Invitrogen) were added into DMEM. For hypoxia treatments, cells were kept in an anaerobic system (Thermo Forma, Marietta, OH, USA) for 12, 24 or 48 h at 37 °C with 1% O2, 5% CO2 and 94% N2. For control group, MCMs were cultivated and operated in a humidified incubator at 37 °C with 95% air and 5% CO2 with the same time. Different concentration gradients of H2O2 (50, 100, 200 or 400 μM) were added into DMEM with the same dose of PBS as control.
Adeno-associated virus (AAV)
AAV (serotype 9) containing anti-miR-467a-5p and the empty AAV (AAV-Mock), purchased from Hanheng Company (Hanheng Biotechnology, Shanghai, China), were injected into (1012 v.g/mL) mice via tail vein to knockdown the level of miR-467a-5p in vivo. AAV injection was conducted immediately after reperfusion. Twenty-eight days later, mice heart was collected for following assays.
Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
RNA was initially extracted from cardiac tissues of mice or MCMs via TRIzol (Takara, Dalian, China). Reverse transcription was conducted with RNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) and RT-qPCR was conducted for assessing gene expression using the TanMan® microRNA Assay Kit (Life Technologies, Carlsbad, CA. USA) and iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA). Results were calculated using 2−ΔΔCt approach with GAPDH or U6 as an internal control. Primer sequences used in this assay were listed as bellow.
mmu-miR-29b-3p: forward 5′-TAG CAC CAT TTG AAA TC-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
mmu-miR-3473a: forward 5′-TAG CAC CAT TTG AAA TC-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
mmu-miR-467a-5p: forward 5′-TAA GTG CCU GCA TGT A-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
mmu-miR-30c-5p: forward 5′-TGT AAA CAT CCT ACA CT-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
mmu-miR-467a-3p: forward 5′-CAT ATA CAT ACA CAC A-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
mmu-miR-34c-3p: forward 5′-AAT CAC TAA CCA CAC A-3′ and reverse 5′-GTG CAG GGT CCG AGG T-3′;
zinc finger E-box binding homeobox 1 (ZEB1): forward 5′-CTG CTG GGA GGA TGA CAC AG-3′ and reverse 5′-GTC CTC TTC AGG TGC CTC AG-3′;
GAPDH: forward 5′-CTC AGA CAC CAT GGG GAA GGT GA-3′ and reverse 5′-ATG ATC TTG AGG CTG TTG TCA TA-3′;
U6: forward 5′-CTC GCT TCG GCA GCA CA-3′ and reverse 3′-AAC GCT TCA CGA ATT TGC GT-5′.
Echocardiographic study
Mice echocardiographic study were examined 28 days (4 weeks) after delivery of AAVs. Left ventricular end-diastolic diameter (LVEDD) as well as left ventricular end-systolic diameter (LVESD) were calculated from the parasternal long-axis view. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were studied to evaluate left ventricular systolic function.
Histopathology assay
The heart tissues were rapidly resected from anesthetized mice 28 days after delivery of AAV vectors, and then maintained in a 4% paraformaldehyde solution. Subsequently, tissues were dehydrated, cleaned, and embedded with paraffin. After that, the paraffin blocks were cut into 4 μm sections with a microtome (Leica M650; Leica Microsystems GmbH, Wetzlar, Germany). Hematoxylin and eosin (H&E, Solarbio) was used for histopathology analysis. A light microscopy (Olympus, Tokyo, Japan) was utilized to observe the histology of the myocardium.
Triphenyl tetrazolium chloride (TTC) staining
Triphenyl tetrazolium chloride (TTC) staining was utilized to measure the size of the myocardial infarction as described before (Luo et al. 2016). Twenty-eight days after injection of AAV, the mice cardiac tissues were harvested. The slices of mice heart were cultured in 1% TTC solution after being washed with ice-cold PBS (Invitrogen). The pale white areas (cannot be stained) were infarcted and red areas (stained) were not infracted. ImageJ v1.8.0 (National Institutes of Health, Bethesda, USA) was used to calculate the infarct sizes.
The measurement of area at risk (AAR)
To measure area at risk in mice’s myocardium, the heart was isolated and stained with Evan’s blue. In detail, after cannulation and washing, the aorta was infused with Evan’s blue (2 mL, 0.5%) with ligation of left anterior descending coronary artery at the same site again. The areas cannot be stained were area at risk (ischemic area), and the areas-stained blue were ischemic areas.
Myocardial enzyme determination
Twenty-eight days after injection of AAV, the bloods from the inner canthus vein were collected and kept at 4 °C for 4 h. The serum of mice was separated by centrifugation and maintained at − 80 °C. The commercial kits (Sangon-Biotech, Shanghai, China; Solarbio, Beijing, China) were applied to measure the contents of malondialdehyde (MDA), lactate dehydrogenase (LDH), creatine kinase (CK) and the activity of superoxide dismutase (SOD) in serum.
Cell transfection
Specific siRNAs against ZEB1 (si-ZEB1) with si-NC as negative control, miR-467a-5p inhibitor (anti-miR-467a-5p) with NC inhibitor as negative control, miR-467a-5p mimics (miR-467a-5p) with NC mimics as negative control were synthesized by GenePharma (Shanghai, China). Hypoxia or H2O2-treated MCM cardiomyocytes were transfected with these plasmids via Lipofectamine 2000 (Invitrogen). The oligo sequences were as follows: si-ZEB1: 5′-AACUGAACCUGUGGAUUAU-3'; si-NC: 5′-AACAGGCACACGUCCCAGCGU-3′; miR-467a-5p inhibitor: 5′-UACAUGCAGGCACUUA-3′; miR-467a-5p mimics: 5′-UAAGUGCCUGCAUGUAUAUGCG-3′.
Western blot
Total protein was extracted, followed by separation using SDS-PAGE (Beyotime, Jiangsu, China), and sequentially transferred onto PVDF membranes (Millipore, Billerica, MA, USA). Membranes were then blocked for 2 h utilizing 5% skimmed milk. Primary antibodies against Bax (ab32503, 1:1000), Bcl-2 (ab182858, 1:1000), Cleaved caspase-3 (ab49822, 1:1000), ZEB1 (ab87280, 1:1000), cTnI (ab209809, 1/2500), cTnT (ab209813, 1/5000), and GAPDH (ab245356, 1:1000) from Abcam (Cambridge, UK) were added to membranes. Membranes were subsequently incubated using secondary antibody (1:5000). Finally, proteins were visualized via ECL (Beyotime).
Luciferase reporter assay
The wild type or mutant interacting sequences of miR-467a-5p on 3’-UTR of ZEB1 were sub-cloned into the pmirGLO dual-luciferase vector (Promega, Madison, WI, USA) to generate wild type or mutant pmirGLO-ZEB1-3’UTR vectors, which were co-transfected into MCMs with miR-467a-5p inhibitor (anti-miR-467a-5p), miR-467a-5p mimics (miR-467a-5p). Luciferase activities were evaluated via Dual-Luciferase reporter assay system (Promega) upon 48 h of co-transfection.
Flow cytometer
An FITC-Annexin V/PI kit (BD Biosciences, Franklin Lakes, NJ, USA) was applied in this assay. Cardiomyocytes were washed thrice using PBS (Sigma-Aldrich), followed by being dyed for 30 min using 5 μL of FITC-Annexin V, and added with 1 μL of PI working solution for 5 min without light. Apoptotic cells were assessed with a flow cytometer (BD Biosciences).
Cell counting kit-8 assay (CCK-8) assay
The CCK-8 assay and colony formation assays were conducted to assess cell proliferation. MCMs (1,000 cells/well) were cultivated in 96-well plates. 10 µL of CCK-8 solvent was supplemented to each well. The absorbance of each well was assessed at the wavelength of 450 nm using a microplate reader.
Statistical analysis
Data were showed as means ± standard deviation. Statistical analysis was progressed using the SPSS (Chicago, IL, USA) and GraphPad Prism 5 software (San Diego, CA). Significance of the difference between 2 groups was evaluated through Student’s test, and difference among more than two groups was assessed by analysis of variance (ANOVA) followed by Tukey’s post hoc test. P < 0.05 was regarded as statistical significance. Experiments have been done at least three times.
Result
MiR-467a-5p is up-regulated in MI tissues and injured MCMs
Previous studies have identified several up-regulated miRNAs in MI patients or mice model (Zhang et al. 2019b; Zhong et al. 2018; Zhu et al. 2019; Zile et al. 2011). As depicted in Fig. 1a, miR-467a-5p displayed the most up-regulation among those miRNAs in MI mice compared with sham-operated mice. Additionally, the expression level of miR-467a-5p was upregulated in cardiac tissues of MI mice at a time dependent way (Fig. 1b). To mimic cardiomyocyte injury in vitro, H2O2 and hypoxia condition were used to treat MCMs. According to the result of CCK-8 assay and flow cytometry analysis, the treatment with increasing concentration of H2O2 triggered decreased proliferation and increased apoptosis of MCMs (Fig. 1c, d). In addition, RT-qPCR analysis indicated that the treatment of H2O2 (100 or 200 μM) enhanced the level of miR-467a-5p in MCMs, and miR-467a-5p level reached the highest in response to 100 μM of H2O2 (Fig. 1e). Hereafter, CCK-8 assay and flow cytometry analysis illustrated that hypoxia induction for 24 or 48 h reduced proliferation and promoted apoptosis of MCMs (Fig. 1f, g). Similarly, miR-467a-5p level was elevated by hypoxia treatment for 12, 24 and 48 h, and miR-467a-5p showed the highest expression after 24 h of hypoxia treatment (Fig. 1h).

MiR-467a-5p is up-regulated in MI tissues and injured MCMs. a The levels of miR-29b-3p, miR-3473a, miR-30c-5p, miR-467a-5p, miR-467a-3p and miR-34c-3p were examined by RT-qPCR.#P < 0.01 compared with sham. b MiR-467a-5p level was examined by RT-qPCR in cardiac tissues from mice undergoing ischemia reperfusion for 30 or 60 min. #P < 0.01 compared with control. &P< 0.01 compared with MI 0 min. c and f Cell proliferation was assessed by CCK-8 assay. #P < 0.01 compared with control. d and g Cell apoptosis was evaluated by flow cytometry analysis.#P < 0.01 compared with control. e and h The detection of miR-467a-5p level was carried out in RT-qPCR analysis in MCM.#P < 0.01 compared with control
MiR-467a-5p deficiency hinders MI in mice
To figure out the role of miR-467a-5p in MI mice, a series of experiments were implemented. To begin with, the level of miR-467a-5p was prominently displayed no significant change 24 h after AAV-anti-miR-467a-5p injection (Supplementary Fig. 1A), and decreased in cardiac tissues of MI mice 4 weeks after AAV-anti-miR-467a-5p injection (Fig. 2a). No significant difference was found in the heart rates of mice in different groups (Fig. 2b). Ratio of heart weight to body weight was lower in MI mice than in sham mice and was higher in MI mice injected with AAV-anti-miR-467a-5p than in MI mice (Fig. 2c). Figure 2d revealed that serum protein levels of MI markers including cTnT and cTnI were increased in MI mice and were downregulated by AAV-anti-miR-467a-5p injection. Additionally, compared to the sham-operated mice, the levels of LDH, CK and MDA were elevated in serum of MI mice while the treatment of AAV-anti-miR-467a-5p reduced LDH, CK and MDA concentrations in serum of MI mice. On the contrary, the activity of SOD was decreased in serum of MI mice and then elevated by miR-467a-5p inhibition (Fig. 2e). At last, we evaluated the cardiac function of mice using echocardiography and discovered that LVEDD and LVESD were significantly enhanced in the MI mice, while LVEF and LVES were decreased in MI mice. However, all these influences were offset by miR-467a-5p knockdown (Fig. 2f).

MiR-467a-5p deficiency hinders MI in mice. a The influence of AAV-anti-miR-467a-5p on the expression of miR-467a-5p in mice cardiac tissues (n = 8 in each group). # P< 0.01 compared with AAV-Mock. b Heart rates of mice in the Sham/AAV-Mock, MI/AAV-Mock, MI/AAV-anti-miR-467a-5p groups. c Ratio of heart weight to body weight of mice in the Sham/AAV-Mock, MI/AAV-Mock, MI/AAV-anti-miR-467a-5p groups. d Western blotting of cardiac troponin T (cTnT) and cardiac troponin I (cTnI) proteins in serum of mice in the Sham/AAV-Mock, MI/AAV-Mock, MI/AAV-anti-miR-467a-5p groups. e The changes of serum concentrations of LDH, CK, SOD and MDA were determined by myocardial enzyme assay (n = 8 in each group). #P< 0.01 compared with Sham/AAV-Mock and φP < 0.01 compared with MI/AAV-Mock. f The determination of LVEED, LVESD, LVEF and LVES were performed by cardiac function assay (n = 8 in each group). #P < 0.01 compared with Sham/AAV-Mock and φ P< 0.01 compared with MI/AAV-Mock
MiR-467a-5p knockdown prevents cardiac damages of MI mice
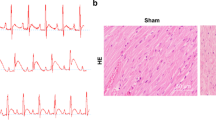
H&E staining was used to detect the pathological morphologies of myocardial tissue. Based on Fig. 3a, the clear left ventricular area with the arranged neatly myocardial fibers without inflammatory cell infiltration in myocardial was observed in sham-operated mice. Meanwhile, a significantly increased infiltration of inflammatory cells, the dissolution myocardial fibers and the missing of myocardial stripes in the infarcted area were noticed in the heart tissues of MI mice. However, all these pathological changes were counteracted by injecting AAV-anti-miR-467a-5p into MI mice. Moreover, compared with sham-operated mice, the I/R operation induced a significant myocardial infarction, and the infarction was then mitigated by miR-467a-5p inhibition (Fig. 3b). The ratio of myocardial infarct size/area at risk (AAR) was lessened by miR-467a-5p in MI mice, while the ratio of AAR/heart displayed no significant difference in MI mice injected with AAV-anti-miR-467a-5p compared to AAV-Mock (Fig. 3c, d).

MiR-467a-5p knockdown prevents cardiac damages of MI mice. a H&E staining assay was used to observe the histopathological characteristics of MI mice heart tissues (n = 8 in each group). b The evaluation of myocardial infarct size by TTC staining among the groups (n = 8 in each group). c The comparison of myocardial infarct size and area at risk among the groups (n = 8 in each group). *P < 0.01 compared with sham. @P < 0.01 compared with sham/AAV-Mock. φP < 0.01 compared with MI/AAV-Mock. d The ratio of area at risk to total myocardial area among the groups (n = 8 in each group). *P < 0.01 compared with sham. @P < 0.01 compared with sham/AAV-Mock
MiR-467a-5p promotes cardiomyocyte apoptosis in MI mice
We also assessed the impact of miR-467a-5p on cardiomyocyte apoptosis. According to the result of western blot analysis, the reduction of Bcl-2 protein level, the increase of Bax and cleaved caspase-3 protein levels in MI mice were countervailed by miR-467a-5p deficiency (Fig. 4a, b).

MiR-467a-5p promotes apoptosis of MCMs. a-b Western blot analysis was utilized to supervise the protein levels of Bax, Bcl-2 and cleaved caspase-3 in mice (n = 8 in each group). #P < 0.01 compared with Sham/AAV-Mock and φP < 0.01 compared with MI/AAV-Mock
MiR-467a-5p directly targets and negatively regulates ZEB1
Then, we explored the regulatory mechanism of miR-467a-5p in MCMs. According to starBase website (http://starbase.sysu.edu.cn/), miR-467a-5p possesses binding site on several mRNAs. To further specify the target gene for miR-467a-5p, we operated RT-qPCR to detect these mRNA levels. The result sowed that zinc finger E-box binding homeobox 1 (ZEB1) displayed the most upregulation in response to miR-467a-5p knockdown and showed the most downregulation in response to miR-467a-5p overexpression, which suggested that miR-467a-5p negatively regulated ZEB1 levels (Supplementary Fig. 1B). As depicted in Fig. 5a–c, relative to sham-operated group, ZEB1 displayed lower protein and mRNA levels in MI group. ZEB1 has been discovered to regulate MI by targeting the JAK/STAT/PI3K/AKT pathway (Shi et al. 2019). Additionally, the decrease of ZEB1 protein and mRNA levels in tissues of MI mice were reversed by miR-467a-5p silence (Fig. 5d–f). With the assistance of Targetscan (http://www.targetscan.org/vert_71/), miR-467a-5p was predicted to harbor binding site on ZEB1 3’UTR (Fig. 5g). Hereafter, we explored the interaction between miR-467a-5p and ZEB1. RT-qPCR analysis revealed that miR-467a-5p expression was down-regulated by transfection of anti-miR-467a-5p and up-regulated by transfection of miR-467a-5p mimics. Furthermore, the luciferase activity of wild type pmirGLO-ZEB1 3’UTR was elevated by miR-467a-5p inhibition and repressed by miR-467a-5p overexpression (Fig. 5h). At last, the ZEB1 protein and mRNA levels were reduced by anti-miR-467a-5p and elevated by miR-467a-5p mimics in MCMs (Fig. 5i–k).

MiR-467a-5p directly targets and negatively regulates ZEB1. a-b The examination of ZEB1 protein level in MCMs was carried out by western blot analysis. #P < 0.01 compared with Sham. c We used RT-qPCR analysis to monitor the mRNA level of ZEB1 in MCMs. #P < 0.01 compared with Sham. d–e The exploration of ZEB1 protein level in mice was conducted in western blot analysis. #P < 0.01 compared with Sham/AAV-Mock and φP < 0.01 compared with MI/AAV-Mock. f We used RT-qPCR analysis to monitor the mRNA level of ZEB1 in mice. #P < 0.01 compared with Sham/AAV-Mock and φP < 0.01 compared with MI/AAV-Mock. g The binding sequences between miR-467a-5p and ZEB1 3’UTR (position 2357–2364) were predicted by Targetscan. h The knockdown and overexpression efficiency of miR-467a-5p in MCMs were assessed by RT-qPCR analysis, and a luciferase reporter assay was performed to verify the binding capacity between miR-467a-5p and ZEB1. #P < 0.01 compared with Mock. i–k The influence of anti-miR-467a-5p and miR-467a-5p mimics on protein and mRNA expression of ZEB1 was measured through western blot and RT-qPCR analysis. #P < 0.01 compared with Mock
MiR-467a-5p promotes MCM cell apoptosis by modulating ZEB1 expression
Eventually, rescue assays were performed to explore whether miR-467a-5p promotes MCM cell apoptosis by targeting ZEB1. CCK-8 assay disclosed that the knockdown of ZEB1 not only facilitated the cell apoptosis but also reversed the inhibitory effect of anti-miR-467a-5p on cell apoptosis in H2O2 or hypoxia-induced MCMs (Fig. 6a, b). In addition, anti-miR-467a-5p-mediated the decline of Bax, cleaved caspase-3 protein levels and the increase of Bcl-2 protein levels were counteracted by ZEB1 deficiency. Similarly, ZEB1 silence induced a rise of Bax, cleaved caspase-3 protein levels and a decline of Bcl-2 protein levels (Fig. 6c–e). Moreover, we detected the effects of ZEB1 in MI mice. ZEB1 expression was upregulated in MI mice by injection of AAV-ZEB1, and was downregulated by injection of AAV-miR-467a-5p (Fig. 7a). ZEB1 alleviated MI in mice and its effects in MI mice were offset by AAV-miR-467a-5p, indicating that miR-467a-5p regulates MI in mice by ZEB1 (Fig. 7b–i).

MiR-467a-5p promotes MCM cell apoptosis by modulating ZEB1 expression. a–b MCM cell apoptosis induced by 100 μM H2O2 or hypoxia treatment for 24 h among different groups was tested by flow cytometry analysis. #P < 0.01 compared with Mock/si-NC and φP < 0.01 compared with anti-miR-467a-5p/si-NC. ΔP < 0.01 compared with anti-miR-467a-5p/si-ZEB1. c–e Western blot analysis was utilized to supervise the protein levels of Bax, Bcl-2 and cleaved caspase-3 in MCMs. #P < 0.01 compared with Mock/si-NC and φP < 0.01 compared with anti-miR-467a-5p/si-NC. Δ P< 0.01 compared with anti-miR-467a-5p/si-ZEB1

ZEB1 inhibits MI in mice and miR-467a-5p rescued the effects of ZEB1 in MI mice. a Relative ZEB1 levels in MI mice injected with AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1. b Heart rates of MI mice in the AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups. c Ratio of heart weight to body weight of MI mice in the AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups. d Western blotting of cardiac troponin T (cTnT) and cardiac troponin I (cTnI) protein levels of MI mice in the AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups. e The changes of serum concentrations of LDH, CK, SOD and MDA were determined by myocardial enzyme assay (n = 8 in each group). #P < 0.01 compared with Sham/AAV-Mock and φP < 0.01 compared with MI/AAV-Mock. f LVEED, LVESD, LVEF and LVES of MI mice in the AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups (n = 8 in each group). g H&E staining images of mice cardiac tissues in AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups. h–i Relative levels of Bax, Bcl-2, Cleaved Caspase-3 proteins in mice cardiac tissues in AAV-Mock, AAV-ZEB1, AAV-miR-467a-5p + AAV-ZEB1 groups. #P < 0.01 compared with AAV-Mock and φP < 0.01 compared with AAV-ZEB1
Discussion
Cardiovascular diseases are regarded as the leading causes of mortality in all death cases globally (Van Camp 2014). Additionally, the most mortality of patients with cardiovascular diseases were caused by MI (Fredman and Spite 2017). Modern therapies like reperfusion treatment and medical (aspirin and clopidogrel) treatment have been widely applied to treat MI patients(Lewis et al. 1983; Yu et al. 2018), while the prognosis of MI patients remains unfavorable (Lisowska et al. 2016). Yet, the molecular mechanisms underlying MI need a thorough investigation.
MiRNAs, a group of small, non-coding RNA molecules, have been identified to be differentially expressed in diverse diseases (Lu and Rothenberg 2018). Upregulation of miR-183 represses neuropathic pain by inhibiting MAP3K4 level in CCI rat models (Huang and Wang 2019). MiRNA-126 knockdown suppresses IL-23R mediated TNF-α or IFN-γ production in collagen-induced rheumatoid arthritis mice model (Gao et al. 2018). A multitude of biomarkers such as miR-498, miRNA-181a and miR-16b have been identified to be dysregulated in MI (Zhu et al. 2016). Recently, miR-467a-5p was discovered to present high expression in MI mice model (Zhang et al. 2019a, b). Similarly, in our research, miR-467a-5p was also up-regulated in MI mice tissues and MCMs treated by hypoxia or H2O2. A previous study proposed that miR-221-3p results in cardiomyocyte injury by targeting p57 in MI rat model and H2O2-treated H9c2 cells (Meng et al. 2018). Our findings revealed that miR-467a-5p exacerbated the progression of MI by regulating myocardial enzyme activity and cardiac function. In detail, knockdown of miR-467a-5p triggered a decrease on LDH, CK, MDA, LVEDD and LVESD, but an increase of SOD, LVEF and LVFS. Importantly, miR-467a-5p suppression mitigated cardiomyocyte apoptosis, histological change, myocardial infract size of heart tissues in MI mice.
ZEB1 has been reported to act as a transcription factor and play an oncogenic role in a variety of tumors (Llorens et al. 2016). In addition, ZEB1 modulates several diseases including diabetic kidney disease, arthritis and cardiac hypertrophy (Tang et al. 2018; Ye et al. 2015). Most recently, ZEB1 has been indicated to be targeted by miR-34a-5p, and activate the JAK/STAT and PI3K/AKT pathways to alleviate the damage of hypoxia-induced H9C2 cells (Shi et al. 2019). In addition, ZEB1 was reported to facilitate cell proliferation, cell migration and collagen production in cardiac fibroblast (Yuan et al. 2020). In our research, we confirmed that ZEB1 was directly targeted and negatively regulated by miR-467a-5p in MI mice and MCMs. ZEB1 overexpression limited cell apoptosis and rescue assays revealed that ectopic ZEB1 neutralized the promoting effect of miR-467a-5p overexpression on MCM apoptosis. A previous study proposed that ZEB1 exerts effects on extracellular matrix remodeling and myocardial fibrosis by interacting with LOX1 (He et al. 2018). ZEB1 is involved in the regulation of endothelial-to-mesenchymal transition and cardiac fibrosis in H5V cells (Xu et al. 2019). Importantly, ZEB1 functions as a transcription factor to activate the transcription of a variety of genes including rs7692387, and thereby modulates atherosclerosis (Kessler et al. 2017). However, all these potential downstream mechanisms of ZEB1 have not been fully explored in MI. We will further investigate the downstream mechanism of ZEB1 in the future.
In summary, for the first time, we explored the role of miR-467a-5p in MI and validated that miR-467a-5p aggravated MI by promoting apoptosis via modulating ZEB1 expression in mice, which implied a potential biomarker for MI treatment. However, our research had limitations. For instance, the effects of ZEB1 on MI and oxidative stress have not been fully investigated. In addition, the pathology of MI is complicated and other regulatory mechanisms of miR-467a-5p remain to be investigated in the future.
Data availability
The datasets used during the current study are available from the corresponding author on reasonable request.
References
Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488. https://doi.org/10.1038/nrm3611
Chen C et al (2019) MicroRNA let-7-TGFBR3 signalling regulates cardiomyocyte apoptosis after infarction. EBioMedicine 46:236–247. https://doi.org/10.1016/j.ebiom.2019.08.001
Chiang M et al (2020) miR-26a attenuates cardiac apoptosis and fibrosis by targeting ataxia-telangiectasia mutated in myocardial infarction. J Cell Physiol 235:6085–6102. https://doi.org/10.1002/jcp.29537
Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X (2013) MicroRNA: function, detection, and bioanalysis. Chem Rev 113:6207–6233. https://doi.org/10.1021/cr300362f
Fredman G, Spite M (2017) Specialized pro-resolving mediators in cardiovascular diseases. Mol Aspects Med 58:65–71. https://doi.org/10.1016/j.mam.2017.02.003
Gao J, Kong R, Zhou X, Ji L, Zhang J, Zhao D (2018) MiRNA-126 expression inhibits IL-23R mediated TNF-alpha or IFN-gamma production in fibroblast-like synoviocytes in a mice model of collagen-induced rheumatoid arthritis. Apoptosis 23:607–615. https://doi.org/10.1007/s10495-018-1474-7
He Q, Wang F, Honda T, James J, Li J, Redington A (2018) Loss of miR-144 signaling interrupts extracellular matrix remodeling after myocardial infarction leading to worsened cardiac function. Sci Rep 8:16886. https://doi.org/10.1038/s41598-018-35314-6
Huang L, Wang L (2019) Upregulation of miR-183 represses neuropathic pain through inhibiton of MAP3K4 in CCI rat models. J Cell Physiol. https://doi.org/10.1002/jcp.29276
Kessler T et al (2017) Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 136:476–489. https://doi.org/10.1161/circulationaha.116.024152
Lee ES et al (2016) Quality improvement for cardiovascular disease care in low- and middle-income countries: a systematic review. PLoS ONE 11:e0157036. https://doi.org/10.1371/journal.pone.0157036
Lewis HD Jr et al (1983) Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 309:396–403. https://doi.org/10.1056/nejm198308183090703
Li P, Yao Y, Ma Y, Chen Y (2019) MiR-150 attenuates LPS-induced acute lung injury via targeting AKT3. Int Immunopharmacol 75:105794. https://doi.org/10.1016/j.intimp.2019.105794
Liang T et al (2016) Cardio-protecteffect of qiliqiangxin capsule on left ventricular remodeling, dysfunction and apoptosis in heart failure rats after chronic myocardial infarction. Am J Transl Res 8:2047–2058
Lim M et al (2018) Intravenous injection of allogeneic umbilical cord-derived multipotent mesenchymal stromal cells reduces the infarct area and ameliorates cardiac function in a porcine model of acute myocardial infarction. Stem Cell Res Ther 9:129. https://doi.org/10.1186/s13287-018-0888-z
Lisowska A, Makarewicz-Wujec M, Filipiak KJ (2016) Risk factors, prognosis, and secondary prevention of myocardial infarction in young adults in Poland. Kardiol Pol 74:1148–1153. https://doi.org/10.5603/KP.a2016.0098
Liu Z, Yang Q, Wei Q, Chang Y, Qu M, Yu L (2019) The protective effect of miR-377 inhibitor against renal ischemia-reperfusion injury through inhibition of inflammation and oxidative stress via a VEGF-dependent mechanism in mice. Mol Immunol 106:153–158. https://doi.org/10.1016/j.molimm.2018.12.028
Llorens MC et al (2016) Phosphorylation regulates functions of ZEB1 transcription factor. J Cell Physiol 231:2205–2217. https://doi.org/10.1002/jcp.25338
Lu TX, Rothenberg ME (2018) MicroRNA. J Allergy Clin Immunol 141:1202–1207. https://doi.org/10.1016/j.jaci.2017.08.034
Luo SY, Chen S, Qin YD, Chen ZW (2016) Urotensin-receptor antagonist SB-710411 protects rat heart against ischemia-reperfusion injury via RhoA/ROCK pathway. PLoS ONE 11:e0146094. https://doi.org/10.1371/journal.pone.0146094
Mathiew-Quiros A, Salinas-Martinez AM, Guzman de la Garza FJ, Garza-Sagastegui MG, Guzman-Delgado NE, Palmero-Hinojosa MG, Oliva-Sosa NE (2017) Myocardial infarction in young mexicans associated to metabolic syndrome. Gac Med Mex 153:297–304
Meng Q, Liu Y, Huo X, Sun H, Wang Y, Bu F (2018) MicroRNA2213p contributes to cardiomyocyte injury in H2O2treated H9c2 cells and a rat model of myocardial ischemiareperfusion by targeting p57. Int J Mol Med 42:589–596. https://doi.org/10.3892/ijmm.2018.3628
Pedrinelli R et al (2012) Hypertension and acute myocardial infarction: an overview. J Cardiovasc Med (hagerstown, Md) 13:194–202. https://doi.org/10.2459/JCM.0b013e3283511ee2
Rozado J et al (2017) Extracorporeal membrane oxygenation system as a bridge to reparative surgery in ventricular septal defect complicating acute inferoposterior myocardial infarction. J Thorac Dis 9:E827-e830. https://doi.org/10.21037/jtd.2017.08.164
Shi K, Sun H, Zhang H, Xie D, Yu B (2019) miR-34a-5p aggravates hypoxia-induced apoptosis by targeting ZEB1 in cardiomyocytes. Biol Chem 400:227–236. https://doi.org/10.1515/hsz-2018-0195
Sotillo E, Thomas-Tikhonenko A (2011) Shielding the messenger (RNA): microRNA-based anticancer therapies. Pharmacol Ther 131:18–32. https://doi.org/10.1016/j.pharmthera.2011.04.006
Su Q, Lv X, Sun Y, Ye Z, Kong B (2019) MicroRNA-494 Inhibits the LRG1 expression to induce proliferation and migration of VECs in rats following myocardial infarction. Mol Ther Nucelic Acid 18:110–122. https://doi.org/10.1016/j.omtn.2019.08.007
Tang WB et al (2018) miR302a-3p may modulate renal epithelial-mesenchymal transition in diabetic kidney disease by targeting ZEB1. Nephron 138:231–242. https://doi.org/10.1159/000481465
Van Camp G (2014) Cardiovascular disease prevention. Acta Clin Belg 69:407–411. https://doi.org/10.1179/2295333714y.0000000069
Venetsanos D et al (2017) Association between gender and short-term outcome in patients with ST elevation myocardial infraction participating in the international, prospective, randomised Administration of Ticagrelor in the catheterisation Laboratory or in the Ambulance for New ST elevation myocardial Infarction to open the Coronary artery (ATLANTIC) trial: a prespecified analysis. BMJ Open 7:e015241. https://doi.org/10.1136/bmjopen-2016-015241
Wincewicz A, Sulkowski S (2017) Stat proteins as intracellular regulators of resistance to myocardial injury in the context of cardiac remodeling and targeting for therapy. Adv Clin Exp Med 26:703–708. https://doi.org/10.17219/acem/62693
Xu L et al (2019) Endothelial-specific deletion of Ets-1 attenuates Angiotensin II-induced cardiac fibrosis via suppression of endothelial-to-mesenchymal transition. BMB Rep 52:595–600. https://doi.org/10.5483/BMBRep.2019.52.10.206
Ye H et al (2015) CD4 T-cell transcriptome analysis reveals aberrant regulation of STAT3 and Wnt signaling pathways in rheumatoid arthritis: evidence from a case-control study. Arthr Res Ther 17:76. https://doi.org/10.1186/s13075-015-0590-9
Yu HR, Wei YY, Ma JG, Geng XY (2018) Beneficial effects of combined administration of Clopidogrel and Aspirin on the levels of proinflammatory cytokines, cardiac function, and prognosis in ST-segment elevation myocardial infarction: a comparative study. Medicine 97:e13010. https://doi.org/10.1097/md.0000000000013010
Yuan X et al (2020) MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J Cell Mol Med 24:227–237. https://doi.org/10.1111/jcmm.14704
Yusuf S et al (2004) Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet (Lond, England) 364:937–952. https://doi.org/10.1016/s0140-6736(04)17018-9
Zhang DY, Wang BJ, Ma M, Yu K, Zhang Q, Zhang XW (2019) MicroRNA-325-3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice. BMC Mol Biol 20:17. https://doi.org/10.1186/s12867-019-0133-z
Zhang DY, Wang BJ, Ma M, Yu K, Zhang Q, Zhang XW (2019) Correction to: MicroRNA-325-3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice. BMC Mol Biol 20:18. https://doi.org/10.1186/s12867-019-0136-9
Zhao Z, Du S, Shen S (2020) microRNA-132 inhibits cardiomyocyte apoptosis and myocardial remodeling in myocardial infarction by targeting IL-1β. J Cell Physiol 235:2710–2721. https://doi.org/10.1002/jcp.29175
Zhong Z et al (2018) Circulating microRNA expression profiling and bioinformatics analysis of dysregulated microRNAs of patients with coronary artery disease. Medicine 97:e11428. https://doi.org/10.1097/md.0000000000011428
Zhu J et al (2016) Circulating miR-181a as a potential novel biomarker for diagnosis of acute myocardial infarction. Cell Physiol Biochem 40:1591–1602. https://doi.org/10.1159/000453209
Zhu L, Chen T, Ye W, Wang JY, Zhou JP, Li ZY, Li CC (2019) Circulating miR-182-5p and miR-5187-5p as biomarkers for the diagnosis of unprotected left main coronary artery disease. J Thorac Dis 11:1799–1808. https://doi.org/10.21037/jtd.2019.05.24
Zile MR, Mehurg SM, Arroyo JE, Stroud RE, DeSantis SM, Spinale FG (2011) Relationship between the temporal profile of plasma microRNA and left ventricular remodeling in patients after myocardial infarction. Circ Cardiovasc Genet 4:614–619. https://doi.org/10.1161/circgenetics.111.959841
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, W., Cao, Y., Chen, C. et al. MiR-467a-5p aggravates myocardial infarction by modulating ZEB1 expression in mice. J Mol Histol 52, 767–780 (2021). https://doi.org/10.1007/s10735-021-09978-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-021-09978-w