
Abstract
As important plant-specific transcription factors, GRAS family members play significant roles in plant development and diverse stress responses. The identification of GRAS genes in many species has been explored; however, little is known about the evolutionary origin of GRAS genes of Brassica napus (BnGRAS genes or BnGRASs) Brassica napus (BnGRAS genes or BnGRASs) in the Brassica genus. Here, 56, 53 and 96 GRAS genes were identified in Brassica rapa, Brassica oleracea, and B. napus, respectively, which were classified into 13 subfamilies and 17 orthologous groups by phylogenetic analysis. Duplication analysis showed that the Brassica-α whole genome triplication event contributed more to the expansion of BnGRAS genes than allopolyploidization and tandem duplication. Moreover, all Brassica GRAS genes were estimated to have undergone purifying selection during their evolution. Additionally, structural feature and expression profile analyses indicated that GRAS genes were conserved in sequence characters within the same subfamily, suggesting similar functions in plant development and biotic and abiotic stress responses. Finally, the evolutionary origin of BnGRASs in the Brassica genus was proposed. Taken together, this study not only deduces the evolutionary origin of BnGRASs in the Brassica genus but also provides important candidate BnGRAS genes for further functional analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As plant-specific transcription factors (TFs), GRAS genes play an indispensable role in plant growth and development, as well as in stress responses (Ito and Fukazawa 2021). The name of GRAS is derived from the first three identified members: GAI (gibberellic acid insensitive), RGA (repressor of GA1-3 mutant), and SCR (scarecrow) (Pysh et al. 1999; Bolle 2004). Due to fast advances in genomics, transcriptomics, and other omics in planta, GRAS family members have been widely identified and analyzed in over 30 mono- and dicotyledonous plants, such as Arabidopsis thaliana, Oryza sativa (Tian et al. 2004; Lee et al. 2008), Vitis vinifera (Grimplet et al. 2016; Sun et al. 2016), Brassica rapa, Brassica juncea, and Brassica napus. napus (Song et al. 2014; Guo et al. 2019; Li et al. 2019). Previous studies have classified GRAS TFs into 8–13 subfamilies, such as DELLA, HAM, LS, DLT, LISCL, NSP1, NSP2, PAT1, SCR, SCL3, and SCL4/7. Recently, GRAS members were divided into 17 distinct subfamilies and 29 orthologous groups (OGs) in angiosperm species, suggesting that at least 29 ancestor genes existed before the angiosperm lineage evolutionary split from Amborella trichopoda (Cenci and Rouard 2017).
Generally, GRAS proteins, ranging from 400 to 700 amino acid (aa) residues (Bolle 2004), are regarded as intrinsically disordered proteins (IDPs) because the amino (N-) termini contain intrinsically disordered regions (IDRs), implying that the N-terminal domains of GRAS proteins harbor various structural variations (Peng et al. 1997; Sun 2011). Additionally, the N-terminal regions of some GRAS proteins may include other motifs, such as the DELLA domain in the DELLA subfamily, indicating the diverse N-termini of GRAS proteins (Peng et al. 1997). In contrast, the carboxyl (C-) termini of GRAS TFs appears to be highly conserved (the so called GRAS domain) and commonly contains five motifs: leucine heptad repeat I (LHR I), VHIID, leucine heptad repeat II (LHR II), PFYRE and the SAW motifs (Tian et al. 2004). The VHIID motif, a core structure, can combine with LHR I and II to form the complex LHR I–VHIID–LHR II, which might play an important role in DNA and protein binding in protein–protein interactions (Richards et al. 2000). However, for functional specificity, the localization of SAW and PFYRE motifs has not yet been clearly elucidated.
Not only are GRAS members structurally diverse, but they also perform multiple functions in plant development and stress responses. SHR (SHORT-ROOT) is involved in the bundle sheath and mesophyll cell fate by regulating the expression of SCR and SCL23 in A. thaliana (Cui et al. 2014). HAM is essential and specific for maintaining the shoot apical meristem in Petunia hybrida (Stuurman et al. 2002). HAMII-3 (SCL6), HAMII-2 (SCL22), and HAMII-1 (SCL27) have been shown to negatively regulate chlorophyll biosynthesis by inhibiting protochlorophyllide oxireductase C (PORC) gene expression in light-grown plants (Ma et al. 2014). DELLA proteins not only function as repressors of gibberellin responses (Sun 2011) but also constitute a main hub in hormone (jasmonate, auxin, brassinosteroid, and ethylene) signaling (Bai et al. 2012; Wild et al. 2012). PAT1-1 (PAT1), PAT1-2 (SCL21), and PAT1-4 (SCL13), which are three members of the PAT subfamily, have been shown to be downstream of the phytochrome A (phyA) signal transduction pathway in A. thaliana (Torres-Galea et al. 2006, 2013). LISCL is involved in meiosis-associated gene regulation in Lilium longiflorum (Morohashi et al. 2003), while LISCL6 (SCL14) is essential for the activation of stress-inducible promoters, especially SA- and 2,4-D-inducible promoters (Fode et al. 2008).
Apart from the important characterized structures and functions of GRAS proteins, genome-wide studies of GRAS genes from A. thaliana, Brachypodium distachyon, O. sativa, Glycine max, Selaginella moellendorffii, and Physcomitrella patens have shown that segmental duplications mainly exist in the soybean genome, and tandem duplication (TD) greatly promotes the expansion of group II GRAS genes (Wu et al. 2014). In addition, both whole genome duplication (WGD) and TD events have been verified as important contributors to the expansion of GmGRAS family members. Syntenic and evolutionary constraint analyses of GRAS proteins among soybean and distinct species (two monocots and four dicots) provided more detailed evidence of GmGRAS gene evolution (Wang et al. 2020a). Moreover, the genome-wide identification of GhGRASs in cotton indicated that the origin of the GRAS family might have occurred due to bacterial infection in the stage between algae and moss. Furthermore, duplication analysis also showed that segmental duplication events played the main role in GhGRAS family expansion, and purifying selection worked on these duplicated homologous gene pairs. TD events led to the increase of introns and expansion of SHR genes (Zhang et al. 2018).
To date, three rounds (i.e., γ, β and α) of WGD or whole genome triplication (WGT) have been suggested through evolutionary process analysis in Brassicaceae species (Bowers et al. 2003). Furthermore, the ancestors of the Brassica species experienced a specific WGT event (Brassica-α WGT, Bra-α WGT) after splitting from a common ancestor with A. thaliana. The allotetraploid B. napus (2n = 4x = 38) was formed by the hybridization (0.038 to 0.051 Million Years Ago, MYA) between B. rapa and Brassica oleracea that occurred approximately 7500 years ago, followed by chromosomal doubling. Due to the high sequence similarity of the two subgenomes in B. napus, subtle structural changes and incipient gene loss were very common in B. napus, including abundant homoeologous exchanges (Allender and King 2010; Chalhoub et al. 2014). Therefore, Brassica plants have been very important model plants for the evolutionary study of polyploidization (Lysak et al. 2005; Chalhoub et al. 2014; Yang et al. 2016). According to the gene density difference, the three orthologous genomic regions in B. rapa or B. oleracea were then classified into three subgenomes: MF1 (medium fractionated), MF2 (most fractionated), and LF (least fractionated) (Cheng et al. 2014; Liu et al. 2014). One gene in A. thaliana should have three copies in Brassica species after the Bra-α WGT. However, the genome-wide idenfication of GRAS members in B. rapa or B. oleracea was not three times that in A. thaliana or in BnGRAS genes in B. napus (Song et al. 2014; Guo et al. 2019), indicating that GRAS genes are very conservative, yet the underlying mechanism is unclear.
The evolutionary origin of GRAS members in plants proved to be very ancient, and the detailed evolutionary analysis of this family might shed some light on the evolutionary origin of species/genus speciation. However, a comprehensive evolutionary study of the BnGRAS genes in the Brassica genus is still lacking. In this study, we first identified GRAS family members from different angiosperms. We then systematically investigated the phylogenetic relationships, gene structures, motif compositions, chromosomal locations, and gene duplication events of the identified GRAS members in the Brassica genus. Moreover, evolutionary analysis of GRAS genes between B. napus and the two diploid parental lines (B. rapa and B. oleracea), as well as one close relative (A. thaliana) in Brassicaceae, was also carried out. In addition, cis-acting elements and different expression profiles of the BnGRAS genes were explored. Finally, the evolutionary origin of GRAS genes in the Brassica genus was proposed in this study. Collectively, these results not only disclose the expansion patterns and evolutionary origin of BnGRAS family members in the Brassica genus but also lay a foundation for further functional studies of different Brassica GRAS members.
Materials and methods
Sequence retrieval of Brassica GRAS family members
Genomic sequences and gene annotations for B. rapa (v2.0) were retrieved from the Brassica database (http://brassicadb.org/brad/), for B. oleracea (v2.1) from Ensembl (http://useast.ensembl.org/index.html), and for B. napus (v4.1) from the Genoscope database (http://www.genoscope.cns.fr/brassicanapus). Using the latest Hidden Markov Model (HMM) model PF03514.11 downloaded from Pfam (http://pfam.xfam.org/) as a query, the hmm search function from the HMMER 3.0 program was employed for GRAS proteins searching the entire protein fasta files of the three Brassica species, with an E value < 1e−10 and protein length > 100 aa. Subsequently, the corresponding sequences were retrieved for the entire protein and CDS sequence files using an in-house perl script. Finally, all sequences were manually confirmed by the dataset comparison in the EST database by employing BlastN from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Finally, genomic sequences of the identified GRAS genes (All CDS sequences are listed in Supplementary file 1) were obtained in accordance with the gene annotation file specification (Supplementary file 2). To distinguish GRAS genes among subgenomes in Brassica species, the subgenomes were redesignated as follows: B. rapa genome as Br, B. oleracea genome as Bo, and B. napus A subgenome as BnA and C subgenome as BnC (Table S1).
Sequence alignment and phylogenetic analysis of GRAS family members
To explore the evolutionary origin of candidate Brassica GRAS proteins (Table S1), multiple sequence alignment and phylogenic analysis were carried out. Except for the identified GRAS proteins in the three Brassica species, GRAS protein sequences of A. trichopoda (one basal angiosperm, and also outgroup for mono- and dicotyledonous phylogenies) and three studied species (A. thaliana, V. vinifera, and Theobroma cacao) were obtained from a previous study (Cenci and Rouard 2017), while GRAS protein databases of Carica papaya and 2 other Brassicaceae species (Capsella rubella and Thellungiella parvula) were downloaded from PlantTFDB 4.0 (http://planttfdb.cbi.pku.edu.cn/; Jin et al. 2017). All the protein sequences are listed in Supplementary file 3. Multiple sequence alignments were performed using the MAFFT program via the EMBL-EBI bioinformatics interface with default parameters (Li et al. 2015). Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) was used to identify conserved blocks using the following parameters: (i) smaller final blocks, (ii) gap positions within the final blocks, and (iii) less strict flanking positions (Castresana 2000). The phylogenetic tree was built by PhyML (http://phylogeny.lirmm.fr/; Guindon et al. 2010). The approximate likelihood-ratio test (aLRT) was chosen as the statistical test for branch support, and LG (amino acid, aa) was chosen as the substitution model. Finally, the phylogenetic tree file (Supplementary file 4) was visualized with iTOL v4 (http://itol.embl.de/; Letunic and Bork 2016).
Distribution analysis of GRAS family members in angiosperms
Apart from the 10 species described above, GRAS members of 12 other species were also identified from previous studies and treated with the same method to obtain more detailed subfamily classification of GRAS members in angiosperms. The 12 species were O. sativa (Tian et al. 2004), Prunus mume (Lu et al. 2015), Populus trichocarpa (Liu and Widmer 2014), Musa acuminata (Cenci and Rouard 2017), Ricinus communis (Xu et al. 2016), Zea mays (Guo et al. 2017), Malus domestica (Fan et al. 2017), Solanum lycopersicum (Niu et al. 2017), Nelumbo nucifera (Wang et al. 2016), and Medicago truncatula, G. max and Phoenix dactylifera (Cenci and Rouard 2017).
Syntenic analysis of GRAS genes in Brassica
The detailed chromosome location of each GRAS gene was mapped to the chromosomes of B. rapa, B. oleracea, and B. napus, according to the retrieved information from the genome annotation files. The syntenic relationship datasets for the corresponding orthologous gene pairs in three subgenomes, MF1, MF2, and LF of A. thaliana and Brassica species, were obtained from the released data (http://brassicadb.cn/#/syntenic-gene/; Cheng et al. 2012). Putative TD events were then identified from PTGBase (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and analyzed using MCScanX (Wang et al. 2012). The chromosomal locations and orthologous and paralogous relationships of GRAS genes of B. rapa, B. oleracea, and B. napus were visualized with circos-0.69 (http://circos.ca/; Krzywinski et al. 2009).
Evolutionary analysis of Brassica GRAS genes
To estimate the selective pressure acting on GRAS genes among B. napus, B. oleracea, B. rapa, and A. thaliana, the non-synonymous nucleotide substitutions (Ka), the synonymous nucleotide substitutions (Ks), and Ka/Ks ratios of all homologous gene pairs within subgenomes and subfamilies, as well as those generated by TD events, were calculated using KaKs_calculator 2.0 (Zhang et al. 2006). The divergence times of orthologous and paralogous gene pairs were evaluated according to the formula T = Ks/2λ, assuming a clock-like rate (λ) of 1.5 synonymous substitutions per 10−8 years for Brassica species (Koch et al. 2000).
Structural analysis of GRAS family members
The local version of Multiple Em for Motif Elicitation (MEME) v4.12.0 was utilized to identify conserved motifs in Brassica GRAS proteins using the default parameters with minor modifications. The maximum number of motifs was set to 35 to identify as many conserved motifs as possible (Tables S6, S7; Bailey et al. 2006). Then, the exon–intron structures obtained from the.gff genome annotation files and motif distributions identified above were depicted together using the revised genome annotation. The promoter sequences (the 2000 bp region upstream of the ATG start codon) of GRAS genes obtained from the genome database were used to identify cis-acting elements with PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/; Lescot et al. 2002), and then visualized with TBtools software (Plant et al. 2020).
Expression pattern analysis of GRAS family members under different conditions
The published transcriptome datasets of B. rapa, B. oleracea, and B. napus were obtained from the NCBI Short Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra), for B. rapa from SRP017757, SRP064840, SRP082359, and SRP064814, for B. oleracea from SRP017530, and for B. napus from ERP004876, SRP028575, SRP035525, SRP045411, SRP069813, and SRP079682. By performing Tophat v2.0.13, the clean reads from RNA-seq after quality control were mapped onto the B. rapa genome v2.0, B. oleracea genome v2.1, and B. napus genome v4.1. The expression levels in log10-based fragments per kilobase of exon model per million mapped fragments (FPKM) plus 1 of each gene were quantified by Cufflinks (v2.2.1; Table S8; Trapnell et al. 2012). The expression patterns of the GRAS genes were subsequently visualized using the heatmap.2 function in the gplot package for R software.
Plant materials and stress treatment
Brassica napus (‘Zhongshuang 11’, ZS11) seeds were cultivated in pots containing mixed soil (1:3 vermiculite/humus). For stress treatment, 14-day-old ZS11 seedlings were immersed in 17% (weight/volume) PEG6000 (drought) and 200 mM NaCl (salt) for 0, 2, 4, 6, and 8 h. After treatment, roots were collected and submerged immediately in liquid nitrogen, and then stored at –80℃ for further analysis.
RNA extraction and quantitative RT-PCR analysis
Total root RNA was extracted for B. napus using Trizol (TaKaRa, Beijing, China). The first-strand cDNA was synthesized using a PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Beijing, China) and used for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) with SYBR Premix ExTaq™ (TaKaRa, Beijing, China) according to the manufacturer’s instructions on a Roche LightCycler® 480 Real-time PCR system (Roche, Basel, Switzerland). BnCACTIN-7 was used as the reference control, and all gene-specific primers are listed in Table S9. Each experiment was conducted three times with 3 biological replicates to evaluate variation. The relative gene expression levels were calculated using the 2−△△Ct method.
Results
More BnGRASs were derived from genome duplication than from tandem duplication
By performing the HMM search, 56, 52, and 94 candidate GRASs were identified from B. rapa, B. oleracea, and B. napus, respectively. These sequences were revised by comparisons with the whole genome sequence and then corrected and integrated through Blast in the EST and SRA databases. Finally, 56, 53 (a new gene: BoPAT1-3b), and 96 (two new genes: BnALISCL3a and BnASHR-1a) validated GRAS sequences were identified from B. rapa, B. oleracea, and B. napus, respectively (Table S1). The allotetraploid B. napus possessed almost twice the GRAS genes as the two diploid Brassica plants. GRAS proteins from Brassica species varied from 103 to 760 aa residues in length, with an average of 512, 538, 525, and 517 aa in the Br, Bo, BnA, and BnC subgenomes, respectively. The aa lengths of these orthologous GRAS genes exhibited high similarities. Although the chromosome locations of the GRAS genes were highly conserved between the interspecies homologous gene pairs, they were unevenly distributed along the chromosomes in each Brassica species (Table S1).
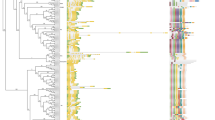
To explore the classification of candidate BnGRASs, a phylogenetic tree with GRAS members from Brassica species and seven other species was constructed (Fig. 1). All GRAS genes were named based on their OGs (Fig. 1; Table S1). The homologous genes from different Brassica species were confirmed by BlastP, and almost all GRAS genes formed interspecies gene pairs. Most gene pairs matched well in the Br-BnA and Bo-BnC groups, whereas BrDELLA1-1 and BrPAT1-3c mapped genes matched the BnC subgenome.

Unrooted ML phylogenetic tree based GRAS sequences from Amborella trichopoda, Vitis vinifera, Theobroma cacao, Carica papaya, Capsella rubella, Thellungiella parvula, Arabidopsis thaliana, Brassica rapa, Brassica oleracea and Brassica napus. The tree was categorized into 17 distinct subfamilies and 30 Orthologous Groups (OGs) with different colors. 13 grey colored OGs were lacked in Brassica species. All GRAS proteins of Brassica genus were emphasized in red
The total GRAS members identified from B. rapa (56) and B. oleracea (53) were greater than those from A. thaliana (33; Fig. 1). Compared to that in A. thaliana, almost every AtGRAS gene had at least one orthologous gene in the three Brassica species, except for SCL4 (AT3G50650) and DELLA1-5 (GAI, AT1G14920), which were lost during the evolution of the Brassica genus. Although many genes retained more than one copy in the Brassica species, there was only one gene copy for 12 GRAS members (PAT1-2, LISCL1, LISCL2, LISCL4, LISCL5, DELLA1-1, DELLA1-3, SCL7, LS, SCR, NSP1, and HAMII-2) in Br, Bo, BnA, and BnC subgenomes (Fig. 1), suggesting that these genes were functional conservative during the Bra-α WGT event. The identified GRAS gene number from each subgenome of B. napus was lower than that from their parental genomes, some of which were lost during B. napus speciation.
As the BnGRAS genes originated from BrGRASs and BoGRASs, most GRAS genes from B. rapa and B. oleracea preserved their homologous pairs in BnA and BnC (Table S1). However, compared with the parental genomes, 9 and 5 GRASs could not be detected in BnA and BnC, respectively. Some of these genes might have been generated individually in B. rapa and B. oleracea after B. napus speciation. Some of them originated from TDs (for example, BoPAT1-1a-td, BrLISCL6-td, BrLISCL7a-td, BrDLTb-tda, BrDLTb-tdb, and BrSCR-2-td), whereas the rest were derived from interspersed repeat events (e.g., BrSCL3d). In addition, some genes could not be observed in B. napus (SCL3c in BnA, NSP1 and HAMII-3 in BnC, and SHR-1b and PAT1-3a in BnA and BnC were missing). In contrast, the homologue of BnAHAMII-3-td could not be detected in the diploid ancestral species, which were generated by TD events in B. napus, or could have disappeared during the evolution of B. rapa after B. napus speciation. In addition, some GRAS genes produced by TDs were detected in both B. napus and its parental species (LISCL3b-td), which might have been generated throughout the evolution of Brassica plants (Fig. 1; Table S1).
Highly close subfamily classification of GRASs in cruciferous species
In total, 477 GRAS protein sequences obtained from 10 species, including A. trichopoda (the basal outgroup), V. vinifera, C. papaya and T. cacao (only experienced γ WGT event), C. rubella, A. thaliana, and T. parvula (experienced γ WGT, α WGD, and β WGD events), together with the GRAS proteins identified from Brassica species (experienced with the specific Bra-α WGT event) were used to construct the phylogenetic tree (Fig. 1). GRAS proteins of Brassica species were classified into 13 distinct subfamilies, namely PAT1, LISCL, HAM, DELLA, DLT, SHR, SCR, SCL4/7, LS, SCL3, SCL32, NSP1, and NSP2, and 17 OGs based on tree topology, as previously described (Cenci and Rouard 2017; Guo et al. 2019). No Brassica-specific subfamily or OG was found in this phylogenetic analysis. Each Brassica species contained at least one member of the above subfamilies. Compared with other species, the members of the SCLA, SCLB, RAM1, and RAD1 subfamilies were absent in all Brassica species, which was the same as in other Brassicaceae species (Fig. 1). These results indicated that the GRAS family was relatively conserved in the Brassica genus, and the SCLA, SCLB, RAM1, and RAD1 subfamilies did not exist in Brassicaceae. Except for orthologous gene copies, no new member was detected after the divergence of B. rapa and B. oleracea from the common ancestor or after B. napus speciation.
Excluding SCL4, LS, NSP1, and NSP2, all other subfamilies enriched their members compared with other cruciferous species, which might have expanded during the Bra-α WGT event, indicating that the number of GRAS genes has increased significantly in each Brassica species (Table S1; Fig. 1). As reported in previous studies, PAT1 and LISCL subfamilies consisted of more GRASs than other subfamilies in Brassica species (Guo et al. 2019). Additionally, the PAT1, SCL3, DLT, SHR, and HAM subfamilies also enlarged their members and had more GRAS genes, while the rest remained comparatively conserved, with few members. Taken together, these results demonstrate that GRASs from the Brassica genus and other cruciferous species, including the model species A. thaliana, share highly close relations in classification, which might facilitate the functional research of GRAS genes in plants.
Expansion resistant patterns of Brassica GRAS genes in angiosperms
To estimate the expansion patterns of GRAS genes in Brassica, GRAS genes from 22 species, including the 3 Brassica species, were further characterized into different subfamilies, and OGs following the same classification method (Fig. 2). A basal angiosperm species, A. trichopoda, the single living representative of the sister lineage to all other extant flowering plants, which originated prior to the split of eudicots and monocots and has not experienced any WGD events, harbors a relatively low number (34) of GRAS genes (Cenci and Rouard 2017). Almost all other species experienced one or more rounds of WGD/WGT after splitting from A. trichopoda. However, the GRAS subfamilies of two different species derived from two close lineages exhibited high differences in gene number because of WGD/WGT events. For instance, the number of GRASs in P. trichocarpa was ~ 2.2-fold of that in R. communis, and the gene number in G. max was ~ 1.79-fold of that in M. truncatula. In addition, when compared with A. trichopoda (34), 19 and 22 more GRAS genes were detected in B. oleracea and B. rapa, which were generated by duplication events. In addition, the number of Brassica GRAS genes in some subfamilies (e.g., PAT1 and LISCL) was dramatically expanded, whereas some subfamilies were expansion resistant (e.g., NSP1) or lost all members of a certain subfamily (e.g., SCLB; Fig. 2).

Distributions of GRAS genes by subfamily in 22 plant species. The left panel: the species phylogenetic tree was inferred from Comparative Genomics, CoGe. Divergence time of these species was estimated by molecular clock dating from TimeTree. Green/red star represents the whole genome duplication (WGD) or triplication (WGT) event. The middle panel: the identified GRAS gene distribution in each subfamily. Numbers in the matrix represent GRAS gene specified by subfamily according each header. The gradient colors from green to red illustrate the abundance of genes. And total GRAS gene numbers in each species were listed on the right panel with blue histogram. MYA: million years ago; Bra-α-WGT: Brassica-α whole genome triplication
Although some cruciferous species (A. thaliana, C. rubella, and T. parvula) experienced one WGT and two WGD events, only 32–34 GRAS genes were identified. When compared with other species, GRAS genes of the Brassica genus were not only absent in 4 subfamilies (RAM1, RAD1, SCLA, and SCLB) and 7 OGs (PAT2, DELLA-2, SCR-3, SHR-2, NSP2-2, NSP2-3, and NSP2-Amb), but also lost many other genes, such as TpDELLA1-5, CrSCL7, and AtSCL32-1–2 during evolution (Fig. 2; Table S1). The scenario of cruciferous species was followed by diploid Brassica species because that they also lacked the same homologues. Compared with other species in Brassicaceae, with the help of various types of duplication events, the GRAS members in B. rapa and B. oleracea were 1.7- and 1.58-fold of that in A. thaliana, respectively. Most AtGRASs retained their corresponding homologues in a single copy, while others were lost, followed by two or three copies in Brassica species. Additionally, when compared with AtGRAS genes, 17 and 19 more GRAS genes were generated by the Bra-α WGT event in B. rapa and B. oleracea, respectively. In addition, 6 and 2 more GRAS genes within B. rapa and B. oleracea were generated by TD events (Table S1), suggesting that TD contributed to the expansion of GRAS family members. Notably, BrDLTb-tda and BrDLTb-tdb were parts of these genes originally derived from TD, but later, each of them was split into two genes. After B. napus speciation, another gene copy of HAMII-3 was produced by a TD event in BnA. Taken together, the Bra-α WGT contributed more to the expansion of Brassica GRAS family members than TD.
Moreover, the GRAS gene locations at each chromosome of the ancestral translocation Proto-Calepineae Karyotype chromosome (tPCK chr1 ~ 7) were also explored. Based on the syntenic relationship datasets of A. thaliana and Brassica species, 30 AtGRAS genes were mined for their corresponding orthologous gene pairs in the three subgenomes of B. rapa or B. oleracea (Table S2). The most retained Brassica GRAS genes were located in the LF subgenome (54.76% in Br and 51.22% in Bo), followed by the MF2 subgenome (26.19% in Br and 29.27% in Bo), and the MF1 subgenome was the least (19.04% in Br and 19.51% in Bo, Table S2), suggesting a relatively similar retention of GRAS genes between the two species. Some subfamilies increased their members during each WGD/WGT event, such as PAT and LISCL, while others (e.g., DLT and NSP1) remained expansion resistant during polyploidization. However, members of some subfamilies (e.g., NSP2) were reduced during the evolutionary process, whereas some others (e.g., SCL3, and DELLA) expanded or shrank their members in different stages (Table S2).
The Bra-α WGT event influenced the purifying selection of BnGRASs
To explore the evolutionary features of GRAS genes among A. thaliana and three other Brassica species, the nucleotide substitutions of orthologous gene pairs from each group were evaluated. In total, 55 and 53 orthologous gene pairs were obtained from the At-Br and At-Bo groups, respectively, and the mean Ka/Ks ratios of orthologous GRAS gene pairs were all less than 1, suggesting that GRAS gene pairs underwent purifying selection during the Bra-α WGT event (Fig. 3; Table S3). Meanwhile, 43 and 48 orthologous gene pairs were obtained from the Br-BnA and Bo-BnC groups, respectively. The mean Ka/Ks ratios of these homologous gene pairs were also less than 1, indicating that purifying selection also existed in BnGRAS genes during the speciation of B. napus (the early period of allopolyploidization). Moreover, the mean Ka/Ks ratios of gene pairs from the At-Br and At-Bo gene pair groups and Br-BnA and Bo-BnC gene pair groups were similar. Nevertheless, the Ka and Ks values of gene pairs from the Br-BnA and Bo-BnC groups were approaching or equal to zero, much less than those from the At-Br and At-Bo groups (Fig. 3; Table S3). These results indicate that the evolution of Brassica GRAS genes were highly conserved, and allopolyploidization had little impact on this process compared with that of the Bra-α WGT event.

Evolutionary selection patterns of homologous GRAS gene pairs in different groups. Non-synonymous nucleotide substitutions (Ka), synonymous nucleotide substitutions (Ks) and Ka/Ks ratios of orthologous GRAS gene pairs from the At-Br, At-Bo, Br-BnA, Bo-BnC, At-BnA, and At-BnC groups were calculated by KaKs_Calculator 2.0. Arabidopsis thaliana (At), Brassica rapa (Br), B. oleracea (Bo), A (BnA) and C (BnC) subgenomes of B. napus. Dots represent the data that were out of range, and standard deviations were also calculated
To further analysis the evolutionary patterns of GRAS genes, the Ka, Ks, and Ka/Ks values of GRAS gene pairs within different subgenomes were also calculated. Most GRAS gene pairs showed purifying selection (the mean Ka/Ks < 1) during species evolution, and the Ka values of GRAS genes all appeared to be 1. However, the mean Ks values within B. rapa and B. oleracea were similar and much higher than those within the BnA and BnC subgenomes (Fig. S1a; Table S4). These results indicated that the GRAS genes exhibited much less selective pressure during the early speciation of B. napus (allopolyploidation), thus further proving the poor influence of allopolyploidation on the evolution or divergence of Brassica GRAS genes (Fig. S1a; Table S4). Similarly, the assessed Ka, Ks, and Ka/Ks values of GRAS gene pairs within PAT, LISCL, HAM, and DELLA subfamilies were almost the same, suggesting purifying selection (Ka/Ks < 1) within the subfamilies of GRAS genes in B. rapa, B. oleracea, and B. napus (Fig. S1b; Table S4). In addition, GRAS gene pairs generated by TD were also subject to purifying selection during evolution (Table S5). Taken together, the evolution of homologous GRAS genes underwent purifying selection in the Brassica genus, and the Bra-α WGT event had a bigger influence than allopolyploidization (and/or TD event) during this process.
Furthermore, the diversity times of different homologous GRAS gene pairs were estimated in Brassica plants. Three peaks roughly emerged with high consistency in all 4 subgenomes, which highly coincided with the corresponding time of the Bra-α WGT event, the Cretaceous–Paleogene extinction event, and the γ-WGT event (Fig. S2). These observations indicate that GRAS genes have experienced consistent purifying selection during the evolution of Brassica plants.
High chromosome location similarity between interspecies homologous GRAS gene pairs in Brassica
Based on the chromosome locations of individual GRAS genes, 56 (100%) BrGRAS genes and 51 (96.23%) of the 53 BoGRAS genes were mapped on 10 (B. rapa, Chr Ar01–10) and 9 (B. oleracea, Chr Co01–09) pseudochromosomes, respectively; and 39 (82.98%) of the 47 GRAS genes in BnA and 40 (81.63%) of the 49 GRAS genes in BnC were anchored on the 10 and 9 pseudochromosomes of BnA (Chr An01–10) and BnC (Chr Cn01–09), respectively (Figs. 4, 5; Table S1). The remaining GRAS genes were mapped onto scaffolds. The distribution of GRAS genes was uneven; some chromosomes (e.g., 20.7% of the BoGRAS genes located on Co07 of B. oleracea) possessed more GRAS genes than others (Table S1), indicating that these GRAS genes were generated by segmental duplication. In addition, some genes (BrLISCL7b-td, BrDLTb-tdb, BrSCR-2-td, and BnAHAMII-3-td) were produced by TD immediately after B. napus speciation instead of others (BrLISCL3b-td, BrDLTb-tda, BoPAT1-1a-td, and BoLISCL3b-td) during the evolution of Brassica species as the coresponding homologous genes could not be found (Fig. 5; Table S1).

Syntenic relations of detected orthologous GRAS gene pairs between A rabidopsis thaliana and Brassica rapa (Green lines), A. thaliana and B. oleracea (blue lines). The detected syntenic orthologous genes in the three sub-genomes, MF1, MF2 and LF of B. rapa from A. thaliana were highlighted in red, yellow and blue, respectively, while the rests were labeled in black. The genes generated by tandem duplication were labeled in green

Syntenic relationships of detected orthologous GRAS gene pairs between Brassica rapa (Br, Ar) and An (BnA) subgenome of B. napus, B. oleracea (Bo, Co), and Cn (BnC) subgenome of B. napus. Green and blue lines indicate the orthologous GRAS genes pairs in the Br-BnA, and Bo-BnC groups, respectively, while grey lines represent GRAS gene pairs located between the unanchored scaffolds and/or pseudo-chromosomes. Red lines indicate that these gene pairs have undergone homoeologous sequence exchanges. Br, BnA, Bo, and BnC subgenomes of Brassica species were colored in green, light green, blue, and light blue, individually
When comparing the chromosome location of BnGRAS genes generated by the Bra-α WGT from Br-BnA with those from Bo-BnC, almost all orthologous BnGRASs and their parental genes were positioned on the same chromosome, indicating that the chromosome location of Brassica GRASs was highly conserved during evolution (Fig. 5; Table S1). However, some BnGRASs still changed their locations; for example, BrSCL3b was located on Ar05, while its orthologous BnASCL3b was detected on An08. Moreover, BrPAT1-3c and BrDELLA1-1 were two GRAS genes on Ar05 and Ar02 of B. rapa, whereas the corresponding orthologous genes, BnAPAT1-3c (BnaCnng71600D(A)), and BnADELLA1-1 (BnaCnng68300D(A)) were detected on the BnC subgenome. Interestingly, this phenomenon was not the same in the C-subgenome (Fig. 5; Table S1), indicating that the GRAS genes from the A-subgenome might have undergone more homoeologous segmental exchanges than those from the C-subgenome. Since the chromosomal location of homologous GRAS gene pairs shared a high similarity between species, the unanchored GRASs could predict the chromosome to which they belong (e.g., BnaCnng71600D (BnA)). However, some GRAS gene pairs did not preserve high location similarity; thus, the accurate locations of those genes could not be fully predicted (BolDLTb (Bo00795s120), BnAPAT1-3c (BnaCnng71600D), and BnADLTb (BnaCnng67210D); Table S1).
Highly structural similarity of BnGRASs
To investigate the sequence diversity among the different Brassica GRAS genes, we compared their gene structures (exon/intron organization). As shown in Fig. 6, 90 (93.75%) BnGRASs, 53 (94.6%) BrGRASs, and 53 (100%) BoGRASs were mono-exon or harbored only one intron, and the gene structures were usually conserved within subfamilies and among orthologues across the three species. The structures of GRASs generated by WGT and TD events had many types of variation; some were highly conserved (e.g., BraPAT1-1a and BnAHAMII-3-td), some had more introns (e.g., BolDELLA1-4b), some exhibited shrunken sequences (e.g., BraPAT3b and BraLISCL7a-td), and some had been divided into two new genes (e.g., BraDLTb-tda and BraDLTb-tdb). Compared with the parental lines, some BnGRAS genes had more introns or deleted partial exons (Fig. 6).

Sequence feature analysis of GRAS members in Brassica species. The exon–intron structures of GRAS genes obtained from genome annotation files and conserved motif patterns of GRAS proteins mined by MEME software were visualized in TBtools. Different colored blocks indicate diverse motifs on exons, while black lines represent the intron regions. Bar = 200 bp
In addition to gene structures, the motif patterns of different GRAS proteins were also explored by MEME (Fig. 6; Tables S6, S7). Overall, 27 distinct motifs were detected in Brassica GRAS proteins: LHRI domain (motifs 7 and 11), VHIID domain (motifs 2 and 3), LHRII domain (motifs 6 and 8), PFYRE domain (motifs 5 and 9), SAW domain (motifs 1, 4 and 12), and motifs 10, 16 and 20 were highly conserved in the C-termini (GRAS domain) of most GRAS proteins. Most GRAS proteins in the same subfamily had conserved motif structures and orders, implying similar functions (Figs. 6, S3; Tables S6, S7). However, not all subfamily members were highly conserved in the GRAS domain; for instance, NSP1 lacked LHRII and PFYRE domains (Fig. 6; Table S6). In addition, some domains even lacked one or two motifs in some subfamilies; for example, motif 1 was not detected in the SAW domain in SCL4/7, SCR, HAM, NSP1, and NSP2 subfamilies. Additionally, motif 16 was situated between LHRI and PFYRE, motif 10 was nested within VHIID and LHRII, and motifs 9, 24 or 25 could be detected between LHRII and PFYRE. Interestingly, motifs 23 and 25 were detected between PFYRE and SAW only in the HAM subfamily. Intriguingly, some motifs appeared to be subfamily-specific; for instance, motif 27 was PAT-specific, motifs 17 and 19 were DELLA-specific, motifs 18 and 23 were HAM-specific, and motifs 13, 15, and 21 were LISCL-specific. Furthermore, we clearly found that the motifs were highly similar between orthologous GRAS gene pairs, especially between homologous proteins from the same subgenome. However, some protein structures contained certain differences in the same orthologous gene pairs; for example, BrSCR-2, BoSCR-2, and BnASCR-2 shared highly similar and conserved C-termini, while BnCSCR-2 lacked the LHRI and VHIID domains (Figs. 6, S3; Table S7). In conclusion, the structural similarity within specific GRAS subfamilies was consistent with the evidence provided by phylogenetic analysis, which documented the conservative functions of GRAS genes in Brassica species.
Diverse cis-acting elements identified on BnGRAS promoters
As cis-acting elements played an essential role in the transcriptional regulation of gene expression (Wang et al. 2020a), promoter sequences (the 2000 bp upstream of the ATG start codon) of the identified BnGRASs were extracted from the B. napus genome, and cis-acting element analysis was carried out using PlantCARE. A total of 24 types of cis-acting elements were observed in the promoter regions of the BnGRAS genes (Fig. S4). Notably, the cis-acting elements related to defense and stress response, hormone response, meristem maintenance, and other regulation mechanisms related to various biological processes were broadly distributed (Fig. S4), suggesting their diverse functions in plant growth and development.
Expression profiling of BnGRAS genes under stress
To explore the functions of Brassica GRAS genes, some published public data downloaded from NCBI were used to further study the expression profiles of Brassica GRAS genes in the leaves and roots (Fig. S5; Table S8). The expression levels of the identified GRAS genes were detected in at least one tissue, suggesting that Brassica GRASs might participate in plant leaf and/or root development. Moreover, similar expression patterns were observed between B. napus and its parental species; most expression patterns of orthologous GRAS gene pairs shared a high similarity not only between B. rapa and B. oleracea but also between the subgenomes of B. napus (Fig. S5a; Table S8-1). However, compared to their parental homologues, some GRASs in B. napus changed their expression patterns, which were derived from the gene redundancy created by allopolyploidization. For instance, there was almost no detected expression of BrPAT1-3b and BoPAT1-3b, while BnAPAT1-3b and BnCPAT1-3b showed relatively high expression levels (Fig. S5a). This might be because the functional loss of BnAPAT1-3a and BnCPAT1-3a was replaced by BnAPAT1-3b and BnCPAT1-3b. In addition, some genes displayed homologous expression dominance, e.g., BnASCL3a and BnASCL3b exhibited expression dominance compared to their homologous genes in BnC, and BnCPAT4b showed expression dominance. Additionally, PAT1-2 was only expressed in the Bo and BnC subgenomes, suggesting that it might be a specific C-subgenome-expressed GRAS memeber. Moreover, some GRAS genes generated by TD (BrLISCL6-td, BrLISCL7a-td, BrDLTb-tdb, and BrSCR-2-td) and interspersed repeats (BrSCL3d) were expressed at a very low level (Fig. S5a).
Plants are subjected to various external environmental stress (Wang et al. 2019), including biotic (such as weeds and diseases) and abiotic stress (such as drought and salinity). Therefore, the expression profiles of BnGRASs in response to diverse biotic and abiotic stressors were also investigated (Fig. S5b, S5c). Phoma stem canker seriously threatens the yield of B. napus and is mainly caused by two fungal viruses: Leptosphaeria biglobosa and Leptosphaeria maculans. As shown in Fig. S5b, BnPAT3a, BnPAT4b, BnLISCL3b, and BnSCL3a showed higher expression levels after infection with L. biglobosa (weakly virulent strain) and L. maculans (strongly virulent strain). Sclerotinia sclerotiorum (Ss) is also a pathogen that harms the growth of B. napus. Pseudomonas chlororaphis strain PA23 can produce compounds to inhibit the growth of Ss. Compared with the mock treatment and the treatment with only PA23 and both PA23 and Ss, the expression levels of BnPAT1-4, BnPAT3, BnLISCL3b, BnLISCL6, and BnDELLA1-3 were higher when exposed to a single Ss treatment (Fig. S5b). For abiotic response, BnPAT1-1c, BnPAT1-3b, BnPAT3a, BnLISCL1, BnLISCL2, BnLISCL6, and BnSCL3 genes had higher expression levels in the roots of 7-day-old B. napus under drought stress (200 g/L PEG6000), while BnPAT1-1a, BnPAT1-3b, BnPAT3a, BnLISCL6, BnSCL3b, and BnCSHR-1a were highly expressed in the roots of 3-week-old B. napus than others under salt stress (200 mM NaCl; Fig. S5c). These results indicate that these BnGRAS genes play an important role in the response to biotic and abiotic stress in roots.
To further confirm this, we investigated the expression patterns of some BnGRASs (BnAPAT1-1a, BnCPAT1-1a, BnAPAT3b, BnCPAT3b, BnASCL3b, BnCSCL3b, BnALISCL6, BnCLISCL6, BnADELLA1-3, BnCDELLA1-3, BnAHAMII-1b, and BnCHAMII-1b) in the roots of 14-day-old B. napus under salt and drought stress treatment (Table S9). The expression of these genes was obviously induced under the abiotic stress treatment and was similar to the transcriptome data, demonstrating that these BnGRAS genes played a role in root development in response to salt and drought stress (Fig. 7).

Real-time quantitative PCR validation of selected BnGRAS genes under salt and drought stress in the 14-day-old B. napus seedling roots. The x-axis represents the different hours (0, 2, 4, 6, and 8 h) upon drought (17% PEG6000) and salt (200 mM NaCl) treatment. BnCACTIN-7 was used as the reference gene. And the values in the y-axis indicate the mean ± SD of three independent biological replicates
Discussion
Structural divergence of BnGRASs
The GRAS transcription factor family constitutes a large number of genes in plants and has been identified throughout the genomes of many species, such as A. thaliana, O. sativa (Tian et al. 2004), B. napus (Guo et al. 2019), and B. rapa (Song et al. 2014). However, the evolutionary comparison of BnGRASs with that of their progenitor species has not yet been studied. Since the released genome sequences and annotations were not flawless, the validity for GRAS genes was manually revised by comprehensive comparison with genomic sequences and the EST and SRA datasets in this study. In previous studies, GRAS genes were named by numbers on the chromosomes, which might cause some misconceptions in their further study and might be inconvenient for later researchers (Song et al. 2014; Guo et al. 2019). Hence, GRAS genes of B. rapa, B. oleracea, and B. napus were not only named or renamed in the same criterion but also subdivided into different subfamilies or OGs according to the orthologous relationships in other Brassicaceae species in the present work (Tian et al. 2004; Table S1). Most GRAS orthologues were detected but not in every Brassicaceae species; for example, HAMI-2 existed in T. parvula but could not be detected in A. thaliana, whereas it was detected in the A subgenome of Brassica species, suggesting that it might be lost during the evolution of A. thaliana.
Gene structure analysis confirmed that most GRAS genes lack introns or contain only one exon in Brassica species (Fig. 6), which is the same as that in most other plant GRAS genes (Song et al. 2014; Guo et al. 2019; Wang et al. 2020a). However, some BnGRASs possessed more introns after allopolyploidization. The divergence of exon–intron structures is mainly shaped by 3 mechanisms: exon/intron gain/loss, exonization/pseudoexonization and insertion/deletion (Xu et al. 2012). Thus, the structural divergence of Brassica GRAS genes might also have been affected by these mechanisms during species evolution. In addition, the gene structures of most Brassica GRAS family members were highly conserved, and TD events contributed to the diversity of exon–intron structures. These cases might be attributed to: (a) some exon sequences were deleted during evolution, such as BnAPAT1-2; (b) certain introns were inserted into the sequence with no other changes, such as BnCSCL32-1–2; and (c) the intron of some genes in diploid Brassica species might emerge after the speciation of B. napus, for instance, one intron was observed in BrDLTa that was lost in BnADLTa (Fig. 6).
Stress response of Brassica GRASs
Biotic and abiotic stress influence the growth and production of plants (Wang et al. 2019). Abiotic stress reduces crop yields by more than 50% compared to a less than 10% reduction caused by biotic stress, including in Brassica plants (Kreps et al. 2002). Many GRAS genes have been revealed to play an important role in improving the biotic and abiotic resistance of Brassica species. VaPAT1 in transgenic Arabidopsis showed enhanced salt and drought stress tolerance (Yuan et al. 2016), and the orthologue of LISCL6 in rice was involved in regulating the drought stress response (Xu et al. 2015). LISCL6 and most of the other genes in the PAT subfamily had higher expression under drought stress, as well as salt stress, in B. napus.
Because gene expression patterns could provide critical clues for their possible functional assessment (Wang et al. 2020b), the transcript levels of Brassica GRAS genes under normal and various types of stress were investigated in this study. Although almost all Brassica GRASs were expressed in at least one tissue, no expression of the Brassica GRAS genes generated by TD and interspersed repeat events was detected in any tissue, thus further confirming their limited effects on the evolution of GRAS genes (Fig. S5a; Table S8). The expression patterns of most GRAS genes were similar in the Br-Bo and BnA-BnC groups. However, the expression profiles between B. napus and its parental species were slightly different; some homologous genes showed decreased expression levels in B. napus to that of its parental species, suggesting the existence of gene redundancy. Expression dominance of homoeologous GRASs did not obviously occur in most cases. Some (PAT, DELLA, and HAM) subfamily genes showed dominant expression and response in B. napus or in the parental lines, regardless of normal or stress treatment (Figs. S5, S6), indicating that they might have undergone certain potential selection biases. Taken together, these findings demonstrate that Brassica GRAS genes play important roles in plant development and stress response, similar to other GRAS members, whereas the subfunctionalization and neofunctionalization of GRAS genes might still proceed in young B. napus; thus, further functional analysis is still needed (Ma et al. 2010; Wang et al. 2020b).
Proposed evolutionary origin of BnGRASs
Previous studies have shown that a wide variety of GRAS numbers exist among different species, which might be related to polyploidy. For instance, 34 GRASs were identified in A. trichopoda, 106 in P. trichocarpa (Liu and Widmer 2014), which is ~ 2.21 times higher than that in R. communis (Xu et al. 2016), and 106 in G. max, which is ~ 1.8 times higher than that in M. trucatula (Wu et al. 2014). After being classified by the latest method, many subfamilies (e.g., PAT1 and LISCL) were found to contain large numbers of GRAS homologues in Brasscia species, which was similar to most other angiosperm species (e.g., Populus simonii, G. max, and Malus pumila). This is probably because many GRAS members of PAT, LISCL, and other subfamilies extensively participate in diverse biotic/abiotic stress response (Liu et al. 2014; Wang et al. 2020a, b). However, compared with other species (Tian et al. 2004; Cenci and Rouard 2017), the total number of Brassica GRASs was not very large, as they lacked all members in SCLA, SCLB, RAD1 and RAM1 subfamilies as other species in Brassicaceae (Fig. 2).
Three rounds (i.e., γ, β, and α) of WGD/WGT and the specific Bra-α WGT event have been suggested through evolutionary process analysis in Brassica species (Bowers et al. 2003). However, most AtGRASs retained their corresponding homologues in a single copy of Brassica plants (Fig. 1; Tables S1, S2). Our evolutionary analysis showed that all GRAS genes in Brassica were subject to purifying seletion, together with the similar and conservative structural features of Brassica GRAS genes, further demonstrating the important functional conservation of GRAS family members and explaining the single copy of AtGRASs (Figs. 1, 2, 3, 4, 5, 6; Tables S1–S7). During the process of evolution, the Bra-α WGT event contributed more to the expansion of GRAS genes in the Brassica genus than TD events (Figs. 2, 3, S1; Tables S3–S5).
After the collinearity and phylogenetic analysis of GRAS genes in multiple species, we found that GRASs in PAT, LISCL, DELLA, and HAM subfamilies were highly expanded, while the members of some subfamilies (SCLA, SCLB, RAD1, and RAM1) were reduced or expansion resistant during evolution (Figs. 1, 2). These observations showed that genome duplication, genome triplication, and TD events played different roles in the expansion of the GRAS gene family in different species, similar to other angiosperms (Cenci and Rouard 2017; Wang et al. 2020a, b). Finally, a possible evolutionary origin of BnGRAS genes in the Brassica genus was proposed (Fig. 8); the GRAS genes from B. rapa and B. oleracea were combined with the help of allopolyploidization to give rise to the BnGRAS genes, and almost all GRAS genes derived from diploid species were retained in the allopolyploid B. napus. In addition, the evolution of the Brassica GRAS genes was highly conserved, and allopolyploidization had little impact on this compared with that of the Bra-α WGT event. However, some GRAS genes of the parental lines were lost after the speciation of B. napus (e.g., BrPAT1-3a, BoPAT1-3a, BoNSP1, BrSHR-1b, and BoSHR-1b), and there was no obvious subgenome-dominant gene. Taken together, the Bra-α WGT event contributed more to the expansion of GRASs than TD and exerted different functions on the evolution of diverse subfamilies in Brassica.

The proposed evolutionary origin of BnGRASs in the Brassica genus. The reference genes were syntenic and orthologous genes from Amborella trichopoda, Vitis vinifera, Theobroma cacao, Carica papaya, Capsella rubella, Thellungiella parvula and Arabidopsis thaliana. The purple, green, red, brown, and blue colored arrows represent the impact of gene expansion from γ-WGT, Cretaceous–Paleogene extinction event (α-WGD and β-WGD event), the Bra-α-WGT, allopolyploidazayion, and tandem duplication event, respectively. Blue colored genes were generated by tandem duplication events, while grey colored genes were lost in the speciation process of Brassica napus. Br, Bo, BnA, BnC each represents the A genome of B. rapa, C genome of B. oleracea and An and Cn subgenomes of B. napus. NF: not found; WGD: whole genome duplication; WGT: whole genome triplication; Bra-α WGT: α-WGT specifically occurred in the Brassica genus
Conclusions
In the present study, 56, 53, and 96 GRAS genes were identified genome-wide in B. rapa, B. oleracea, and B. napus and classified into 13 subfamilies and 17 OGs. The chromosomal locations, gene structures, motif patterns, and gene expression profiles of Brassica GRAS members were highly conserved during speciation. Additionally, polyploidization events (WGD and WGT) contributed more to the expansion and diversity of Brassica GRAS family members than other events, whereas gene loss was also detected during evolution. Furthermore, the early period of allopolyploidization and TD also played a role in the evolution of the BnGRAS genes. Moreover, the evolutionary origin of BnGRAS genes in the Brassica genus, which was strongly affected by duplication events, was first proposed here. In addition, BnGRAS genes might play an important role in stress responses in roots. In summary, our results not only broaden the understanding of the evolutionary mechanism of the BnGRAS family in the Brassica genus but also accelerate the functional research of Brassica GRAS genes.
Data availability
All data generated or analyzed during this study are included as Supplementary Data file.
References
Allender CJ, King GJ (2010) Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol 10:54. https://doi.org/10.1186/1471-2229-10-54
Bai MY, Shang JX, Oh E et al (2012) Brassinosteroid, gibberellin and phytochrome impinge on a common transcription module in Arabidopsis. Nat Cell Biol 14:810–817. https://doi.org/10.1038/ncb2546
Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34:W369-373. https://doi.org/10.1093/nar/gkl198
Bolle C (2004) The role of GRAS proteins in plant signal transduction and development. Planta 218:683–692. https://doi.org/10.1007/s00425-004-1203-z
Bowers JL, Chapman BA, Rong J, Paterson AH (2003) Unraveling angiosperms genome evolution by phylogenitc analysis of chromosomal duplications events. Nature 422:433–438. https://doi.org/10.1038/nature01509.1
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. https://doi.org/10.1093/oxfordjournals.molbev.a026334
Cenci A, Rouard M (2017) Evolutionary analyses of GRAS transcription factors in Angiosperms. Front Plant Sci 8:273. https://doi.org/10.3389/fpls.2017.00273
Chalhoub B, Denoeud F, Liu S et al (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345(6199):950–953
Cheng F, Wu J, Fang L, Wang X (2012) Syntenic gene analysis between Brassica rapa and other Brassicaceae species. Front Plant Sci 3:198. https://doi.org/10.3389/fpls.2012.00198
Cheng F, Wu J, Wang X (2014) Genome triplication drove the diversification of Brassica plants. Hortic Res 1:14024. https://doi.org/10.1038/hortres.2014.24
Cui H, Kong D, Liu X, Hao Y (2014) SCARECROW, SCR-LIKE 23 and SHORT-ROOT control bundle sheath cell fate and function in Arabidopsis thaliana. Plant J 78:319–327. https://doi.org/10.1111/tpj.12470
Fan S, Zhang D, Gao C et al (2017) Identification, classification, and expression analysis of GRAS gene family in Malus domestica. Front Plant Sci 8:253. https://doi.org/10.3389/fphys.2017.00253
Fode B, Siemsen T, Thurow C et al (2008) The Arabidopsis GRAS protein SCL14 interacts with class II TGA transcription factors and is essential for the activation of stress-inducible promoters. Plant Cell 20:3122–3135. https://doi.org/10.1105/tpc.108.058974
Grimplet J, Agudelo-Romero P, Teixeira RT et al (2016) Structural and functional analysis of the GRAS gene family in Grapevine indicates a role of GRAS proteins in the control of development and stress responses. Front Plant Sci 7:353. https://doi.org/10.3389/fpls.2016.00353
Guindon S, Delsuc F, Dufayard J, Gascuel O (2010) Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137. https://doi.org/10.1007/978-1-59745-251-9_6
Guo P, Wen J, Yang J et al (2019) Genome-wide survey and expression analyses of the GRAS gene family in Brassica napus reveals their roles in root development and stress response. Planta 250:1051–1072. https://doi.org/10.1007/s00425-019-03199-y
Guo Y, Wu H, Li X et al (2017) Identification and expression of GRAS family genes in maize (Zea mays L.). PLoS ONE 12:e0185418. https://doi.org/10.1371/journal.pone.0185418
Ito T, Fukazawa J (2021) SCARECROW-LIKE3 regulates the transcription of gibberellin-related genes by acting as a transcriptional corepressor of GAI-ASSOCIATED FACTOR1. Plant Mol Biol 105:463–482. https://doi.org/10.1007/s11103-020-01101-z
Jin J, Tian F, Yang D et al (2017) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45:D1040–D1045. https://doi.org/10.1093/nar/gkw982
Koch M, Haubold B, Mitchell-Olds T (2000) Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol 17:1483–1498. https://doi.org/10.1093/oxfordjournals.molbev.a026248
Kreps JA, Wu Y, Chang H et al (2002) Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol 130:2129–2141. https://doi.org/10.1104/pp.008532
Krzywinski M et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. https://doi.org/10.1101/gr.092759.109.19
Lee M-HMM, Kim B, Song S-K et al (2008) Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol Biol 67:659–670. https://doi.org/10.1007/s11103-008-9345-1
Lescot M, Déhais P, Thijs G et al (2002) PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30:325–327. https://doi.org/10.1093/nar/30.1.325
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. https://doi.org/10.1093/nar/gkw290
Li M, Sun B, Xie F et al (2019) Identification of the GRAS gene family in the Brassica juncea genome provides insight into its role in stem swelling in stem mustard. PeerJ 7:e6682. https://doi.org/10.7717/peerj.6682
Li W, Cowley A, Uludag M et al (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43:W580-584. https://doi.org/10.1093/nar/gkv279
Liu S, Liu Y, Yang X et al (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun 5:3930. https://doi.org/10.1038/ncomms4930
Liu X, Widmer A (2014) Genome-wide comparative analysis of the GRAS gene family in Populus, Arabidopsis and rice. Plant Mol Biol Rep 32:1129–1145. https://doi.org/10.1007/s11105-014-0721-5
Lu J, Wang T, Xu Z et al (2015) Genome-wide analysis of the GRAS gene family in Prunus mume. Mol Genet Genomics 290:303–317. https://doi.org/10.1007/s00438-014-0918-1
Lysak MA, Koch MA, Pecinka A, Schubert I (2005) Chromosome triplication found across the tribe Brassiceae. Genome Res 15:516–525. https://doi.org/10.1101/gr.3531105
Ma HS, Liang D, Shuai P, Xia XL, Yin WL (2010) The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J Exp Bot 61(14):4011–4019. https://doi.org/10.1093/jxb/erq217
Ma Z, Hu X, Cai W et al (2014) Arabidopsis miR171-targeted Scarecrow-Like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. Plos Genet 10:e1004519. https://doi.org/10.1371/journal.pgen.1004519
Morohashi K, Minami M, Takase H et al (2003) Isolation and characterization of a novel GRAS gene that regulates meiosis-associated gene expression. J Biol Chem 278:20865–20873. https://doi.org/10.1074/jbc.M301712200
Niu Y, Zhao T, Xu X, Li J (2017) Genome-wide identification and characterization of GRAS transcription factors in tomato (Solanum lycopersicum). PeerJ 5:e3955. https://doi.org/10.7717/peerj.3955
Peng J, Carol P, Richards DE et al (1997) The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Gene Dev 11:3194–3205. https://doi.org/10.1101/gad.11.23.3194
Plant M, Chen C, Chen H et al (2020) TBtools : an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13:1194–1202. https://doi.org/10.1016/j.molp.2020.06.009
Pysh LD, Wysocka-diller JW, Camilleri C et al (1999) The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J 18:111–119. https://doi.org/10.1046/j.1365-313x.1999.00431.x
Richards DE, Peng J, Harberd NP (2000) Plant GRAS and metazoan STATs: one family? BioEssays 22:573–577. https://doi.org/10.1002/(SICI)1521-1878(200006)22:6%3c573::AID-BIES10%3e3.0.CO;2-H
Song X, Liu T, Duan W et al (2014) Genome-wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 103:135–146. https://doi.org/10.1016/j.ygeno.2013.12.004
Stuurman J, Jäggi F, Kuhlemeier C (2002) Shoot meristem maintenance is controlled by a GRAS-gene mediated signal from differentiating cells. Gene Dev 16:2213–2218. https://doi.org/10.1101/gad.230702
Sun TP (2011) The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr Biol 21:R338–R345. https://doi.org/10.1016/j.cub.2011.02.036
Sun X, Xie Z, Zhang C et al (2016) A characterization of grapevine of GRAS domain transcription factor gene family. Funct Integr Genomics 16:347–363. https://doi.org/10.1007/s10142-016-0479-y
Tian C, Wan P, Sun S et al (2004) Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol Biol 54:519–532. https://doi.org/10.1023/B:PLAN.0000038256.89809.57
Torres-Galea P, Hirtreiter B, Bolle C (2013) Two GRAS proteins, SCARECROW-LIKE21 and PHYTOCHROME A SIGNAL TRANSDUCTION1, function cooperatively in phytochrome A signal transduction. Plant Physiol 161:291–304. https://doi.org/10.1104/pp.112.206607
Torres-galea P, Huang L, Chua N (2006) The GRAS protein SCL13 is a positive regulator of phytochrome-dependent red light signaling, but can also modulate phytochrome A responses. Mol Gen Genomics 276:13–30. https://doi.org/10.1007/s00438-006-0123-y
Trapnell C, Roberts A, Goff L et al (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Wang J, Jiao J, Zhou M et al (2019) Physiological and transcriptional responses of industrial rapeseed (Brassica napus) seedlings to drought and salinity stress. Int J Mol Sci 20:5604. https://doi.org/10.3390/ijms20225604
Wang L, Ding X, Gao Y, Yang S (2020a) Genome-wide identification and characterization of GRAS genes in soybean (Glycine max). BMC Plant Biol 20:415. https://doi.org/10.1186/s12870-020-02636-5
Wang T, Su H, Zhou Y (2020b) Genome-wide analysis of the GRAS gene family and functional identification of GmGRAS37 in drought and salt tolerance. Front Plant Sci 11:604690. https://doi.org/10.3389/fpls.2020.604690
Wang Y, Shi S, Zhou Y et al (2016) Genome-wide identification and characterization of GRAS transcription factors in tomato (Solanum lycopersicum). PeerJ 4:e2388. https://doi.org/10.7717/peerj.2388
Wang Y, Tang H, Debarry JD et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:1–14. https://doi.org/10.1093/nar/gkr1293
Wild M, Daviere J-M, Cheminant S et al (2012) The Arabidopsis DELLA RGA-LIKE3 is a direct target of MYC2 and modulates jasmonate signaling responses. Plant Cell 24:3307–3319. https://doi.org/10.1105/tpc.112.101428
Wu N, Zhu Y, Song W et al (2014) Unusual tandem expansion and positive selection in subgroups of the plant GRAS transcription factor superfamily. BMC Plant Biol 14:373. https://doi.org/10.1186/s12870-014-0373-5
Xu G, Guo C, Shan H, Kong H (2012) Divergence of duplicate genes in exon-intron structure. P Natl Acad Sci USA 109:1187–1192. https://doi.org/10.1073/pnas.1109047109
Xu K, Chen S, Li T et al (2015) OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol 15:1–13. https://doi.org/10.1186/s12870-015-0532-3
Xu W, Chen Z, Ahmed N et al (2016) Genome-wide identification, evolutionary analysis, and stress responses of the GRAS gene family in Castor Beans. Int J Mol Sci 17:1004. https://doi.org/10.3390/ijms17071004
Yang J, Liu D, Wang X et al (2016) The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat Genet 48:1225–1232. https://doi.org/10.1038/ng.3657
Yuan Y, Fang L, Karungo SK et al (2016) Overexpression of VaPAT1, a GRAS atranscription factor from Vitis amurensis, confers abiotic stress tolerance in Arabidopsis. Plant Cell Rep 35:655–666. https://doi.org/10.1007/s00299-015-1910-x
Zhang B, Liu J, Yang ZE et al (2018) Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genomics 19:348. https://doi.org/10.1186/s12864-018-4722-x
Zhang Z, Li J, Zhao XQ et al (2006) KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinform 4:259–263. https://doi.org/10.1016/S1672-0229(07)60007-2
Funding
This work was supported by the National Natural Science Foundation of China (U1904106), the Provincial Natural Science Foundation of Henan (22100002), the Fostering Project for Basic Research of Zhengzhou University (JC21310015), the Postdoctoral Research Grant in Henan Province (202003003) and Youth Innovation Project of Key Discipline of Zhengzhou University (XKZDQN202002).
Author information
Authors and Affiliations
Contributions
BT and GS conceived and designed the project. ZX, ZT and FW wrote the manuscript. ZX, ZT and FW carried out the bioinformatics analysis. ZT and FW conducted the experiments. XW and GC supervised the project. WC and GC performed the date analysis. ZT, ZX, FW and GS revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xie, Z., Tian, Z., Wei, F. et al. Genome-wide bioinformatics analysis reveals the evolutionary origin of BnGRAS genes in the Brassica genus. Genet Resour Crop Evol 70, 587–606 (2023). https://doi.org/10.1007/s10722-022-01448-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-022-01448-w