
Abstract
Tea plant [Camellia sinensis (L.) O. Kuntze] constitutes one of the most important economic crops in many countries. However, in many areas, tea plants are subjected to high salinity, which severely affects the growth and development of these plants. To understand the potential molecular mechanisms of tea plants in response to salt stress, we used RNA-Seq technology to compare the transcriptomes from tea plants treated with and without NaCl and analyzed the differentially expressed genes (DEGs). In total, 470,738 transcripts and 150,257 unigenes were obtained that had average lengths of 1422.09 and 680.40 nt, respectively, and 28,831 of these sequences were annotated in public databases. In addition, 1769 DEGs were identified, including 947 up-regulated and 822 down-regulated ones. Many of these DEGs were involved in Ca2+ signal transduction, the abscisic acid (ABA) pathway, and mitogen-activated protein kinase (MAPK) cascades. Many DEGs were also transcription factors and key functional proteins involved in salt resistance in tea plants; these genes constitute a regulatory network in response to salt stress. qRT-PCR analyses of nine unigenes were performed to confirm the validity of the data, and the results were highly consistent with the RNA-Seq results. Taken together, these findings reveal the underlying molecular mechanism of tea plants in response to salt stress and could provide many candidate genes for additional studies, especially those involving the genetic engineering and breeding of tea plants that are highly resistant to salt stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants are subjected to different kinds of stresses during their life cycles, such as drought, heat, cold, heavy metal and salt stresses (Fedoroff et al. 2010). Among, salt stress is one of the most serious abiotic stresses that affects the normal growth and development of plants and limits crop yield. Hence, it is especially important to understand the mechanisms of salinity tolerance in plants to breed species that exhibit strong resistance and adaptability. Salt stress is perceived by plants as an early-occurring osmotic stress and as a subsequent ionic stress (Munns and Tester 2008), after which a series of protection mechanisms, including the Ca2+-mediated signaling pathway, mitogen-activated protein kinase (MAPK) cascades and the abscisic acid (ABA) signaling pathway, are triggered in plants to reduce the damage of these stresses (Zhu 2002). The most ubiquitous secondary messenger is Ca2+, and Ca2+-dependent signaling networks play crucial roles in the process of plant responses to salt stress. The salt overly sensitive (SOS) pathway is the most studied Ca2+-mediated signaling pathway and is essential for maintaining favorable ion ratios in the cytoplasm in response to salt stress (Ji et al. 2013). Similarly, ABA is a well-known endogenous signal molecule that enables plants to survive severe adverse environmental conditions, especially salt stress (Ryu and Cho 2015). A complex signaling network dominated by ABA, which includes pyrabactin resistance (PYR)/PYR1-like (PYL) receptor proteins; protein phosphatase 2C (PP2C); SNF1-related protein kinase 2s (SnRK2s); ABA-responsive element-binding protein (AREB1); and ABA insensitive 5 (ABI5), has been confirmed in plants in response to salt stress, and this network is vital to the salt tolerance of plants (Julkowska and Testerink 2015). In addition, increasing amounts of transcription factors (TFs) have been reported to play vital roles in regulating the resistance mechanisms of plants under salt stress (Lindemose et al. 2013). For example, Du et al. (2017) reported that many TFs such as bHLH, ERF, bZIP, NAC and WRKY were differentially expressed in maize under salt stress conditions. Furthermore, many functional proteins have become important research topics with respect to plant salt stress tolerance, including heat shock proteins (HSPs), late embryogenesis abundant (LEA) proteins and aquaporin proteins (AQPs), which help plants adapt to and withstand the effect of salt stress (Song et al. 2014; Banerjee and Roychoudhury 2016; Yue et al. 2014).
Tea plant [Camellia sinensis (L.) O. Kuntze] constitute one of the most important economic crops in many countries, such as China, India, Sri Lanka, and Japan (Chen et al. 2007). In many areas, tea plants are subjected to salt stress, which severely affects the quality and yield of tea products; therefore, the development of the tea industry is restricted (Upadhyaya and Panda 2013). Recently, Zhang et al. (2017) carried out a transcriptome analysis of tea plant in response to salt and drought stresses which mainly focused on the comparison of the effects of these two stresses and some metabolites and TFs which were significantly expressed. In addition, other researches on the effects of salt stress in tea plants were related to the mechanisms of physiological responses and the functions of genes. For example, salt stress increases the synthesis of theanine by altering the accumulation of theanine synthetase (Deng et al. 2012), and Wang et al. (2016c) reported that the CsNAC gene family plays an important role in the salt stress response in tea plants. However, there is still a large part of the complex molecular regulatory mechanisms underlying the response to salt stress remains unknown in tea plant.
RNA-Seq high-throughput sequencing technology has been widely used to explore the potential mechanisms associated with important traits in plants, especially changes in gene expression in response to biotic and abiotic stresses (Marguerat and Bahler 2010). Transcriptome sequencing has also been used in many studies of tea plants. For example, Li et al. (2017) elucidated the accumulation mechanism of anthocyanins in ‘Zijuan’ tea leaves using transcriptome analysis, and Wang et al. (2016a) suggested a potential molecular mechanism of the effects of drought stress on the leaf quality of tea plants. In the present study, to understand the potential molecular regulatory mechanisms of tea plants in response to salt stress, we used Illumina RNA sequencing to compare the transcriptomes of tea plants treated with and without NaCl. All the experiments were replicated three times to guarantee the reliability of the results. At the same time, differentially expressed genes (DEGs) involved in signal transduction pathways, TFs and genes encoding functional proteins were identified and analyzed. Several vital salt stress-responsive genes were also selected for qRT-PCR to verify the RNA-Seq results. The obtained results provide a deeper insight into the molecular mechanism of tea plants in response to salt stress, and the identified DEGs could provide candidate genes for breeding salinity resistance into C. sinensis.
Materials and methods
Plant materials and NaCl treatment
Camellia sinensis (L.) O. Kuntze cv. ‘Pingyangtezao’ is a traditional tea plant cultivar in China that exhibits stress tolerance. In this study, 200 one-year-old ‘Pingyangtezao’ cutting seedlings with consistent growing trend were pre-incubated in nutrient solution under standard growth conditions (temperature: 25 °C, air relative humidity: 60–70%, photoperiod: 12 h light/12 h dark) in the greenhouse of Northwest A&F University. The nutrient solution was prepared according to Wan et al. (2012) and contained mass elements: NH4+–N, Ca, K, Al, Mg of 750, 395, 350, 250, 210 μM and trace elements: B, Fe as Fe-EDTA, Zn, Mn, Mo and Cu of 3.33, 2.1, 0.51, 0.50, 0.17 and 0.13 μM, respectively, and the pH of nutrient solution was 5.0. After 2 months of pre-incubation, these tea plants were used for stress assays under the same growth conditions. According to the characteristics of the tea plant materials and the result of a pre-experiment, the concentration and length of time of NaCl treatment were determined as 250 mM and 4 h. In brief, tea plants were treated by soaking the roots in nutrient solution that contained 250 mM NaCl (salt treatment) or standard nutrient solution (control), and fresh leaf samples (first and second leaves) were randomly collected at 4 h after treatment, each treatment was repeated for three times (CK1, CK2, CK3 and Salt1, Salt2, Salt3). The samples were immediately immersed in liquid nitrogen and stored at − 80 °C for RNA sequencing, qRT-PCR validation and further analysis.
RNA extraction, cDNA library construction and sequencing
The total RNA of the six samples (CK1, CK2, CK3, Salt1, Salt2 and Salt3) were extracted using RNAiso Plus (TaKaRa, Dalian, China), and the concentration and quality of the total RNA were examined using both a NanoDrop 2000c spectrophotometer (NanoDrop, Wilmington, DE, USA) and 0.8% agarose gel electrophoresis. The cDNA library construction and sequencing were then performed at Shanxi Breeding Biotech. Co., Ltd. (Yangling, China) using an Illumina HiSeq2500 platform based on sequencing by synthesis (SBS) technology. The raw data were filtered to obtain clean, high-quality data before sequence assembly, the adapter sequences at both ends of the reads were trimmed, and the sequences that had a quality value < 10 or had ambiguous nucleotides were removed.
De novo assembly was performed using the Trinity program as described by Haas et al. (2013). First, the clean reads were broken into short sequence segments called K-mers, and these K-mers were then extended into longer segments (contigs). The contigs were then clustered into components, and a complete de Bruijn graph for each component was constructed. Last, combined with the sequenced reads, transcripts produced by paralogous genes in each component were identified using the de Bruijn graph, and the resulting outputs were termed ‘unigenes.’
Unigene annotation and classification
For unigene functional annotation and classification, a Basic Local Alignment Search Tool X (BLASTX) alignment (E-value < 10−5) (Altschul et al. 1997) between unigene sequences and public databases was performed, which included the NCBI non-redundant protein (NR), SwissProt (Apweiler et al. 2004), Clusters of Orthologous Groups of proteins (COG) (Tatusov et al. 2000), euKaryotic Orthologous Groups (KOG) (Koonin et al. 2004) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Kanehisa et al. 2004). In addition, gene ontology (GO) annotations and classifications were analyzed using Blast2GO and WEGO software (Ashburner et al. 2000), and HMMER software (Eddy 1998) was used to search the Protein Family (Pfam) database using an E-value cutoff of 10−10 (Finn et al. 2014).
Identification of DEGs
The expression abundance of each unigene was estimated in accordance with the fragments per kilobase of transcript per million mapped reads (FPKM) method using RSEM software (Li and Dewey 2011). At the same time, DEGs were identified to analyze the differential expression between the CK and salt-treated samples according to the criteria of a false discovery rate (FDR) < 0.01 and a ratio of stress/control (FC) ≥ 2 (Anders and Huber 2010). GO enrichment analyses and pathway enrichment analyses were performed to analyze the degree of enrichment and to calculate the enrichment significance of GO terms and KEGG pathways using TopGO software and the Fisher accurate testing method, respectively (Chen et al. 2015).
Quantitative real-time PCR (qRT-PCR) analysis
qRT-PCR validation of RNA-Seq results
qRT-PCR was performed in accordance with the methods of Wang et al. (2016b) in order to validate the RNA-Seq results. Total RNA was isolated from another six samples randomly selected from control and salt-treated materials and was treated with DNase I to remove any genomic DNA contamination. First-strand cDNA was synthesized using a PrimeScript™ RT reagent kit with gDNA Eraser (TaKaRa, Dalian, China), after which qRT-PCR detection was completed using an EvaGreen qPCR MasterMix-No Dye Kit (ABM, Richmond, BC, Canada) on a StepOne Plus PCR instrument (ABI, USA) following the manufacturers’ protocols. Briefly, the amplification regimen consisted of an initial denaturation at 95 °C for 10 min followed by 40 cycles of 95 °C for 5 s and 60 °C for 1 min. The Csβ-actin gene of tea plants was chosen as the reference gene in accordance with previous methods (Hao et al. 2014), and the primers used in this assay are shown in Supplementary Table S1. The relative expression levels of the genes were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001), and the data are presented as the mean ± SD from three independent biological replicates. SPSS 20.0 software was used to determine significant differences between the CK and NaCl treatment data.
qRT-PCR analysis of the expression levels of CsSOS1 in tea leaves after NaCl treatment
In order to analyze the expression pattern of CsSOS1 in tea leaves under 0 to 24 h of NaCl treatment, 18 one-year-old tea plants were treated with nutrient solution that contained 250 mM NaCl and the first and second leaves of each plant were collected immediately after 0, 2, 4, 8, 12 and 24 h of treatment, respectively, each treatment was repeated for three times. The methods of RNA extraction, reverse transcription to cDNA, qRT-PCR detection of the 18 samples were the same as mentioned above.
Data deposition
The sequencing data were deposited in the NCBI Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/Traces/sra) under accession number SRP107589.
Results
Transcriptome sequencing and de novo assembly
To analyze the transcriptome response to NaCl stress in tea plants, six cDNA libraries from three control (CK1, CK2, CK3) and three salt-treated (Salt1, Salt2, Salt3) leaf samples were constructed and sequenced using an Illumina HiSeq2500 platform. An overview of the sequencing and mapping results are shown in Table 1. An average of 52,931,130 reads was obtained that had a Q30 percentage of no less than 92.93% in each sample. De novo assembly was performed using the Trinity program, and 470,738 transcripts and 150,257 unigenes were obtained that had mean lengths of 1422.09 and 680.40 nt, respectively, and N50 lengths of 2427 and 1013 nt, respectively (Supplementary Table S2). After assembly, the clean data were mapped onto the transcripts and unigenes, and the average mapped ratio was 80.14%. Together, these data indicated that the quality of the throughput and sequencing was high enough for further analysis.
Unigene annotation and classification in public databases
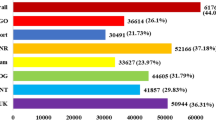
To predict the functions and transcriptional directions of the assembled unigenes, all assembled unigene sequences were queried against the NR, SwissProt, Pfam, COG, GO, KOG, and KEGG databases to obtain annotations; the results are shown in Table S3. A total of 28,831 sequences were successfully annotated in these public databases. Among them, 9059 unigenes were annotated in the COG database and classified into 25 function classes (Fig. 1a): the largest group was ‘general function prediction only’ (2222, 24.53%), followed by ‘replication, recombination and repair’ (1435, 15.84%), ‘transcription’ (1169, 12.90%) and ‘signal transduction mechanisms’ (937, 10.34%). A total of 16,345 unigenes were annotated in the GO database, and the classification results are presented in Fig. 1b. These unigenes were assigned to three categories, including 52 subcategories, and among these classifications, ‘cell part’ (6717, 41.10%) was the largest subcategory in the cellular component category; ‘catalytic activity’ (9092, 55.63%) in the molecular function category and ‘metabolic process’ (11,194, 68.49%) in the biological process category were the most abundant. In addition, 6006 assembled unigenes were assigned to 218 KEGG pathways, which mainly included ‘ribosome’ (361, 6.01%), ‘protein processing in endoplasmic reticulum’ (213, 3.55%), ‘oxidative phosphorylation’ (211, 3.51%), ‘spliceosome’ (183, 3.05%), ‘RNA transport’ (180, 3.00%) which were the top five abundant pathways (Supplementary Table S4).

COG and GO classification of unigenes. a A total of 9059 unigenes were classified into 25 COG function classes. b A total of 16,345 unigenes were assigned into three GO categories (‘cellular component’, ‘molecular function’ and ‘biological process’) and 52 subcategories
Identification and annotation of DEGs
As shown in Fig. 2a, 1769 DEGs were identified, including 947 up-regulated and 822 down-regulated ones, and 1687 DEGs were annotated in public databases (Supplementary Table S5). Among the annotated DEGs, 1046 were classified in the GO database. The results of GO enrichment analysis showed that the eight most significantly enriched (KS < 0.001) GO terms included the following: “photosynthesis, light harvesting”, “chloroplast thylakoid membrane”, “heme binding”, “sequence-specific DNA binding”, “chlorophyll binding”, “2-alkenal reductase [NAD(P)] activity”, “peroxidase activity” and “carboxylic ester hydrolase activity” (Supplementary Fig. S1–S3). In addition, 314 DEGs were assigned in the KEGG database. The pathway enrichment results showed that ‘photosynthesis-antenna proteins’ was the most dominant pathway, followed by ‘photosynthesis’, ‘plant hormone signal transduction’ and ‘isoquinoline alkaloid biosynthesis’ (Supplementary Fig. S4).

Statistics of total DEGs and TFs in DEGs. a Total number of up- and down- regulated DEGs under salt stress. b Up- and down-regulated TFs in DEGs
Key genes involved in the response to salt stress
DEGs involved in major signal transduction pathways
Signal transduction pathways are crucial participants in regulatory networks of plant responses to salt stress. As presented in Table 2, 20 DEGs related to the Ca2+ signaling pathway were identified, including genes encoding calmodulin-like proteins (CMLs), cation/Ca2+ exchangers (CCXs), calmodulin binding-like proteins (CBLs), CBL-interacting protein kinases (CIPKs), Ca2+-dependent protein kinases (CDPKs), calmodulin (CaM) and Ca2+-transporting ATPase. Expectedly, most of these DEGs were up-regulated under salt stress, including CML20, CML18, CML49, CDPK7, CDPK16, CIPK13 and CIPK15, whereas only four were down-regulated, including CBL (c238957.graph_c0), CaM4, CCX1-like and CAX20. Similarly, 11 DEGs associated with the ABA signaling pathway were identified; among them, 2 DEGs, PYR1 and PYL2, were both down-regulated, whereas 5 DEGs, including PP2C2, PP2C3, PP2C14, PP2C51 and PP2C60, were up-regulated. However, it is worth noting that 4 DEGs, including PP2C12, PP2C27, and PP2C54 were down-regulated (Table 3). Furthermore, 8 DEGs involved in the MAPK cascade pathway were identified, and the majority of them were up-regulated, with the exception of MAPKKK2 (c241143.graph_c1) (Supplementary Table S6).
TFs related to salt stress
TF families, including bZIP, HD-Zip, AP2/ERF, WRKY, NAC, MYB, bHLH and zinc finger-TFs (ZF-TFs), play vital roles in regulating plant resistance mechanisms under abiotic stress. In the present study, 150 differentially expressed TFs were identified from the DEGs (111 genes up-regulated and 39 down-regulated), among which 17 genes belong to the bZIP/HD-Zip family and 12, 27, 13, 19, 13 and 49 genes belong to the WRKY, AP2/ERF, NAC, MYB, bHLH and ZF families, respectively (Fig. 2b). Interestingly, the statistical analysis results showed that more DEGs were up-regulated in each TF family than were down-regulated. In addition, some proven key TFs involved in the process of plant responses to salt stress, such as bZIP8, ABI5, WRKY6, ERF4, NAC8, MYB44, bHLH47 and CCCH-type ZF, were differentially expressed. The detailed results of the analysis are shown in Supplementary Table S7 and S8.
DEGs encoding vital functional proteins in response to salt stress
Many genes that encode vital functional proteins such as HSPs, AQPs, LEA proteins, and F-box proteins were differentially expressed during the salt stress response process in tea plants. 10 DEGs encoding HSPs, 5 DEGs encoding LEA proteins, 8 DEGs encoding AQPs and 13 DEGs encoding F-box proteins were identified and are presented in Supplementary Table S9. The majority of these DEGs were up-regulated, but all DEGs encoding HSPs were down-regulated, especially the 70-kDa HSPs.
qRT-PCR validation
To confirm the reliability of the expression levels obtained from the RNA-Seq transcriptome, 9 unigenes that were correlated with the salt stress response, including CDPK1 (c245204.graph_c0), CIPK13 (c247640.graph_c0), WRKY7 (c210400.graph_c0), Ca2+-ATPase (c251091.graph_c0), ABI5 (c236105.graph_c0), PP2C (c236202.graph_c0), LEA (c241433.graph_c0), HSP (c208363.graph_c0) and bZIP8 (c240205.graph_c0), were selected for qRT-PCR assays. The results showed that the expression level of each unigene closely corresponded to the transcript level estimated from the sequence data (Fig. 3), which implies reproducibility and accuracy of the RNA-Seq results.

qRT-PCR validation of 9 unigenes selected from the RNA-Seq transcriptome. qRT-PCR relative expression levels were calculated using the 2−ΔΔCt method presented on the left axis; the expression levels of the RNA-Seq results were calculated using the FPKM presented on the right axis. *Represents a significant difference in the qRT-PCR results between the control and salt-treated individuals (P < 0.05). **Represents a highly significant difference in the qRT-PCR results between the control and salt-treated individuals (P < 0.01)
Discussion
Salt stress is one of the most serious abiotic stresses that affects the yield and quality of crops, and the underlying molecular mechanism of the effects of salt stress on plants has become the focus of current studies (Zhu 2016). Recently, Zhang et al. (2017) analyzed the transcriptome of tea plant in response to continuous salt stress and identified 3715 DEGs. They found most DEGs were down-regulated under salt stress at 24 h but elevated after 48 h and many of them are relative to osmotic products such as proline, sugar and GABA which may play important roles in tea plant adapting to salt stress. And they also speculated that salt stress may affect tea qualities due to the down-regulation of some biosynthetic genes which involved in the synthesis of quality compounds including polyphenol and caffeine. However, there are still some important molecular mechanisms of tea plants in response to salt stress remain unknown, such as signal transduction pathways and some key functional proteins. Here, using RNA-Seq technology, we comprehensively analyzed the changes in gene expression in tea plants treated with NaCl in order to investigate the defense response to salt stress. More than 79.40 Gb of clean data was obtained, and 150,257 unigenes were assembled; these numbers were as high as those reported in similar studies of tea plants (Wang et al. 2016a). In addition, 1769 DEGs were identified, and DEGs related to the Ca2+ signaling pathway, ABA pathway, MAPK cascade, TFs and other crucial functional proteins were the main focus of this study.
DEGs involved in signal transduction pathways
Ca2+ signal transduction pathway
The Ca2+-mediated signal transduction pathway responds to many physiological processes in plants, including plant responses to various abiotic stresses such as salt stress (Steinhorst and Kudla 2014). The Ca2+ signaling pathway starts with a change in cytoplasmic Ca2+ concentration, which occurs through Ca2+ transporters such as cyclic nucleotide-gated ion channels (CNGCs), two-pore Ca2+ channels (TPCs), Ca2+-ATPases and cation/H+ antiporters (CAXs) (Wilkins et al. 2016). In the present study, the expression levels of multiple Ca2+-ATPases and CAX genes were up-regulated following salt stress treatment, which suggests that they are involved in the transportation of Ca2+ in the early stages of salt stress. In addition, a putative group of Ca2+ channels, glutamate receptor-like channels (GLRs), was recently identified and the Ca2+ transport activities of these channel proteins have been confirmed in the pollen tubes of tea plants (Wang et al. 2016b). Interestingly, two GLR genes were identified among the DEGs, which implies that GRLs also participate in the process of Ca2+ transportation in mesophyll cells of tea plants under salt stress. Furthermore, multiple Ca2+ sensors and relay proteins, such as CaM, CMLs, CDPKs, CBLs, and CIPKs, were differentially expressed after salt stress treatment in tea plants. These data suggest that an initial signaling network dependent on Ca2+ is involved in the salt stress response process in tea plants which contains Ca2+ transport and downstream responses of Ca2+ sensors and relay proteins (Boudsocq and Sheen 2009). Unexpectedly, the plasma membrane Na+/H+ antiporter gene SOS1, which is downstream of the Ca2+ pathway, was not differentially expressed in our RNA-Seq results, despite it being essential to the salt stress response (Shi et al. 2000). To explore the reasons for the above results, the expression levels of CsSOS1 in the leaves of tea plants treated with 250 mM NaCl for 0, 2, 4, 8, 12 or 24 h were analyzed using qRT-PCR. The results showed that the expression levels of CsSOS1 were not significant until 8 h and that the levels peaked at 24 h in tea plants (Supplementary Fig. S5). These results are similar to those of SpSOS1 (Zhou et al. 2015), which is a reasonable explanation for the above RNA-Seq results. Furthermore, MAPKs, which are important downstream targets of Ca2+ signals, mediate the stress response in plants (Yamakawa et al. 2004). Previous studies have reported that some MAPK cascade genes can regulate salt stress tolerance in many plants (Cheong and Kim 2010). As expected, three MAPKK and five MAPKKK genes were induced by salt stress in the present study; these results are consistent with those of a previous study that reported how the MAPK signaling pathway participates in plant salinity stress resistance, which may depend on the regulation of Ca2+ (Mehlmer et al. 2010).
ABA signaling pathway
ABA is an important plant stress hormone that functions in plant growth, development and adaptation to various stresses and can be induced under many osmotic stress conditions, including high salinity (Zhang et al. 2006). The early response components in the ABA signaling pathway mainly consist of three protein classes: ABA receptors, PP2Cs and SnRK2s. Briefly, once ABA is produced, ABA-bound receptors bind and inhibit PPC2s, thereby activating SnRK2s (Yoshida et al. 2014). In the present study, two ABA receptors, PYR1 and PYL2, were both significantly down-regulated, which is consistent with the results of Postnikova et al. (2013) that suggests the down-regulation of ABA receptors can reduce plant sensitivity to ABA in response to salt stress and help plants adapt to salt stress. Similarly, multiple PP2C family genes in tea plants were differentially expressed in response to salt stress in the present study; among these genes, PP2C2, PP2C3, PP2C14, PP2C51 and PP2C60 were up-regulated, but PP2C12, PP2C27 and PP2C54 were down-regulated. These results are similar to those of Cao et al. (2016) and indicate that PP2Cs participate in the salt stress response of tea plants through different molecular mechanisms; however, further analysis is needed to determine the specific function of each PP2C. In contrast, no SnRK2s were identified among the DEGs. This may be because the duration of the salinity treatment was too short to elicit a reaction of SnRK2s in the leaves of tea plants (Liu et al. 2016b). However, the ABA pathway was reported to be connected and integrated with the Ca2+ pathway and MAPK pathway in many recent studies (Edel and Kudla 2016; de Zelicourt et al. 2016). Therefore, our findings suggest that close-knit signaling networks such as those of Ca2+, MAPKs, and ABA and the crosstalk among them are stimulated and play important roles in tea plants in response to salt stress.
TFs
TFs have long been an important topic in plant resistance research, and some TF families play significant roles in salt stress adaptation (Lindemose et al. 2013). Similar to the reports of Zhang et al. (2017), the majority of the TFs were significantly up-regulated under salt stress such as bHLH, MYB, NAC and AP2/ERF family, and as mentioned in Zhang’s analysis, some bHLH family TFs are critical in the modulation of the ABA-dependent-signaling pathway. In addition, Some MYB and NAC family genes have been reported to be the regulators of salt stress response, such as MYB20 (Li et al. 2015), and these results are consistent with the results in maize, in which TFs play important positive regulatory roles in response to salt stress (Du et al. 2017). But beyond that, many other families of TFs are also closely associated with the signal transductions. Many bZIP-family TFs are downstream factors of ABA signaling and regulate pivotal cellular processes, especially those of plant responses to salt stress (Jakoby et al. 2002). In the present study, nine bZIP genes, including bZIP14, bZIP27, bZIP53 and ABI5, among others, were identified as DEGs, and eight HD-Zips were also differentially expressed; these HD-Zips affect ABA biosynthesis and regulate salt stress responses through ABA-mediated signal transduction pathways (Zhang et al. 2012). In addition, WRKY TFs are also important regulators in the response to salt stress (Jiang et al. 2017). Among these TFs, WRKY17 was reported to responds to salt stress through ABA signaling in cotton (Yan et al. 2014), and Park et al. (2005) confirmed that some members in WRKY group II participate in the CaM-mediated Ca2+ signaling pathway under salt stress. In our study, multiple WRKY genes, including WRKY7, WRKY33, WRKY18 and WRKY40, were up-regulated in tea plants in response to salt stress and suggest that WRKY TFs may play positive regulatory roles in response to salt stress in tea plants and that this response may depend on Ca2+ and ABA signaling pathways. Moreover, ZF-TFs are another important family of TFs, and ZF-TFs play important roles in the salt stress response of plants (Ciftci-Yilmaz and Mittler 2008). Two CCCH-type ZF-TFs, AtSZF1 and AtSZF2, which can regulate salt stress responses have been identified in Arabidopsis (Sun et al. 2007). Li and Chen (2001) reported that two C2H2-type ZFs in cotton may be involved in the plant response to salt stress and that this process depends on the regulation of ABA signaling. Here, six different types of ZF-TFs, including C2H2, CCCH, C3H4, Dof, RING-H2 and GATA, were identified in our DEGs, which imply that ZF-TFs are also essential in the salt stress response process of tea plants. In short, these data show that TFs participate in the molecular regulation of tea plants under salt stress and that this participation may be closely linked to multiple signaling pathways, such as those of Ca2+, MAPKs and ABA. However, additional studies are needed to confirm this linkage.
Key functional proteins related to salt resistance in tea plants
In addition to signal transduction and TFs, many downstream functional proteins involved in salt stress were identified in our DEGs, including HSPs, LEA proteins, AQPs and F-box proteins. HSPs are a kind of stress protein that can be induced in response to heat stress, and the synthesis of most HSPs is not induced under salt stress (Wang et al. 2004). In the present study, the HSPs of the DEGs were down-regulated during the process of the salt stress response in tea plants; these results contrast with those of recent studies using RNA-Seq, in which HSPs in barley were up-regulated in response to salt stress (Liu et al. 2016a). This finding implies that HSP involvement in the response to salt stress may differ among plant species. In addition, some LEA proteins are vital proteins that function in resistance to salt stress, such as DHN1 and DHN5 (Banerjee and Roychoudhury 2016). Similarly, our data revealed that numerous LEA proteins are differentially expressed and that the majority of them are up-regulated, which indicates that these genes are possibly salt resistance genes in tea plants. Also, LEA proteins are regulated by some TFs under salt stress, including bZIPs, MYBs and DREBs (Bies-Ethève et al. 2008). It is reasonable to speculate that salt stress, combined with the analysis of the TFs among the DEGs, promotes the accumulation of LEA proteins, which depend on the regulation of partial TFs, especially bZIPs and MYBs. Furthermore, AQPs and F-box proteins are considered to subserve the enhancement of salt stress tolerance in plants. Twenty CsAQP genes that belong to five subfamilies were recently isolated and analyzed from tea plants, and the majority of these AQPs, especially PIPs and TIPs, were up-regulated in response to salt stress, with the exception of SIPs (Yue et al. 2014). Similar results were obtained in our data, as the majority of the differentially expressed AQPs were from the PIP and TIP subfamilies. Additionally, all differentially expressed F-box proteins significantly increased in the salt-treated tea plants in the present study, this result is consistent with a previous study that reported F-box proteins both act as key elements and play positive roles in salt tolerance in Arabidopsis (Jia et al. 2015). Taken together, our data reveal that many functional proteins are involved in the salt stress response process in tea plants. This process may be dependent upon the regulation of respective upstream TFs and is necessary for improving the salt tolerance of tea plants.
Conclusion
An RNA-Seq technology-based transcriptomic analysis of tea plants in response to salt stress has been performed in this study. Based on the results and those of previous studies, an inferred salt-responsive molecular regulatory network in tea plants was put forward, presented in Fig. 4. The process can be divided into three stages: signal sensing and transduction, transcriptional regulation and the synthesis of downstream functional proteins. Briefly, when tea plants are exposed to high salinity, Na+ enters the cell and simultaneously triggers relevant signal transduction pathways, especially those of Ca2+ and ABA. These pathways further inspire secondary and tertiary regulatory networks, including those comprising Ca2+ sensors, relay proteins, MAPK cascades and TFs. TFs then activate or inhibit the expression of downstream vital functional genes, such as HSPs, LEA proteins, AQPs and F-box proteins, to produce a response to salt stress and improve the salt tolerance of tea plants. In summation, our study revealed the molecular mechanism of tea plants that operates in response to salt stress and can provide many candidate genes for a series of additional studies, especially those involving the genetic engineering and breeding of tea plants that are highly resistant to abiotic stress.

Regulatory model predicted from the DEGs under salt stress in C. sinensis. High salinity damages the biomembranes of plant cells, and Na+ enters the cell. At the same time, salinity-induced Ca2+ and ABA act as secondary messengers to transfer the salt stress signal to downstream TFs and target genes. This process depends on a signaling network that includes various proteins, such GLRs, Ca2+-ATPases, CAXs, CBLs, CIPKs, CaM, CDPKs, MAPKs, and PP2Cs, among others. Many TFs, including WRKYs, MYBs, NACs, bZIPs, AP2/ERFs and ZFs, are then activated in response to salt stress, and the Na+/H+ antiporter SOS1, which is downstream of the Ca2+ pathway, is triggered to discharge excess Na+ from the cell. Afterward, many functional proteins such as HSPs, LEA proteins, F-box proteins and AQPs are regulated in response to salt stress. Together, a molecular regulatory mechanism mediated by Ca2+ and ABA signaling is triggered in response to salt stress in tea plants. See the main text for details
References
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Apweiler R, Bairoch A, Wu CH et al (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:D115-D119
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29
Banerjee A, Roychoudhury A (2016) Group II late embryogenesis abundant (LEA) proteins: structural and functional aspects in plant abiotic stress. Plant Growth Regul 79:1–17
Bies-Ethève N, Gaubier-Comella P, Debures A et al (2008) Inventory, evolution and expression profiling diversity of the LEA (late embryogenesis abundant) protein gene family in Arabidopsis thaliana. Plant Mol Biol 67:107–124
Boudsocq M, Sheen J (2009) Stress signaling II: calcium sensing and signaling. In: Pareek A, Sopory S, Bohnert H (eds) Abiotic stress adaptation in plants. Springer, Dordrecht, pp 75–90
Cao JM, Jiang M, Li P, Chu ZQ (2016) Genome-wide identification and evolutionary analyses of the PP2C gene family with their expression profiling in response to multiple stresses in Brachypodium distachyon. BMC Genom 17:175
Chen L, Zhou ZX, Yang YJ (2007) Genetic improvement and breeding of tea plant (Camellia sinensis) in China: from individual selection to hybridization and molecular breeding. Euphytica 154:239–248
Chen L, Chu C, Lu J, Kong XY, Huang T, Cai YD (2015) Gene ontology and KEGG pathway enrichment analysis of a drug target-based classification system. PLoS ONE 10(5):e0126492
Cheong YH, Kim MC (2010) Functions of MAPK cascade pathways in plant defense signaling. Plant Pathol J 26(2):101–109
Ciftci-Yilmaz S, Mittler R (2008) The zinc finger network of plants. Cell Mol Life Sci 65:1150–1160
de Zelicourt A, Colcombet J, Hirt H (2016) The role of MAPK modules and ABA during abiotic stress signaling. Trends Plant Sci 21:677–685
Deng WW, Wang S, Chen Q, Zhang ZZ, Hu XY (2012) Effect of salt treatment on theanine biosynthesis in Camellia sinensis seedlings. Plant Physiol Biochem 56:35–40
Du XL, Wang G, Ji J, Shi LP, Guan CF, Jin C (2017) Comparative transcriptome analysis of transcription factors in different maize varieties under salt stress conditions. Plant Growth Regul 81:183–195
Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763
Edel KH, Kudla J (2016) Integration of calcium and ABA signaling. Curr Opin Plant Biol 33:83–91
Fedoroff NV, Battisti DS, Beachy RN et al (2010) Radically rethinking agriculture for the 21st century. Science 327:833–834
Finn RD, Bateman A, Clements J et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222-D230
Haas BJ, Papanicolaou A, Yassour M et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Hao XY, Horvath DP, Chao WS, Yang YJ, Wang XC, Xiao B (2014) Identification and evaluation of reliable reference genes for quantitative real-time PCR analysis in tea plant (Camellia sinensis (L.) O. Kuntze). Int J Mol Sci 15:22155–22172
Jakoby M, Weisshaar B, Droge-Laser W et al (2002) bZIP transcription factors in Arabidopsis. Trends Plant Sci 7:106–111
Ji HT, Pardo JM, Batelli G, Van Oosten MJ, Bressan RA, Li X (2013) The salt overly sensitive (SOS) pathway: established and emerging roles. Mol Plant 6:275–286
Jia FJ, Wang CY, Huang JG, Yang GD, Wu CG, Zheng CC (2015) SCF E3 ligase PP2-B11 plays a positive role in response to salt stress in Arabidopsis. J Exp Bot 66:4683–4697
Jiang JJ, Ma SH, Ye NH, Jiang M, Cao JS, Zhang JH (2017) WRKY transcription factors in plant responses to stresses. J Integr Plant Biol 59:86–101
Julkowska MM, Testerink C (2015) Tuning plant signaling and growth to survive salt. Trends Plant Sci 20:586–594
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277-D280
Koonin EV, Fedorova ND, Jackson JD et al (2004) A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol 5:R7
Li ZY, Chen SY (2001) Isolation, characterization and chromosomal location of a novel zinc-finger protein gene that is down-regulated by salt stress. Theor Appl Genet 102:363–368
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323
Li CN, Ng CKY, Fan LM (2015) MYB transcription factors, active players in abiotic stress signaling. Environ Exp Bot 114:80–91
Li J, Lv XJ, Wang LX, Qiu ZM, Song XM, Lin JK, Chen W (2017) Transcriptome analysis reveals the accumulation mechanism of anthocyanins in ‘Zijuan’ tea (Camellia sinensis var. asssamica (Masters) kitamura) leaves. Plant Growth Regul 81:51–61
Lindemose S, O’Shea C, Jensen MK, Skriver K (2013) Structure, function and networks of transcription factors involved in abiotic stress responses. Int J Mol Sci 14:5842–5878
Liu CH, Lu RJ, Guo GM et al (2016a) Transcriptome analysis reveals translational regulation in barley microspore-derived embryogenic callus under salt stress. Plant Cell Rep 35:1719–1728
Liu JY, Chen NN, Cheng ZM, Xiong JS (2016b) Genome-wide identification, annotation and expression profile analysis of SnRK2 gene family in grapevine. Aust J Grape Wine R 22:478–488
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25:402–408
Marguerat S, Bahler J (2010) RNA-seq: from technology to biology. Cell Mol Life Sci 67:569–579
Mehlmer N, Wurzinger B, Stael S et al (2010) The Ca2+-dependent protein kinase CPK3 is required for MAPK-independent salt-stress acclimation in Arabidopsis. Plant J 63:484–498
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59:651–681
Park CY, Lee JH, Yoo JH et al (2005) WRKY group IId transcription factors interact with calmodulin. FEBS Lett 579:1545–1550
Postnikova OA, Shao J, Nemchinov LG (2013) Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol 54:1041–1055
Ryu H, Cho YG (2015) Plant hormones in salt stress tolerance. J Plant Biol 58:147–155
Shi HZ, Ishitani M, Kim CS, Zhu JK (2000) The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc Natl Acad Sci USA 97:6896–6901
Song AP, Zhu XR, Chen FD, Gao HS, Jiang JF, Chen SM (2014) A chrysanthemum heat shock protein confers tolerance to abiotic stress. Int J Mol Sci 15:5063–5078
Steinhorst L, Kudla J (2014) Signaling in cells and organisms—calcium holds the line. Curr Opin Plant Biol 22:14–21
Sun JQ, Jiang HL, Xu YX, Li HM, Wu XY, Xie Q, Li CY (2007) The CCCH-type zinc finger proteins AtSZF1 and AtSZF2 regulate salt stress responses in Arabidopsis. Plant Cell Physiol 48:1148–1158
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Upadhyaya H, Panda SK (2013) Abiotic stress responses in tea [Camellia sinensis L (O) Kuntze]: an overview. Rev Agric Sci 1:1–10
Wan Q, Xu RK, Li XH (2012) Proton release by tea plant (Camellia sinensis L.) roots as affected by nutrient solution concentration and pH. Plant Soil Environ 58:429–434
Wang WX, Vinocur B, Shoseyov O, Altman A (2004) Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci 9:244–252
Wang WD, Xin HH, Wang ML et al (2016a) Transcriptomic analysis reveals the molecular mechanisms of drought-stress-induced decreases in Camellia sinensis leaf quality. Front Plant Sci 7:385
Wang WD, Sheng XY, Shu ZF et al (2016b) Combined cytological and transcriptomic analysis reveals a nitric oxide signaling pathway involved in cold-inhibited Camellia sinensis pollen tube growth. Front Plant Sci 7:456
Wang YX, Liu ZW, Wu ZJ, Li H, Zhuang J (2016c) Transcriptome-wide identification and expression analysis of the NAC gene family in tea plant [Camellia sinensis (L.) O. Kuntze]. PLoS ONE 11:e0166727
Wilkins KA, Matthus E, Swarbreck SM, Davies JM (2016) Calcium-mediated abiotic stress signaling in roots. Front Plant Sci 7:1296
Yamakawa H, Katou S, Seo S, Mitsuhara I, Kamada H, Ohashi Y (2004) Plant MAPK phosphatase interacts with calmodulins. J Biol Chem 279:928–936
Yan HR, Jia HH, Chen XB, Hao LL, An HL, Guo XQ (2014) The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol 55:2060–2076
Yoshida T, Mogami J, Yamaguchi-Shinozaki K (2014) ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr Opin Plant Biol 21:133–139
Yue C, Cao HL, Wang L et al (2014) Molecular cloning and expression analysis of tea plant aquaporin (AQP) gene family. Plant Physiol Biochem 83:65–76
Zhang JH, Jia WS, Yang JC, Ismail AM (2006) Role of ABA in integrating plant responses to drought and salt stresses. Field Crop Res 97:111–119
Zhang SX, Haider I, Kohlen W et al (2012) Function of the HD-Zip I gene Oshox22 in ABA-mediated drought and salt tolerances in rice. Plant Mol Biol 80:571–585
Zhang Q, Cai MC, Yu XM, Wang LS, Guo CF, Ming R, Zhang JS (2017) Transcriptome dynamics of Camellia sinensis in response to continuous salinity and drought stress. Tree Genet Genomes 13:78
Zhou Y, Yin XC, Duan RJ, Hao GP, Guo JC, Jiang XY (2015) SpAHA1 and SpSOS1 coordinate in transgenic yeast to improve salt tolerance. PLoS ONE 10:e0137447
Zhu JK (2002) Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53:247–273
Zhu JK (2016) Abiotic stress signaling and responses in plants. Cell 167:313–324
Acknowledgements
This work was supported by the earmarked fund for Modern Agro-industry Technology Research System (CARS-23), the Agricultural Special Fund Project of Shaanxi Province, the China Postdoctoral Science Foundation (Grant No. 2016M602873).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wan, S., Wang, W., Zhou, T. et al. Transcriptomic analysis reveals the molecular mechanisms of Camellia sinensis in response to salt stress. Plant Growth Regul 84, 481–492 (2018). https://doi.org/10.1007/s10725-017-0354-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10725-017-0354-4