
Abstract
To get rice varieties with specific germination activity (GA) suitable for production and breeding, the genetic characteristics of GA were revealed by measuring the seed weight (SW), germination rate (GR), coleoptile length (CL) and radicle length (RL) of seeds at 5, 6 and 7 weeks after heading with the recombinant inbred line (RIL) as the material. A total of 10 unconditional QTLs were detected, and the phenotypic variation explained by a single QTL ranged from 3.97 to 14.16%. Among them, 10 unconditional QTLs were detected at three different developmental stages, and the phenotypic variation explained by a single QTL ranged from 3.97 to 14.16%. Among them, qGR-7, qSW7-1, qSW7-2, qCL7-1, and qRL3-1 played an important role in seed activity. In addition, a total of 295 epistatic QTLs were detected, of which 4 were located in the qRL3-1 and qGR-7 intervals. Three RIL lines (RIL18, RIL106, and RIL155) were further screened through genotype and phenotype identification. These three lines contain 3–6 additive QTLs, which can aggregate 6–8 excellent alleles, and have an important theoretical value of resistance to pre-harvest sprouting breeding. In addition, a total of 41 Mqtls was detected by meta-analysis, of which 15 Mqtls had physical intervals less than 1.0 Mb. And qSW2-2, qSW2-3, qSW2-1, qCL2-1, and qRL3-1 were located in the Mqtl2-2, Mqtl2-4, Mqtl3-3, and Mqtl4-2 intervals. A further prediction of candidate genes, expression level determination, and variation analysis showed that Os01g0813100 was a candidate gene affecting seed activity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The germination activity (GA) of mature seeds is low, which can prevent the occurrence of pre-harvest sprouting, to ensure that crop yields are unaffected. However, in the process of rice breeding and domestication, varieties with rapid and uniform germination characteristics were gradually selected, which led to the partial or total loss of low GA during the seed development stage (Gu et al. 2008; Harlan et al. 1973). Therefore, the GA of rice seeds should be maintained at a suitable equilibrium level, that is, low GA in the development stage and high GA in the sowing stage.
The variation of seed GA is controlled by many genetic factors, called quantitative trait loci (QTL), which are significantly influenced by the environment and continuous phenotypic variation during seed development (Baskin and Baskin 2004). About 220 QTL linked to the GA of rice seeds have been detected, of which only four (qSdn-1, qSdn-5, qSD12, and Sdr1) have been investigated through fine-mapping (Gu et al. 2010; Lu et al. 2011; Takeuchi et al. 2003), and only three genes (Sdr4, Sd7-1/qPC7, and qSD1-2) have been cloned that is related to GA (Sugimoto et al. 2010; Xing-You et al. 2011; Ye et al. 2015). These results enrich the understanding of the genetic mechanism of seed GA but fail to make a breakthrough in solving the trouble of pre-harvest sprouting in rice.
According to developmental genetics, genes are selectively expressed at different stages of seed germination. QTL analysis can be adapted to include the effects of developmental stages (Zhu 1995). Therefore, the QTL detected at specific growth stages (Han et al. 2011) has conditional genetic effects (Sen and Churchill 2001). Previous studies have neglected the different gene roles of seed GA at different developmental stages, but to develop molecular marker-assisted breeding, conditional and unconditional QTL require equal attention (Han et al. 2011). GA is an important phenomenon in the growth and development of plants. The reasons for GA are complex and diverse. Seeds with low GA must differentiate and grow before germination. In addition, the development of the embryo will directly affect GA, and seed GA can be evaluated by germination rate (GR), coleoptile length (CL), and radicle length (RL). Moreover, when mature rice seeds are exposed to adverse germination conditions, the seeds will be induced to enter low germination levels (Graeber et al. 2012). Therefore, when studying the genetic GA characteristics of rice, adverse germination conditions must also be considered.
The purpose of this study was to investigate the developmental behavior of GA in rice seeds, identify conditional and unconditional additive QTL, and detect epistatic QTL of GA in rice seeds. In addition, meta-analysis was used to mine candidate genes in meta-QTL (MQTL) with fine physical intervals. Through genetic and phenotypic identification, the lines with low GA levels at seed development stage and high GA level at the sowing stage were screened out. The screened recombinant inbred lines (RILs) and identified QTLs were used to predict the new parental combination of rice GA. The candidate genes obtained from the meta-analysis may be used for marker-assisted selection (MAS) and pyramiding. The results obtained from this research may provide valuable information for improving seed GA in rice breeding.
Materials and methods
Plant materials
Two rice (Oryza sativa L.) varieties, Dongnong422 (DN422) and Kongyu131 (KY131), and 190 RILs derived from DN422 × KY131 were used in this study (Yang et al. 2018). All the experimental materials were planted at the experimental station of Northeast Agricultural University (Heilongjiang Province, China; 47° 98′ N, 128° 08′ E; 128 m above sea level). When the main spike appears from the sheath of the flag leaf, it is considered as the beginning of the heading (Lu et al. 2011). At the onset of heading, nine main panicles with the same flowering date as the sample were selected from each line. Samples were collected from the 5th, 6th, and 7th week after heading. Three panicles of each line were used to identify seed GA by storing them at 4 °C to maintain seed dormancy.
Evaluation of germination activity
Fresh seeds of different maturity were weighed, and were surface sterilized in 20% diluted bleach (5% NaClO) for 15 min and then thoroughly rinsed with water. The sterilized seeds were placed in a 9 cm Petri dish with double filter paper, 10 ml of distilled water was added, and seed germinability was in an incubator at 30 °C for 7 days. The GR, CL, and RL of each line were measured. GR = accumulated number of germinated seeds/total seeds tested. CL and RL were measured using a standard ruler (Hsu and Tung 2015), and seed maturity (SM) is evaluated by the degree of seed filing, that is seed weight.
QTL mapping
A genetic linkage map with 155 markers (Yang et al. 2018) was used for QTL analysis, which was carried out in ICIMapping 4.2 (http://www.isbreeding.net) with composite interval mapping (ICIM) and selective genotyping mapping (SGM). For each trait, the significant QTL, identified by 1000 permutation datasets (Churchill and Doerge 1994), was declared by genome-wide threshold values of P = 0.05, and QTL with LOD (logarithm of the odds) values larger than 2.5 was considered. Epistasis analyses were performed by analyzing pair-wise QTL interactions (Wang et al. 2012).
Prediction for novel parental combination
In crop breeding, the varieties with the best phenotypic value and excellent alleles can be used in the design of parental combinations (Niu et al. 2013). According to the major QTL genotypes linked to GA, we compared the genotypes of the RIL population and investigated germination activity-related traits in seed development stage and mature seed germination stage. Lines with a low level of GA during the developmental stage and a high level of GA during mature seed germination were selected. According to the Monte Carlo simulation experiment, a cross combination was determined to achieve the maximization of the number of excellent alleles (Niu et al. 2013). In addition, to evaluate the existence of secondary dormancy, after seven weeks of flowering, the mature seeds were exposed to 30 °C for 36 h to break dormancy. Then, these selected target strains were identified to study the germination of seeds under three biological stresses, namely low temperature (15 °C), NaCl (100 mM), and PEG (10%), to evaluate secondary dormancy.
Meta-analysis of seed GA-related traits in rice
The consensus QTL information of seed GA-related traits, including name, chromosome, trait, LOD score, phenotypic variance (R2), and position was collected from a public database (http://www.gramene.org) and detected in this study. Overall, the QTLs of 164 seed GA-related traits were integrated into an individual experiment (Table S4). Genetic maps were created by integrating six maps (Table S5), on which the integrated QTLs with different methods and backgrounds were projected by BioMercator V4.2. To determine the existence of consistent QTL and locate the accurate and effective confidence intervals (CI) of original QTLs, a meta-analysis for seed GA-related traits was used. The Akaike information criterion (AIC) model revealed the most likely model, and each model provides the physical location of the most likely consistent QTL with the maximum likelihood estimation of the Gaussian function. The nodulation traits consensus QTL variance was calculated using the following formula: Var(QTL) = 1/Σσi2, in which σi2 is the position of the variance for each of the QTLs on the linkage group. A 95% CI of the “real QTLs” was calculated from the var(QTL): CI = 3.92 × var (QTL)1/2.
Searching for proteins homologous to seed GA in rice
To obtain candidate genes related to seed GA, three seed GA genes, DOG1 (Bentsink et al. 2006), TRAB1 (Hobo et al. 1999), and Sdr-4 (Sugimoto et al. 2010) were targeted for homologous alignment. We first download all the protein sequences in the candidate interval using batch download tools (http://www.plantgdb.org/) and then upload a FASTA-format file containing sequences to search for matching Pfam families using the HMMER website (http://pfam.xfam.org/search). Finally, the candidate genes in the target region were obtained by homologous alignment.
RNA isolation and qRT-PCR analysis
Total RNA was extracted from KY131 and DN422 seeds at 7, 14, 21, 28, 35, 42, and 49 days after heading, using the TRIzol method (Thermo Fisher Scientific, Waltham, MA, USA) and treated with DNase I to eliminate any DNA contamination. RNA quality was assessed by electrophoresis and stored at − 80 °C until use. First-strand cDNA (10 µL) was synthesized according to the instructions for the PrimeScript™ RT Master Mix (Takara Biomedical Technology (Beijing) Co., Ltd., Beijing, China). Primers were designed with Primer Premier v. 5.0 (PREMIER Biosoft International, Palo Alto, CA, USA) and were based on the candidate gene transcript sequence. Primer amplification specificity was verified in the rice genome database using BLAS from NCBI (Coordinators 2017).
Results
Phenotypic evaluations
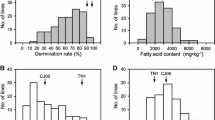
There was no significant difference in GR between the two parents at 5 weeks after heading, but there was a significant difference after 6 and 7 weeks. However, the GR of KY131 is faster than that of DN422 with the increase in seed growth and development, which indicates that KY131 has a better GA. The statistical results of SW, CL, and RL between the two parents were similar to those of GR (Table 1). There was continuous frequency distribution and transgressive segregation among RILs in SW, GR, CL, and RL (Fig. 1). In addition, there was a significant positive correlation between SD, GR, CL, and RL in the 5th week of seed development, while there was no significant correlation between SW and CL or RL in the 6th week of seed development. In the 7th week after heading, when the seeds were fully mature, the SW was no longer the influencing factor of GR, CL, or RL. In addition, there was a very significant positive correlation between GR, CL, and RL (Table 2, Fig. S1).

Frequency distribution of seed germination activity-related traits in rice in RILs
Additive QTL analysis based on ICIM
Ten unconditional additive QTL for seed GA-related traits were identified at three developmental stages (Table 3; Fig. 2). The phenotypic variance explained by each QTL ranged from 3.97 to 14.16%. The positive alleles of five QTL (qSW2-1, qSW2-2, qSW2-3, qSW4-1, and qCL2-1) from DN422 and five QTLs (qGR-7, qSW7-1, qSW7-2, qCL7-1, and qRL3-1) from KY131 contributed to the increase in GA.

Genetic linkage map showing QTLs for seed germination activity. The black font represents the detected QTLs, the red font represents the seed germination activity-related QTLs detected by previous studies, and the blue font represents the non-seed germination activity-related QTLs reported by previous studies
An additive QTL (qGR-7) linked to final germination percentage was located on RM1362-RM1306 in the early, middle, and late stages of development. The mean phenotypic variation of the QTL was 10.76%. This interval contains reported QTL, namely qPLLN7 (Yan et al. 2003) and S23(t)) (Sobrizal 2000) related to fertility, and qDTH-7 (Fujino and Sekiguchi 2005) and hd7a (Yu et al. 2002) associated with the heading, but no QTL related to seed GA was found. It is suggested that there may be new gene loci linked to GA in this region. In addition, six additive QTLs linked to seed weight were identified, namely qSW2-1, qSW2-2, qSW7-1, qSW2-3, qSW4-1, and qSW7-2. qSW2-2 and qSW4-1 were identified at two different developmental stages, and the average phenotypic variation of QTL between them was 7.54%. The marker interval of qSW2-1 was a physical distance of 113.6 Kb, and five rice spike-related QTL (Marri et al. 2005; Wang et al. 2014; Xing et al. 2001; Yan et al. 2003) and one heading date QTL (Yamamoto et al. 2000) was reported, indicating that qSW2-1 might be a site that is related to the economical character of rice. In addition, qSW4-1 and gwt4a (Lin et al. 1995) are co-located, but qSW4-1 is located in the physical interval of 24.9 Mb, so it is impossible to determine whether qSW4-1 and gwt4a are the same QTL. However, a seed GA QTL, qSDS-4 (Gu et al. 2004) was included in this interval. Two additive QTL (qCL2-1 and qCL7-1) linked to coleoptile length were identified, and qCL7-1 was located on RM1362-RM1306 during the early, middle, and late developmental stages. Two QTL related to CL were detected, namely qCL2-1 and qCL7-1, but only qCL7-1 was detected at different stages of seed development, while qCL2-1 was detected only in the early stage of seed development, but not in the middle and late stages. One additive QTL (qRL3-1) linked to radicle length was detected during the middle and late developmental stages, and the interval of qRL3-1 has been reported for six seed GA genes (Cai and Morishima 2002; Cheng et al. 2014; Long et al. 2013; Wan et al. 1997; Ye et al. 2010). The other QTL are newly found sites related to seed GA.
However, only three conditional additives QTL (qRL3-1, qCL6-1, and qSW6-1) were identified in the 6th to 7th weeks after heading. The phenotypic variance explained by each QTL ranged from 6.10 to 18.65%. The positive alleles of three QTLs in KY131 lead to an increase in GA. Furthermore, qRL3-1 was detected in both conditions and non-conditions.
Additive QTL analysis based on SGM
By selecting genotypic mapping, 28 linkage markers were found in the three different developmental stages of the three traits (Table S1, Fig. S2), of which eight markers (RM1267, RM12938, RM1347, RM1362, RM1306, RM20261, RM207, and RM417) were detected 38 times, These eight markers were also detected in ICIM mapping. Unfortunately, the remaining 20 linkage markers were not detected in ICIM.
Epistatic QTL analysis
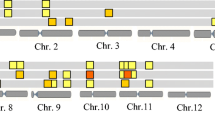
Through the epistasis analysis of GA-related traits in different development stages of rice seeds, it was found that GR, SW, and RL had 295 QTL interaction intervals (Fig. 3), of which 74 pairs of QTL were derived from the results detected using genotypic mapping (Table S2). Only four pairs of epistatic QTL were mapped by ICIM, SGM, and EPI, the main effects of these four epistatic QTLs were 0.3148, 0.3101, 0.1570, and 2.1141, respectively. However, there was no epistatic QTL identified for CL.

Epistatic QTLs of seed germination activity related traits in rice. The dotted line in each circle diagram represents both a pair of epistatic QTL
According to RAPD (http://rapdb.dna.affrc.go.jp), 13 genes were mapped in four pairs of epistatic QTL intervals, which had the same function as the cloned seed germination activity-related genes in three marker intervals (Fig. S3). Three and eight genes were mapped in the major QTL qRL3-1 and qGR-7 intervals, respectively. The co-expression relationship among these genes was further predicted by http://expression.ic4r.org/, and it was found that LOC_Os03g16480 and LOC_Os07g44950 (Pearson's r = 0.816594), and LOC_Os03g49730 and LOC_Os05g32760 (Pearson's r = 0.902182) had co-expression characteristics. It is suggested that the reason why the additive QTL is involved in the epistatic effect may be related to the existence of co-expressed genes in the interaction interval.
Mining of excellent lines
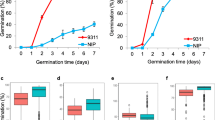
Through genotype and phenotype identification, the GA level of the three lines (RIL18, RIL106, and RIL155) was low during the developmental stage and high in the seed germination (Fig. 4A). The selected lines had three to six QTL loci (Table S3), and except for DN422, none of the three lines and KY131 had secondary dormancy under abiotic stress (Fig. 4B), and their CL and RL performed well (Fig. 4C, D). Using these three lines, it was predicted that three cross combinations of six to eight alleles could be pyramids (Table S3).

Seed germination activity-related traits of selected lines. A the GA level of the three lines (RIL18, RIL106, and RIL155) was low during the developmental stage and high in the seed germination; B Seeds of three selected RILs collected at 7 weeks after heading were used to evaluate seed germination activity after post-ripening stage under abiotic stress conditions (cold, drought and salt); C, D three selected RILs performed normally in coleoptile length and radicle length
Meta-analysis
We then collected 164 QTL for rice GA-related traits from 28 QTL maps (Table S4). Pre-consensus maps for all 12 chromosomes suitable for meta-analysis were created by the integration of six genetic maps (Table S5). Meta-analysis showed that 136 QTLs of GA were projected on different chromosomes (Fig. 5), chromosome 1 had the largest number of QTLs (20) and chromosome 10 had the least number of major QTLs (4) for GA. A total of 41 MQTL were detected based on meta-analysis, and the physical distance of these MQTL varied from 0.11 to 22.52 Mb. 15 MQTLs spanned physical intervals < 1.0 Mb. Among them, six QTLs, namely qSW2-2, qSW2-3, qSW2-1, qCL2-1, qRL3-1, and qSW4-1 were included in Mqtl2-2, Mqtl2-4, Mqtl3-3, and Mqtl4-2, respectively (Table 4). This MQTL with fine genetic and physical intervals might be regarded as important regions for MAS, fine mapping, and candidate gene identification.

Meta-analysis result for seed germination activity-related traits. All the QTLs for the same color on the left of the chromosome stick are defined as the same MQTL, the information of the original QTL is listed in Table S4
Putative GA genes in the MQTL regions
To ascertain whether some GA genes or their homologous genes are located in the detected GA MQTL regions, we searched the rice protein database with BLASTP for proteins homologous to DOG1 (Bentsink et al. 2006), TRAB1 (Hobo et al. 1999), and Sdr-4 (Kazuhiko et al. 2010). The protein sequences of 41 MQTL regions were downloaded for homology comparison. A total of 46 loci had homology with TRAB1 in the whole rice genome (Fig. S4, Table S6), and six conserved domains of zinc-finger proteins had homology with seed GA TRAB1 in MQTL regions (Table 5, Fig. S4). Three of the candidate genes have been cloned and functional verification, Os02g0266800 plays a role in the regulation of rice genes expressed in developing rice seeds (Izawa et al. 1994), Os01g0859300 and Os06g0211200 are two-locus associated with ABRE-binding protein responding to ABA (Hong et al. 2011; Tang et al. 2012).
qRT-PCR and Sanger sequencing
To further understand the expression pattern of candidate genes involved in seed GA in two rice varieties, six differentially expressed genes were selected for qRT-PCR analysis. First, cDNA was synthesized from total RNA extracted from seeds at 7, 14, 21, 28, 35, 42, and 49d after heading and qRT-PCR was carried out with the gene-specific primers shown in Table S7. As shown in Fig. 6, the expression levels of Os01g0813100 and Os06g0211200 in DN422 were significantly higher than that of KY131 in different development stages. However, the relative expression of the other four genes was low in different developmental stages of the seeds of the two varieties. The sequence analysis of two genes showed that there were three SNPs (34568271-bp, 34568272-bp, and 34568273-bp) in the CDS region of the Os01g0813100 of DN422, resulting in a change in two amino acids (GAG/GGC, CGG/GGG), on the contrary, Os06g0211200 exhibited no significant sequence differences between DN422 and KY131 (Fig. 7). We then referred to the data of the 3010 Rice Genomes Project and found that those three SNPs were not recorded in the RFGB v2.0 database at the Minor Allele Frequency (MAF) > 0.01 level. Thus these three SNPs were a rare natural variation that could be used as a molecular marker, and as a special functional variation in DN422 to restrict the GA of rice varieties.

Expression pattern of candidate genes in two rice varieties for the different development stage

Sequence alignment of genes Os01g0813100 and Os06g0211200 on DN422, KY131 and reference genomes, respectively, showing only the first 360 bp of the CDS region
Discussion
The GA characteristics of rice are regulated by both genetic and environmental factors (Takahashi 1997). The level of GA in rice is strongly influenced by the maturity of the seed (Takeuchi et al. 2003), Therefore, by studying the dynamic characteristics of seeds in different growth stages, the relationship between GA and maturity can be revealed. In rice, the heading date also affects the maturity level of the seed (Cheng et al. 2014; Takeuchi et al. 2003), however, the opposite conclusion was observed in previous studies (Lin et al. 1998; Miura and Araki 1999). In our previous research, a major QTL qe3HD1-7-2 of HD was reported in the RM1362-RM1306 marker interval, which was detected to be related to GA in this study (Yang et al. 2018). Here, a RIL population with 15 days difference in parent heading date (Yang et al. 2018) was used to elucidate the GA characteristics of seeds with different maturity levels. In Harbin (Heilongjiang Province, China; lat. 47° 98′ N, long. 128° 08′ E; 128 m above sea level), the seed doesn't mature until seven weeks after heading, so the seeds used for GA investigation are selected at the 5th, 6th and 7th weeks after heading. According to the phenotypic data, the seeds without post-ripening treatment had low GA, the germination rate of seeds from either line did not exceed 70%, and only a few lines had strong GA (Fig. 1). In addition, the maturity of seeds determines their weight. During the middle and late stages of seed development, there were no significant correlations between seed weight and seed GA, but there is a significant correlation in the early stage of seed development (Table 2, Fig. S1), which indicates that seed maturity is time-sensitive when determining seed GA characteristics.
Generally, there are two possibilities to induce seed GA, one is that the seed itself has not reached physiological maturity i.e. embryo development has not been completed, and the other is that the seed still lacks the conditions required for germination after reaching physiological maturity. To investigate the GA of rice seeds at different maturities, the GR, SW, CL, and RL were measured. The phenotypic value of the seed germination-related traits was very low during the developmental stages of the seeds and the GA was significantly increased in mature seeds treated with the post-ripening process (Fig. 4). Finally, through the detection of unconditional, conditional, and epistatic QTLs, we found a major QTL interval (RM1362-RM1306), in which phenotypic variance explained more than 10%, and it controlled the expression of GR, SW, and CL. Phenotypic correlation analysis showed that the phenotype and genetic correlation of GR, SW, and CL were consistent. According to the theory of developmental genetics, genes are selectively expressed at different developmental stages (Cheng et al. 2014). The number of QTLs detected at different stages of development was almost the same, and five QTLs (qGR-7, qSW2-2, qSW4-1, qCL7-1, and qRL3-1) were expressed at different stages of development, indicating that the expression of GA gene is highly correlated with different developmental processes of seeds. In addition, different traits have a different genetic loci, and the detected QTL that is linked to the RL is not coincident with the position of the QTL detected for the other three traits. Moreover, the QTL of RL was not detected in the early stage of development. However, the epistatic QTL was the least in the early stage of seed development (77 pairs), followed by the late stage (106 pairs), and the highest in the middle stage of development (112 pairs). Among these three developmental stages, the maximum phenotypic variance explained, appeared in the middle stage of development (0.98%), followed by the early stage (0.90%) and the late stage (0.88%). These results suggested that epistasis as a genetic factor was much more important for GA.
Crops need to maintain low GA in equilibrium (Graeber et al. 2012). If seeds have a low level of GA in the development stage, they can avoid pre-harvest germination, while mature seeds have a high level of GA, which is very important to ensure the growth of seedlings and yield formation. When some physiologically mature seeds are in an environment that is not conducive to germination, they can be induced to a low level of GA again (Graeber et al. 2012). Therefore, in the process of breeding and selection, we should aim at retaining those varieties with low GA levels in the development stage and increase GA levels after external stimulation. Here, through phenotypic selection and genotyping, three GA QTL-carrying lines were obtained during the seed development. Before physiological maturity, the value of GA-related traits of these three lines was lower (Fig. 4A), but we found that the germination rates of these three lines under three abiotic stresses had no significant difference compared with the control (Fig. 4B). Therefore, these three lines could be used as our breeding materials. Although we haven't found the functional gene that controls GA in rice, the superior lines will produce isolates in offspring through hybridization, and the recombinant lines will be identified by molecular-assisted selection. As long as the phenotype meets the breeding objectives, we have successfully applied this major QTL to breeding.
Moreover, we found that seven of the reported additive QTL were mapped in or near QTL identified by meta-analysis (Tables 3 and 4). For example, the qSW2-2 and qSW2-3 identified here was near the region of sd2 (Lee et al. 2005), qSW2-1 and qCL2-1 was near the region of qSD2.1 (Wang et al. 2014), qRL3-1 was near the region of qSD3.1 (Cheng et al. 2014), qSD3 (Ye et al. 2010), DOR (Cai and Morishima 2002), Pgi-1 (Wan et al. 1997), and qSW4-1 were near the region of qSDS-4 (Gu et al. 2004). Moreover, the epistatic QTL for GR-linked markers RM7, RM21, and RM254 identified here were located near the region of additive QTL qSD-3 (Wan et al. 2005), qSdn-11 (JM Wan et al. 2006), and qSD11BR (Chen et al. 2006), respectively. However, there were no seed GA QTLs previously reported to be close to the major QTL, qGR-7, qSW7-1, qSW7-2, or qCL7-1, which indicates that these four QTLs might be novel loci. In addition, it would be beneficial to identify a greater number of candidate genes from MQTL, as six conserved domains of zinc-finger proteins had homology with seed GA TRAB1 in five MetaQTL regions (Table 5). As shown by qRT-PCR sequence analysis, there were three SNPs in the CDS region of the Os01g0813100 of DN422. This may be a genetic reason for the low GA value of DN422 seeds. However, this is not enough to assign a certain phenotype to a single SNP. On the contrary, we believe that a favorable allelic variation in one key gene is not enough to provide GA, the final GA phenotype of the seed should be considered as the combined result of favorable allelic variations from different key genes. The biological functions of Os01g0813100 and its related genome variations need to be further confirmed by gene editing and high-efficiency over-expression transformation systems.
Abbreviations
- GA:
-
Germination activity
- RIL:
-
Recombinant inbred line
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphisms
- Meta QTL:
-
Meta-QTL
References
Baskin JM, Baskin CC (2004) A classification system for seed dormancy. Seed Sci Res 14(1):1–16
Bentsink L, Jowett J, Hanhart CJ, Koornneef M (2006) Cloning of DOG1, a quantitative trait locus controlling seed dormancy in Arabidopsis. Proc Natl Acad Sci USA 103(45):17042–17047
Cai H, Morishima H (2002) QTL clusters reflect character associations in wild and cultivated rice. Theor Appl Genet 104(8):1217–1228
Chen HS, Tao LX, Wang X, Huang XL, Zhuang JY, Zheng KL (2006) Identification of QTL associated with pre-harvest sprouting traits in rice. Chin J Rice Sci 20:253–258
Cheng J, Wang L, Du W, Lai Y, Huang X, Wang Z, Zhang H (2014) Dynamic quantitative trait locus analysis of seed dormancy at three development stages in rice. Mol Breed 34(2):501–510
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 3:963–971
Coordinators NR (2017) Database resources of the national center for biotechnology information. Nucleic Acids Res 45(Database issue):D12
Fujino K, Sekiguchi H (2005) Identification of QTLs conferring genetic variation for heading date among rice varieties at the northern-limit of rice cultivation. Breed Sci 55(2):141–146
Graeber K, Nakabayashi K, Miatton E, Leubner-Metzger G, Soppe WJ (2012) Molecular mechanisms of seed dormancy. Plant Cell Environ 35(10):1769–1786
Gu X-Y, Kianian SF, Foley ME (2004) Multiple loci and epistases control genetic variation for seed dormancy in weedy rice (Oryza sativa). Genetics 166(3):1503–1516
Gu X, Turnipseed EB, Foley ME (2008) The qSD12 locus controls offspring tissue-imposed seed dormancy in rice. Genetics 179(4):2263–2273
Gu XY, Liu T, Feng J, Suttle JC, Gibbons J (2010) The qSD12 underlying gene promotes abscisic acid accumulation in early developing seeds to induce primary dormancy in rice. Plant Mol Biol 73(1–2):97
Han Y, Xie D, Teng W, Zhang S, Chang W, Li W (2011) Dynamic QTL analysis of linolenic acid content in different developmental stages of soybean seed. Theor Appl Genet 122(8):1481–1488
Harlan JR, De Wet JMJ, Price EG (1973) Comparative evolution of cereals. Evolution 27(2):311–325
Hobo T, Kowyama Y, Hattori T (1999) A bZIP factor, TRAB1, interacts with VP1 and mediates abscisic acid-induced transcription. Proc Natl Acad Sci USA 96(26):15348–15353
Hong JY, Chae MJ, Lee IS, Lee YN, Nam MH, Kim DY, Yoon IS (2011) Phosphorylation-mediated regulation of a rice ABA responsive element binding factor. Phytochemistry 72(1):27–36
Hsu SK, Tung C (2015) Genetic mapping of anaerobic germination-associated QTLs controlling coleoptile elongation in rice. Rice 8(1):38–38
Huang J, Cai M, Long Q, Liu L, Lin Q, Jiang L, Chen S, Wan J (2014) OsLOX2, a rice type I lipoxygenase, confers opposite effects on seed germination and longevity. Transgenic Res 23(4):643–655
Izawa T, Foster R, Nakajima M, Shimamoto K, Chua N (1994) The rice bZIP transcriptional activator RITA-1 is highly expressed during seed development. Plant Cell 6(9):1277–1287
Kazuhiko S, Yoshinobu T, Kaworu E, Akio M, Hirohiko H, Naho H, Tsukaho H (2010) Molecular cloning of Sdr4, a regulator involved in seed dormancy and domestication of rice. Proc Natl Acad Sci 107(13):5792–5797
Lee SJ, Oh CS, Suh JP, McCouch S, Ahn SN (2005) Identification of QTLs for domestication-related and agronomic traits in an Oryza sativa× O. rufipogon BC1F7 population. Plant Breed 124(3):209–219
Lin H, Qian H, Zhuang J, Lu J, Min S, Xiong M, Zheng K (1995) Interval mapping of QTLs for yield and other related characters in rice. Rice Genet Newsl 12:251–253
Lin S, Sasaki T, Yano M (1998) Mapping quantitative trait loci controlling seed dormancy and heading date in rice, Oryza sativa L., using backcross inbred lines. Theor Appl Genet 96(8):997–1003
Long Q, Zhang W, Wang P, Shen W, Zhou T, Liu N, Wang Y (2013) Molecular genetic characterization of rice seed lipoxygenase 3 and assessment of its effects on seed longevity. J Plant Biol 56(4):232–242
Lu BY, Xie K, Yang CY, Wang SF, Liu X, Zhang L, Wan JM (2011) Mapping two major effect grain dormancy QTL in rice. Mol Breed 28(4):453–462
Marri PR, Sarla N, Reddy LV, Siddiq E (2005) Identification and mapping of yield and yield related QTLs from an Indian accession of Oryza rufipogon. BMC Genet 6(1):33
Miura K, Araki H (1999) Effect of temperature during the ripening period on the lnduction of secondary dormancy in rice seeds (Oryza sativa L.). Breed Sci 49(1):7–10
Niu Y, Xu Y, Liu X-F, Shi-Ping Y (2013) Association mapping for seed size and shape traits in soybean cultivars. Mol Breeding 31(4):785–794
Sen S, Churchill GA (2001) A statistical framework for quantitative trait mapping. Genetics 159(1):371–387
Sobrizal Y (2000) Mapping of F_1 pollen semi-sterility gene found in backcross progeny of Oryza sativa L. and Oryza glumaepatula Steud. Rice Genet Newsl 17:61–63
Sugimoto K, Takeuchi Y, Ebana K, Miyao A, Hirochika H, Hara N, Hattori T (2010) Molecular cloning of Sdr4, a regulator involved in seed dormancy and domestication of rice. Proc Natl Acad Sci 107(13):5792–5797
Takahashi N (1997) Inheritance of seed germination and dormancy. Sci Rice Plant 5:348–359
Takeuchi Y, Lin ST, Yano M (2003) Fine linkage mapping enables dissection of closely linked quantitative trait loci for seed dormancy and heading in rice. Theor Appl Genet 107(7):1174–1180
Tang N, Zhang H, Li X, Xiao J, Xiong L (2012) Constitutive activation of transcription factor OsbZIP46 improves drought tolerance in rice. Plant Physiol 158(4):1755–1768
Wan J, Nakazaki T, Kawaura K, Ikehashi H (1997) Identification of marker loci for seed dormancy in rice (Oryza sativa L.). Crop Sci 37(6):1759–1763
Wan J, Cao Y, Wang C, Ikehashi H (2005) Quantitative trait loci associated with seed dormancy in rice. Crop Sci 45(2):712–716
Wan J, Jiang L, Tang J, Wang C, Hou M, Jing W, Zhang L (2006) Genetic dissection of the seed dormancy trait in cultivated rice (Oryza sativa L.). Plant Sci 170(4):786–792
Wang ZF, Cheng JP, Chen ZW, Huang J, Bao YM, Wang JF, Zhang HS (2012) Identification of QTLs with main, epistatic and QTL× environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor Appl Genet 125(4):807–815
Wang L, Cheng J, Lai Y, Du W, Huang X, Wang Z, Zhang H (2014) Identification of QTLs with additive, epistatic and QTL× development interaction effects for seed dormancy in rice. Planta 239(2):411–420
Xing Y-Z, Xu C-G, Hua J, Tan Y-F (2001) Analysis of QTL x environment interaction for rice panicle characteristics. Yi Chuan Xue Bao Acta Genet Sin 28(5):439–446
Xing-You G, Foley ME, Horvath DP, Anderson JV, Jiuhuan F, Lihua Z, Koh-Ichi K (2011) Association between seed dormancy and pericarp color is controlled by a pleiotropic gene that regulates abscisic acid and flavonoid synthesis in weedy red rice. Genetics 189(4):1515–1524
Yamamoto T, Lin H, Sasaki T, Yano M (2000) Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 154(2):885–891
Yan CJ, Liang GH, Gu SL, Yi CD, Lu JF, Li X, Tang SZ, Gu MH (2003) Molecular marker analysis and genetic basis for sterility of typical indica/japonica hybrids. Yi Chuan Xue Bao Acta Genet Sin 30(3):267–276
Yang LM, Wang JX, Lei L, Wang JG, Muhammad JS, Liu HL, Zou DT (2018) QTL mapping for heading date, leaf area and chlorophyll content under cold and drought stress in two related recombinant inbred line populations (Japonica rice) and meta-analysis. Plant Breed 137(4):527–545
Ye H, Foley ME, Gu X-Y (2010) New seed dormancy loci detected from weedy rice-derived advanced populations with major QTL alleles removed from the background. Plant Sci 179(6):612–619
Ye H, Feng J, Zhang L, Zhang J, Mispan MS, Cao Z, Gu XY (2015) Map-based cloning of seed dormancy1-2 identified a gibberellin synthesis gene regulating the development of endosperm-imposed dormancy in rice. Plant Physiol 169(3):2152
Yu S, Li J, Xu C, Tan Y, Li X, Zhang Q (2002) Identification of quantitative trait loci and epistatic interactions for plant height and heading date in rice. Theor Appl Genet 104(4):619–625
Zhu J (1995) Analysis of conditional genetic effects and variance components in developmental genetics. Genetics 141(4):1633–1639
Acknowledgements
And we would like to thank the instrumental analysis center of Northeast Agricultural University and to thank Editage (www.editage.cn) for English language editing.
Funding
This research was financially supported by the Major Science and Technology Project of Heilongjiang Province, China (Grant No. 2020ZX16B01) and by the China Postdoctoral Science Foundation (Grant No. 2019M651249).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Li, P., Liu, H., Wen, H. et al. Comprehensive genetic analysis reveals seed germination activity-related QTL and meta-QTL in rice (Oryza sativa L.). Genet Resour Crop Evol 70, 1007–1022 (2023). https://doi.org/10.1007/s10722-022-01484-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-022-01484-6