
Abstract
Diversity and structure of populations may differ substantially between morphological traits and molecular markers. Explanations of such discrepancies are crucial for further progress in breeding as well as for the maintenance of genetic resources. Our objective was to compare indices of among-cultivars differentiation for morphological traits (QST) and molecular markers (FST) in alfalfa (Medicago sativa), a legume forage species. Ten cultivars representing the Northern and Southern types commonly grown in Europe were investigated. For each cultivar, 40 genotypes were analysed with 16 SSR markers and four morphological traits measured in two locations and several cuts. QST values were in general high (0.02–0.39) compared to the differentiation observed with molecular markers (FST = 0.01), especially for growth habit, indicating that morphological traits were more efficient to structure the diversity than molecular markers. For morphological traits, a clear separation of Northern and Southern cultivars was observed, whereas for molecular markers, no clear structure was detected. Nevertheless, the grazing type cultivar Luzelle was separated from the rest of the cultivars for both morphological traits and molecular markers. Although pairwise differences between cultivars were significant for both morphological traits and molecular markers, the main part of the variation was found within cultivars. This large within-cultivar variation may be explained, besides the outcrossing reproductive mode and autotetraploid genetics, by the recent history of M. sativa domestication in Europe and the frequent seed exchanges. Selection for morphological traits (QST > FST) was achieved without modification of within-cultivar neutral diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Information about germplasm diversity and genetic relationships among accessions or cultivars is of fundamental importance for breeding and the management of germplasm (Becker 1993). This knowledge is also needed to implement genetic studies aiming at deciphering the genetic determinism of breeding traits. Besides the among-population diversity, the within-population diversity is an additional source of variation that may be large in outcrossing species.
Levels of genetic variation in cultivated plant species depend on the same interplay between evolutionary forces as natural populations. This includes mutation, genetic drift characterized by random changes in allele frequencies among generations due to the finite size of populations, gene exchanges or gene flow among populations and selection (both natural and artificial selection). The theory of population structure that has been developed to analyse the evolutionary mechanisms at work in natural populations can be used to study cultivated populations and to identify the main forces at work in breeding programs and human practices. Heterogeneous distribution of genetic variation within and among populations is provided by mutation, genetic drift, and spatially variable natural selection, while gene flow and homogeneous directional selection tend to produce genetic homogeneity among-populations (Endler 1977; Slatkin 1987). Molecular markers, such as SSR loci are usually considered to be selectively neutral (Kimura 1983) so that the genetic level and population genetic structure revealed with molecular markers should mostly reflect the effect of demographic factors such as migration and genetic drift. Genetic variation and between populations differentiation observed for a quantitative trait is expected to reflect both demographic factors and selective constraints. Comparing the patterns of population differentiation for molecular markers to the one observed on morphological traits thus provides a way to identify selective pressures acting on morphological traits.
The degree of population genetic differentiation can be measured by FST, which is a standardized measure of allele frequency variation for a genetic locus. For neutral loci that are not influenced by natural selection, the degree of differentiation among populations depends largely on their effective size and the amount of migration between them: small, isolated populations are expected be more differentiated from each other than large populations that are connected by gene flow (Wright 1951). QST is a quantitative genetics analogue of FST that measures the amount of genetic variance among populations relative to the total genetic variance in the trait. The value of QST for a neutral quantitative trait that has an additive genetic basis is expected to be equal to the FST for a neutral genetic locus. FST measured from neutral molecular markers can be used as a null expectation for the degree of population divergence due to drift and migration. In cases in which QST ≈ FST, the inference is that trait divergence among subpopulations could have been achieved by genetic drift alone. If QST > FST, trait divergence exceeds neutral expectation, and is likely to have been caused by directional selection. If QST < FST, trait divergence among populations is less than expected by genetic drift alone; this pattern is suggestive of uniform selection or stabilizing selection across the populations. QST–FST comparisons have been used in an increasing number of studies to infer the action of natural selection on complex phenotypic traits, as well as to quantify the degree of spatial genetic structuring in quantitative traits among populations (Spitze 1993).
Alfalfa (Medicago sativa) is an important forage crop in temperate regions and is widely used for animal feeding. As a legume crop able to fix atmospheric nitrogen, it has a renewed interest to decrease the energy costs related to nitrogen fertilization and pollutions caused by the synthesis of chemical nitrogen fertilizer and by nitrogen escapes in the soils. M. sativa subsp. sativa was domesticated presumably in the near East (Iran, the Mesopotamia plain) and/or in Central Asia approximately in 5000 B.C. (Muller et al. 2003; Small 2011). Its cultivation was then twice introduced into the Mediterranean region, first with the Roman Empire via Turkey, Greece to Italy and Spain and second with the Moors via Maghreb into Spain. The crop might have been introduced from Italy and Spain into France shortly after the two introductions in the Mediterranean region but it was mainly introduced from Spain to France in the sixteenth century (Muller et al. 2003). The introgression of M. sativa subsp. falcata, which natural populations originated from Siberia, resulted in a crop more resistant to cold and with a higher dormancy, which allowed the expansion to northern Europe. In France, M. sativa was cultivated in every region and numerous landraces were used. Modern varieties are mainly based on two landraces: Flamande (Flemish from the North of France) and Provence (a Mediterranean type from the South of France). Whereas Southern type is mainly based on M. sativa subsp. sativa, Northern type was introgressed with M. sativa subsp. falcata (Julier 1996). Modern varieties are synthetic populations, usually obtained through three or four generations of panmictic reproduction of a set of various numbers of parents (clones, half-sib or full-sib families). Breeding programs mainly focus on pest and disease resistance, lodging resistance and forage yield combined to forage quality (Lonnet 1996).
Numerous studies have described the diversity and population structure in M. sativa (Annicchiarico et al. 2016; Crochemore et al. 1998; Li et al. 2014a; Mengoni et al. 2001; Musial et al. 2002; Qiang et al. 2015), as well as gene flow among natural and cultivated populations (Jenczewski et al. 1999b; Prosperi et al. 2006) or relationship between diversity and yield or combining ability (Kidwell et al. 1994b; Maureira et al. 2004; Riday et al. 2003). In most cases, morphological studies were conducted at the population level. The assessment of genetic differentiation of populations using both neutral marker loci (FST) and quantitative traits (QST) scored on the same genotypes was never performed in M. sativa.
The objectives of this study were to compare molecular markers (SSR) and morphological traits to evaluate population diversity and structure. FST and QST were compared in order to investigate the degree of selection for the morphological traits and thus to explain the discrepancies and similarities in population structure for these two marker types.
Materials and methods
Plant material
Ten M. sativa cultivars or ecotypes divided into two groups were investigated with morphological traits and molecular markers: the Southern type with poor winter hardiness and low dormancy adapted to the Mediterranean region and the Northern type with a higher winter hardiness and dormancy adapted to northern France and Europe. The Northern group was represented by the landrace Flamande and six modern varieties (Luzelle, Mercedes, Alpha, Symphonie, Cannelle and Harpe) and the Southern group was represented by the landrace Provence and two modern varieties (Zenith and Barmed), as described in Online Resource 1.
Experimental conditions and morphological traits
In March 2003, 40 germinated seeds of each of the ten cultivars were established in pots in the greenhouse at INRA, Lusignan (France) under natural light conditions. The 400 pots were randomized and first measurements were performed in summer 2003 in the greenhouse. In winter 2003/04, six clonal replicates were produced for each genotype. Three clonal replicates of the 400 genotypes were planted in the field in three randomized blocks with a space of 0.7 m among the plants in two environments in France: INRA, Lusignan (46°26′N, 0°7′W) and Barenbrug Tourneur Recherches, Connantre (48°43′N, 3°55′E). The soil is a deep clay silt soil in Lusignan and a rendzina soil in Connantre.
Four traits were measured in 2003 in the greenhouse and in the first two growth cycles (c1 or c2) in 2004–2007 in Lusignan (Lus) and 2004–2006 in Connantre (Con): maximum stem height and rate of stem growth that are forage yield components, growth habit (erect varieties are selected for cultivation in pure stands), flowering date that is used to describe and differentiate the varieties. The height of the tallest stem was measured four to five times per growth cycle and rate of stem growth (RG) was calculated by regression between these height measurements and sums of degree-days. Maximum stem height (MH) was defined as height of the tallest stem of the last height measurement per growth cycle. Date of flowering (DF) was assessed as flowering date transformed into sum of degree-days. Sums of degree-days with a temperature basis of 0 °C were measured from January 1st to measurement date for the first growth cycle and from date of first cut to the measurement date for the second growth cycle. Growth habit (GH) was scored visually using a scale from 1 (erect) to 5 (prostrate).
Statistical analysis of morphological data
The experimental design corresponds to a “common garden” experiment used in population genetics when studying natural populations. To estimate QST, we performed an analysis of variance for each measurement separately using the GLM procedure of SAS statistical package version 8.1 (SAS Institute Inc. 2000). The model included the effects of block (fixed), cultivar, genotype nested within cultivar and error (random). Components of variance for random effects were calculated for each measurement using the option REML of the procedure VARCOMP of SAS. QST was then calculated using the formula:
where \(V_{c}^{2}\) is the variance of the cultivar effect and \(V_{g}^{2}\) the variance of the genotype nested within cultivar effect (Spitze 1993). Significative difference of QST at a threshold of 95, 99 and 99.9% was estimated after a bootstrap procedure on the cultivars and the genotypes, with 1000 iterations.
Canonical discriminant analysis was performed to estimate within-cultivar diversity and to graphically represent the within and between-population diversity. The CANDISC procedure of SAS was used by using the mean value of the genotypes. For each of the ten cultivars, the standard deviation of each of the first five canonical axes was estimated. Each standard deviation was weighted by the proportion explained by the respective axes and the five weighted values were summed resulting in an estimation of within-cultivar diversity (DS). The genotypes were represented on the first two axes of the canonical discriminant analysis. The CANDISC procedure also calculated the Mahalanobis distances among pairs of populations.
Genotyping
DNA was extracted from young leaves following the method described in Cheung et al. (1993). Sixteen SSR markers (Online Resource 2) were selected in order to have two markers on each chromosome (Julier et al. 2003). Thirteen markers were analysed using an automated DNA sequencer LI-COR IR2 (LI-COR Inc.) and 3 markers were scored on an ABI Prism 3700 genetic analyzer (Applied Biosystems Inc.). PCR analyses were conducted in a total volume of 10 µL containing about 15 ng DNA, 1 × PCR buffer, 2 mM MgCl2, 0.1 µM of primers, 0.2 mM of each dNTP and 0.25 U Taq Polymerase (Invitrogen Inc.). PCR conditions consisted of 4 min at 94 °C, 35 cycles of 30 s 94 °C, 30 s at 55 °C and 30 s at 72 °C followed by a final extension of 3 min at 72 °C. Markers which were examined on a LI-COR DNA sequencer were analysed and scored as described in Flajoulot et al. (2005). Among them, the allele dosage was coded for nine markers (co-dominant coding). For the four other markers, it was not possible to code the allele dosage, each allele was scored as presence or absence (dominant coding). For markers which were analysed on ABI Prism genetic analyzer, alleles were scored for presence and absence (dominant coding), using the Genotyper software (Applied Biosystems Inc.).
To enhance the accuracy of alleles coding, two parents of an alfalfa mapping population were integrated in the analysis of molecular markers, for which data of allelic dosage were available (Julier et al. 2003).
Statistical analysis of molecular data
For the nine markers that were coded as co-dominant, we checked if the dose of the fragments was scored accurately. Assuming that the populations were at Hardy–Weinberg equilibrium, departure from equilibrium for the markers in each cultivar was tested using AUTOTET software (Thrall and Young 2000) under the hypothesis of random chromosomal segregation. Resulting significant values of the test were adjusted using Bonferroni correction. The frequency of putative null alleles were estimated from the heterozygote deficiency (Brookfield 1996). One of these nine markers (MTIC365) did not fulfil the criteria for accuracy of codominant coding. For all cultivars, it clearly departed from Hardy–Weinberg equilibrium with an average value of 0.60 for the fixation index and significant difference between observed and expected heterozygosities (P < 0.05). Furthermore, the estimated frequency of null alleles was 0.271 while the frequencies for the other eight codominantly coded markers ranged from 0.001 to 0.026. Therefore, MTIC365 was further considered in calculations as a dominantly coded marker.
To estimate within-cultivar diversity, the average number of alleles per marker (A) and the average number of alleles per genotype (Ai) were calculated for the 8 codominantly coded markers and for all 16 markers. For the codominantly coded markers, the expected heterozygosity (HE) assuming random chromosome reduction was computed using AUTOTET (Thrall and Young 2000).
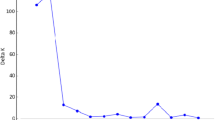
The genetic structure among cultivars was evaluated with different methods. FST was calculated over all 10 cultivars with the 8 codominantly coded markers, by using the software Gene4x (Ronfort et al. 1998). In NTSYS (Rohlf 2000), Euclidean squared distances were calculated from frequencies per cultivar of the 279 alleles of all 16 markers. In Structure (Pritchard et al. 2000), a admixture model with correlated allele frequencies model was applied. Calculations were performed with the eight codominantly coded and the eight dominantly coded markers and a ploidy level of four. Twenty times calculations were performed for each of K = 1–11 with a length of burnin period of 20,000 and 50,000 MCMC reps. With the resulting values, optimal K was identified based on LnP(D).
The distribution of variation within and between the cultivars was tested with an analysis of molecular variance (AMOVA) in which the autotetraploidy of the genotypes was taken into account (Jenczewski et al. 1999a). For this calculation, we excluded rare alleles, i.e. alleles with less than four fragments over 400 genotypes (Lynch and Milligan 1994), and markers with more than 5% missing values, i.e. MTIC107, MTIC365, MTIC58, Enod20 and MTIC134 locus 2.
The FST was compared to QST values. QST was noted as different from FST if FST was not included in the confidence interval of QST. The limitations in comparisons between FST and QST (Edelaar et al. 2011) were not applicable in our conditions because gene flow (here, exchanges between breeding pools) is expected to be high compared to the mutation rate of SSR markers.
In this study, we used a small set of markers compared to the genotyping methods that recently emerged (Li et al. 2014b). However, we used codominant SSR markers whose dosage could be read for some of them, offering a better evaluation of within-population diversity. Furthermore, more markers would not drastically change the estimation of genetic parameters such as FST, Euclidian distances or AMOVA, nor their comparison to the calculations carried out on morphological traits.
Results
Morphological analyses
Forty-five measurements were performed for the four following traits: rate of stem growth, growth habit, maximum stem height and date of flowering over four cuts in 3 years and two locations (Table 1). Analysis of variance revealed highly significant effects for cultivars and/or genotypes within-cultivars for 41 of the measurements. For further calculations, only these 41 significant measurements were taken into account.
Most QST values were significantly different from 0 and explained up to 39% of the genetic variation. On average, QST was low for date of flowering (0.04), intermediate for maximum stem height and rate of stem growth (0.06 and 0.07, respectively) and high for growth habit (0.19) meaning that growth habit was the trait that most differentiated the cultivars (Table 1).
Squared Mahalanobis distances between varieties were on average 11.3; they ranged from 3.6 between Cannelle and Harpe to 27.2 between Luzelle and Barmed and were significant (P < 0.05) for all other pairs of cultivars (Table 2). The average distance between one cultivar and the other nine was larger for Luzelle (19.4) than for the other cultivars (7.7–13.0).
The first two axes of the canonical discriminant analysis for the ten cultivars explained 59% of the total variation (34 and 25%; Fig. 1). The first axis was mainly explained by growth habit of the first growth cycle in Connantre in 2005 (GHc1Con05), maximum stem height of the first growth cycle in Lusignan in 2006 (MHc1Lus06) and growth habit of the first growth cycle in Lusignan in 2004 (GHc1Lus04). The second axis was mainly explained by date of flowering of the second growth cycle in Lusignan in 2007 (DFc2Lus07), growth habit of the first growth cycle in Connantre in 2005 (GHc1Con05) and rate of stem growth of the first growth cycle in Lusignan in 2005 (RGc1Lus05). The first and second axes separated the Southern group (i.e. Provence, Barmed, Zenith), the “Northern without Luzelle” group (i.e. Flamande, Mercedes, Alpha, Symphonie, Cannelle and Harpe) and Luzelle cultivar (Fig. 1). Canonical discriminant analysis for the three groups “Northern without Luzelle”, Southern and Luzelle showed squared Mahalanobis distances of 6.2 (P < 0.001) between “Northern without Luzelle” and Southern, of 13.8 (P < 0.001) between “Northern without Luzelle” and Luzelle and of 19.6 (P < 0.001) between Southern and Luzelle. Canonical discriminant analyses performed separately for the two groups “Northern without Luzelle” and Southern revealed a separation of the cultivars within the Southern group whereas within the “Northern without Luzelle” group, no further separation was observed (data not shown).

Canonical discriminant analysis of the ten cultivars based on morphological traits. Analysis was based on 41 measurements of the four traits: date of flowering, rate of stem growth, growth habit and maximum stem height
The index for within-cultivar diversity (DS) ranged from 0.76 for Alpha to 0.93 for Luzelle with an average of 0.85. It was not higher for the two landraces than for the cultivars (Table 3).
Molecular analyses
The 16 markers were equally distributed with two markers per chromosome, with one on each of the two parts of the chromosome. A total of 279 alleles was identified ranging from 5 (MTIC124) to 40 (Enod20) alleles per marker (Online Resource 2) and an average of missing values of 4.6% per marker. Frequencies of alleles ranged from 0.003 (one genotype in 400 with this fragment), to 0.938 (375 genotypes with this fragment). Thirty cultivar-specific alleles were observed but they always had a low frequency: 23 alleles had a frequency of 0.025 (one genotype in the 40 genotypes of the cultivar) and among the other specific alleles, the maximal frequency was of 0.175 (seven genotypes with this fragment in the cultivar). This type of cultivar-specific alleles cannot be used for cultivar identification.
Eight of the 16 markers were codominantly coded, i.e. presence and absence of fragments were scored and the dose of each fragment was also taken into account. They were distributed with one marker per chromosome but no marker was located on chromosome 8 and two markers were located on chromosome 7 (Online Resource 2). For these eight markers, all ten cultivars were in Hardy–Weinberg equilibrium (P > 0.05), with the exception of MTIC451 and MTIC343 for Mercedes (P < 0.05). The other eight markers were dominantly coded (Online Resource 2), i.e. only for presence or absence of fragments, as intensity differences of fragments was visually too difficult to estimate or existence of null alleles was assumed.
For the eight codominantly coded markers, the three diversity indices, namely average number of alleles per marker (A), average number of alleles per genotype (Ai) and expected heterozygosity (HE), were similar for the ten cultivars. Index A ranged from 10.9 (Luzelle) to 13.9 (Zenith), Ai from 2.7 (Mercedes/Cannelle) to 2.9 (Symphonie) and HE from 0.726 (Cannelle) to 0.762 (Symphonie, Zenith, Luzelle). When considering all 16 markers, A and Ai were lower than for the eight codominantly coded markers, but also similar among the ten cultivars (Table 3).
FST value for ten cultivars calculated with the eight codominantly coded markers was only 0.0127 (P < 0.0001, standard error of 0.0000). Pairwise FST values ranged from 0.003 between Flamande and Provence to 0.028 between Cannelle and Barmed and were significant with P < 0.01 (Table 4). Average difference of FST to the other cultivars was higher for Luzelle (0.0197) and for Barmed (0.0171) than for the other cultivars ranging from 0.0090 for Flamande to 0.0140 for Cannelle. Similarly, with Euclidean squared distances based on frequencies per cultivar for all 279 alleles (all loci), the lowest distances were observed among Provence, Flamande and Harpe (1.7), whereas the highest value was identified between Luzelle and Barmed (4.7, Table 4). On average, Euclidean squared distances between a cultivar and the other nine cultivars were higher for Luzelle (3.7) and Barmed (3.6) and lower for the other cultivars (2.3–3.3).
Using Structure software with the eight codominantly coded markers and the eight dominantly coded markers, six clusters were found. The highest proportion of membership to a cluster was observed for Luzelle and Barmed. The other cultivars contained a similar proportion of each of the six clusters (Online Resource 3).
FST values were calculated for two allocations of groups. The first allocation included the three groups identified with morphological traits: “Northern without Luzelle” (Flamande, Mercedes, Alpha, Symphonie, Cannelle and Harpe), Southern (Provence, Barmed, Zenith) and Luzelle. FST value between “Northern without Luzelle” and Southern was 0.003 (P < 0.001) and Luzelle showed similar FST values to the other two groups (0.016 with “Northern without Luzelle” and 0.014 with Southern, both P < 0.001) (Online Resource 4). The second allocation included three differently defined groups, as found with molecular markers: Luzelle, Barmed and the other eight cultivars. FST values were 0.012 (P < 0.001) between the eight cultivars group and Barmed, 0.016 (P < 0.001) between the eight cultivars and Luzelle and 0.019 (P < 0.001) between Barmed and Luzelle. When performing AMOVA based on dominantly coded markers and 147 alleles, group effect accounted for 0.13% of total variance in the first allocation (“Northern without Luzelle”, Southern and Luzelle) and 0.25% of the total variance in the second allocation (Luzelle, Barmed and the other eight cultivars; Online Resource 4). However, in both cases, the major part of the variation (more than 99.5%) was observed within cultivars.
Comparisons between FST and QST
All global QST values (over the whole set of cultivars) for the growth habit measurements were highly different from FST (Table 1), indicating a strong selective pressure on this trait. For the maximum stem height and the rate of growth, most QST values were significantly higher than FST. Contrastingly, for the date of flowering, QST was slightly significantly different from the FST value. In pairwise comparisons of cultivars, QST (not shown) and FST (Table 4) values were different, this excluded simple genetic drift.
Discussion
This study analysed not only the same cultivars but also the same genotypes with both morphological and molecular methods. Furthermore, the genotypes were investigated over several years and two environments. This design, which had never been experimented for M. sativa, allowed the direct comparison of morphological traits and molecular markers.
Comparison of structure of cultivars
In addition to the overall significant difference among the 10 cultivars, differences between pairs of cultivars were also significant but small for molecular markers and for morphological traits. These significant differences were confirmed by a study investigating French cultivars of M. sativa with SSR markers in which mainly significant FST values were observed (Flajoulot et al. 2005). In contrast, no significant difference among cultivated populations based on allozyme markers and only a small part of the pairwise differences was significant for RAPD markers and for morphological traits (Jenczewski et al. 1999a, b). This discrepancy may point out that, although significant differences among M. sativa populations were often observed, these differences were in general based on a very small part of total variance. All cultivars, either landraces or varieties, had similar within-population diversity (number of alleles and heterozygosity) showing little effect of selection on within-population diversity, as already found in a set of varieties originating from a single breeding program (Flajoulot et al. 2005). Qiang et al. (2015) found more diversity in landraces than in varieties or wild populations, but the number of genotypes was also higher for landraces than for other types, possibly causing a bias in diversity estimation.
Considering morphological traits, Luzelle was clearly different from the other cultivars. The growth habit contributed most to this separation as shown by the high QST value. This strong contribution of growth habit to separate M. sativa populations was confirmed in other studies with morphological traits (Crochemore et al. 1998; Julier et al. 1995; Prosperi et al. 2006). The largest difference between Luzelle and the rest of the cultivars was also observed for molecular analyses. Luzelle is the cultivar with the highest part of falcata genome as one parent of the cultivar belongs to subsp. falcata (P. Guy, pers. comm.). This results in a prostrated growth habit for grazing purposes. In general, when populations originating from the whole M. sativa species complex were studied, a clear separation of subsp. falcata was reported for molecular markers (Crochemore et al. 1996; Maureira et al. 2004; Musial et al. 2002; Segovia-Lerma et al. 2003). The main dormancy classes were also recovered (Li et al. 2014a).
Barmed was also different from the rest of cultivars. Barmed was selected from genetic resources of United States of America and North Africa (D. Noël, pers. comm.) and may therefore largely differ from the resources of other French cultivars. However, this separation was less pronounced than for Luzelle.
Even if similarities were noticed between structure given by morphological and that given by molecular analyses, major discrepancies were observed. Morphological traits clearly separated “Northern without Luzelle” group (i.e. Flamande, Mercedes, Alpha, Symphonie, Cannelle and Harpe) from Southern group (Provence, Zenith and Barmed), whereas no clear structure was observed with molecular markers. The two cultivars with the highest molecular similarity were Flamande and Provence, i.e. the two landraces representing the archetypes of the Northern and Southern groups. In a large set of cultivars, a clear separation of Northern and Southern groups with morphological traits was also reported although this structure was not detectable with RAPD markers (Crochemore et al. 1996, 1998). Poor correlations between structure of populations obtained by morphological traits and molecular markers have also been reported by other authors, for example in Lolium perenne (Roldan-Ruiz et al. 2001) or in six-rowed Hordeum vulgare (Manninen and Nissila 1997). Authors concluded that discrepancies came from the fact that molecular markers represent neutral diversity, whereas morphological traits represent selected diversity. However, in most studies, selection for morphological traits was not studied (Crochemore et al. 1998; Roldan-Ruiz et al. 2001) because experimental designs did not include replicates on individual genotypes. In our study, selective effects were demonstrated by the differences between QST and FST values, thus explaining the discrepancies between the structures given by morphological traits and molecular markers.
Role of selection in the differences among cultivars
Both QST and FST were globally different from zero, indicating difference among cultivars. For comparison of morphological traits and molecular markers, neutrality of the latter was assumed but we hypothesized that morphological traits were not neutral. When populations are strongly differentiated with respect to morphological traits, but not to molecular markers (QST > FST), a predominant role for heterogeneous selection among populations is predicted. In contrast, when population differentiation is similar for morphological traits and molecular markers, genetic drift alone can explain the differentiation among populations (Spitze 1993). In natural populations, QST values generally are higher than FST values suggesting a predominant role of natural selection in among population differentiation (Leinonen et al. 2008).
In this study, we observed that QST values for the four traits were generally higher than the FST. This indicates that selection is acting on morphological traits. This was particularly pronounced for growth habit, for which all QST values were significantly (P < 0.05) different from FST values, and to a lower extent for stem height and stem growth rate. In cultivars, besides natural selection, a strong artificial selection influences diversity among populations. This artificial selection could act in the same direction as natural selection since high fitness is important for natural and agricultural purposes. Nevertheless, natural and artificial selections may have contrasted effects if breeding traits do not contribute to a highly competitive population. Farmers may require an alfalfa variety with erect growth for silage/haying or a more prostrate growth for grazing. Therefore, growth habit is a trait considered in breeding (Lonnet 1996) and showed a high QST value. Stem elongation is also a breeding criterion because it was shown to be correlated to forage yield (Annicchiarico et al. 2015). QST values higher than FST for stem height and stem growth rate are consistent to the selection for these traits. In contrast, the low QST values for the date of flowering is in accordance with the fact that this trait is not included in breeding criteria list (Lonnet 1996). In most species, date of flowering usually is a major breeding or adaptative trait, as it determines the coincidence between reproduction and favourable environmental conditions (Anderson et al. 2011). Date of flowering and reproduction are less important for adaptation in a perennial forage species such as alfalfa than for an annual seed crop. Indeed, little is known on genetic architecture of flowering date in perennial species (Anderson et al. 2011; Friedman and Rubin 2015). Most experiment data [reviewed by Le Corre and Kremer (2012)] also reported QST values higher than FST. These authors suggest that adaptative traits may be controlled by large number of genes resulting in multiple but possibly low allele frequency changes.
Comparison of within-cultivar diversity with traits and markers
Although differences between cultivars were identified with molecular markers and morphological traits, QST and FST values were in general small and the main part of variation was present within cultivars. For molecular markers, this high within-cultivar diversity was further confirmed by the average number of alleles per genotype and expected heterozygosity. Although values between studies varied due to different marker systems, calculation methods and number of genotypes per cultivar, several authors observed a high within-cultivar diversity for M. sativa based on molecular markers (Flajoulot et al. 2005; Jenczewski et al. 1999a, b; Kidwell et al. 1994a; Mengoni et al. 2001; Musial et al. 2002; Qiang et al. 2015). For morphological traits, the high spread of genotypes within each cultivar in canonical discriminant analysis further confirmed the high within-cultivar diversity. Only very few other diversity studies with genotype replications have been reported and all have verified a high within-cultivar diversity (Bolaños-Aguilar et al. 2000; Julier et al. 2000). In addition, within-cultivar diversity was similar in the eight varieties and the two landraces. Comparable results were obtained with French varieties using SSR markers (Flajoulot et al. 2005) and a set of Australian varieties using RAPD markers (Musial et al. 2002).
A large part of within-population diversity is expected in tetraploid and outcrossing species such as M. sativa, but several other facts contribute to this lack of differentiation between cultivars. Principally, European landraces mainly come from populations introduced in the sixteenth century from Spain to the rest of Europe (Muller et al. 2003). This time offered limited opportunities for population differentiation, especially for this perennial species. In addition, as M. sativa mostly exists as cultivated populations in Europe, their evolution relies on selection pressures or genetic drift handled by farmers or breeders. Furthermore, seed exchange was often conducted in modern breeding (e.g. Barmed see above) and during the establishment of landraces. For example, farmers of the North of France (Northern type) used to buy seeds from farmers of the South (Southern type) as conditions in the North were often not appropriate for seed production (Julier 1996). Similarly gene flow between the originally introduced germplasms is hypothesized in the USA (Li et al. 2014a). Considered as a whole, traditional and modern breeding was achieved without strong modification of the neutral genetic background.
The genetic structure available in alfalfa is favorable to the use of specific design to decipher genetic architecture of agronomic traits. The large within-population diversity, for both traits and markers, and the low level of structure are adequate to conduct association genetic studies. Indeed, the plant material presented here was used to assess the effect of a candidate gene of Constans-like family in flowering date and stem length variation (Herrmann et al. 2010; Julier 2012).
Conclusions
This study allowed a direct comparison of M. sativa structure and diversity using morphological traits and molecular markers. Selection for morphological traits was shown (QST > FST), especially for growth habit. This higher part of diversity among cultivars identified for morphological traits explained the clearer structure between cultivars observed for morphological traits compared to molecular markers. Furthermore, our study helped to explain discrepancies observed between the population structures obtained with the two marker types. It verified the hypothesis that morphological traits represent selected characteristics and molecular markers neutral diversity. However, the main part of variation was observed within M. sativa cultivars, which could be explained, beside the reproductive mode and its genetic characteristics, by the history of domestication and breeding. The obtained results broadened the basic understanding of trait selection and of structure of M. sativa populations.
References
Anderson JT, Willis JH, Mitchell-Olds T (2011) Evolutionary genetics of plant adaptation. Trends Genet 27:258–266. doi:10.1016/j.tig.2011.04.001
Annicchiarico P, Barrett B, Brummer EC, Julier B, Marshall AH (2015) Achievements and challenges in improving temperate perennial forage legumes. Crit Rev Plant Sci 34:327–380. doi:10.1080/07352689.2014.898462
Annicchiarico P, Nazzicari N, Ananta A, Carelli M, Brummer EC (2016) Assessment of cultivar distinctness in alfalfa: a comparison of genotyping-by-sequencing, simple-sequence repeat marker, and morphophysiological observations. Plant Genome 9:1–12. doi:10.3835/plantgenome2015.10.0105
Becker H (1993) Plant breeding. UTB Verlag Eugen Ulmer, Stuttgart
Bolaños-Aguilar ED, Huyghe C, Julier B, Ecalle C (2000) Genetic variation for seed yield and its components in alfalfa (Medicago sativa L.) populations. Agronomie 20:333–346
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygous deficiency. Mol Ecol 5:453–455
Cheung W, Hubert N, Landry B (1993) A simple and rapid DNA microextraction method for plant, animal, and insect suitable for RAPD and other PCR analyses. PCR Methods Appl 3:69–70
Crochemore ML, Huyghe C, Kerlan MC, Durand F, Julier B (1996) Partitioning and distribution of RAPD variation in a set of populations of the Medicago sativa complex. Agronomie 16:421–432
Crochemore ML, Huyghe C, Ecalle C, Julier B (1998) Structuration of alfalfa genetic diversity using agronomic and morphological characteristics. Relationship with RAPD markers. Agronomie 18:79–94
Edelaar P, Burraco P, Gomez-Mestre I (2011) Comparisons between Q(ST) and F-ST-how wrong have we been? Mol Ecol 20:4830–4839
Endler JA (1977) Geographic variation, speciation, and clines vol 10. Monographs in population biology. Princeton University Press, Princeton
Flajoulot S, Ronfort J, Baudouin P, Barre P, Huguet T, Huyghe C, Julier B (2005) Genetic diversity among alfalfa (Medicago sativa) cultivars coming from a single breeding program, using SSR markers. Theor Appl Genet 111:1420–1429
Friedman J, Rubin MJ (2015) All in good time: understanding annual and perennial strategies in plants. Am J Bot 102:497–499. doi:10.3732/ajb.1500062
Herrmann D, Barre P, Santoni S, Julier B (2010) Association of a CONSTANS-LIKE gene to flowering and height in autotetraploid alfalfa. Theor Appl Genet 121:865–876. doi:10.1007/s00122-010-1356-z
Jenczewski E, Prosperi JM, Ronfort J (1999a) Differentiation between natural and cultivated populations of Medicago sativa (Leguminosae) from Spain: analysis with random amplified polymorphic DNA (RAPD) markers and comparison to allozymes. Mol Ecol 8:1317–1330
Jenczewski E, Prosperi JM, Ronfort J (1999b) Evidence for gene flow between wild and cultivated Medicago sativa (Leguminosae) based on allozyme markers and quantitative traits. Am J Bot 86:677–687
Julier B (1996) Traditionnal seed maintenance and origins of the French lucerne landraces. Euphytica 92:353–357. doi:10.1007/BF00037119
Julier B (2012) Alfalfa breeding benefits from genetic analyses on M. truncatula. In: Bushman S (ed) Molecular breeding of forage and turf, June 4–7, 2012. Utah State University, Utah, pp 17–19
Julier B, Porcheron A, Ecalle C, Guy P (1995) Genetic variability for morphology, growth and forage yield among perennial diploid and tetraploid lucerne populations (Medicago sativa L.). Agronomie 15:295–304
Julier B, Huyghe C, Ecalle C (2000) Within- and among-cultivar genetic variation in alfalfa: forage quality, morphology, and yield. Crop Sci 40:365–369. doi:10.2135/cropsci2000.402365x
Julier B, Flajoulot S, Barre P, Cardinet G, Santoni S, Huguet T, Huyghe C (2003) Construction of two genetic linkage maps in cultivated tetraploid alfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol 3:9. doi:10.1186/1471-2229-3-9
Kidwell KK, Austin DF, Osborn TC (1994a) RFLP evaluation of nine Medicago accessions representing the original germplasm sources for North American alfalfa cultivars. Crop Sci 34:230–236
Kidwell KK, Bingham ET, Woodfield DR, Osborn TC (1994b) Relationship among genetic distance, forage yield and heterozygosity in isogenic diploid and tetraploid alfalfa populations. Theor Appl Genet 89:323–328
Kimura M (1983) Rare variant alleles in the light of the neutral theory. Mol Biol Evol 1:84–93
Le Corre V, Kremer A (2012) The genetic differentiation at quantitative trait loci under local adaptation. Mol Ecol 21:1548–1566
Leinonen T, O’hara RB, Cano JM, Merila J (2008) Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis. J Evol Biol 21:1–17
Li XH et al (2014a) Development of an alfalfa SNP array and its use to evaluate patterns of population structure and linkage disequilibrium. PLoS ONE. doi:10.1371/journal.pone.0084329
Li XH, Wei YL, Acharya A, Jiang QZ, Kang JM, Brummer EC (2014b) A saturated genetic linkage map of autotetraploid alfalfa (Medicago sativa L.) developed using genotyping-by-sequencing is highly syntenous with the Medicago truncatula genome. G3-Genes Genomes Genetique 4:1971–1979. doi:10.1534/g3.114.012245
Lonnet P (1996) Objectifs et critères actuels de la sélection des luzernes pérennes. Fourrages 147:303–308
Lynch M, Milligan BG (1994) Analysis of popualtion genetic structure with RAPD markers. Mol Ecol 3:91–99. doi:10.1111/j.1365-294X.1994.tb00109.x
Manninen O, Nissila E (1997) Genetic diversity among Finnish six-rowed barley cultivars based on pedigree information and DNA markers. Hereditas 126:87–93. doi:10.1111/j.1601-5223.1997.00087.x
Maureira I, Ortega F, Campos H, Osborn T (2004) Population structure and combining ability of diverse Medicago sativa germplasms. Theor Appl Genet 109:775–782
Mengoni A, Gori A, Bazzicalupo M (2001) Use of RAPD and microsatellite (SSR) variation to assess genetic relationships among populations of tetraploid alfalfa, Medicago sativa. Plant Breed 119:311–317
Muller MH, Prosperi JM, Santoni S, Ronfort J (2003) Inferences from mitochondrial DNA patterns on the domestication history of alfalfa (Medicago sativa). Mol Ecol 12:2187–2199
Musial JM, Basford KE, Irwin JAG (2002) Analysis of genetic diversity within Australian lucerne cultivars and implications for future genetic improvement. Aust J Agric Res 53:629–636
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Prosperi JM, Jenczewski E, Angevain M, Ronfort J (2006) Morphologic and agronomic diversity of wild genetic resources of Medicago sativa L. collected in Spain. Genet Resour Crop Evol 53:843–856
Qiang H, Chen Z, Zhang Z, Wang X, Gao H, Wang Z (2015) Molecular diversity and population structure of a worldwide collection of cultivated tetraploid alfalfa (Medicago sativa subsp. sativa L.) germplasm as revealed by microsatellite markers. PLoS ONE 10:e0124592. doi:10.1371/journal.pone.0124592
Riday H, Brummer EC, Campbell TA, Luth D, Cazcarro PM (2003) Comparison of genetic and morphological distance with heterosis between Medicago sativa subsp. sativa and subsp. falcata. Euphytica 131:37–45
Rohlf RF (2000) NTSYS-pc: numerical taxonomy and multivariate analysis system vol Exeter software, 2.1st edn. Applied Biostatistics Inc., New York
Roldan-Ruiz I et al (2001) A comparative study of molecular and morphological methods of describing relationships between perennial ryegrass (Lolium perenne L.) varieties. Theor Appl Genet 103:1138–1150
Ronfort J, Jenczewski E, Bataillon T, Rousset F (1998) Analysis of population structure in autotetraploid species. Genetics 150:921–930
Segovia-Lerma AM, Cantrell RG, Conway JM, Ray IM (2003) AFLP-based assessment of genetic diversity among nine alfalfa germplasms using bulk DNA templates. Genome 46:51–58
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792. doi:10.1126/science.3576198
Small E (2011) Alfalfa and relatives: evolution and classification of Medicago. NRC Research Press of Canada, Ottawa
Spitze K (1993) Population structure in Daphnia obtusa—quantitative genetic and allozymic variation. Genetics 135:367–374
Thrall PH, Young A (2000) AUTOTET: a program for analysis of autotetraploid genotypic data. J Hered 91:348–349
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Acknowledgements
We thank the ACVF (Association des Créateurs de Variétés Français) section Alfalfa for contributing to genotyping and phenotyping. JF Bourcier, F Durand, C Ecalle, A Gilly and J Jousse who contributed to genotyping or phenotyping are greatly acknowledged. This work was supported by Contrat de Branche from the French Ministry of Agriculture (2005–2008). DH received a grant from the Plant Breeding department of INRA.
Author information
Authors and Affiliations
Contributions
BJ, JR, CH conceived and designed the experiments; SF, BJ, PB performed the experiments; DH, BJ, SF analysed the data; DH, BJ, PB, JR, CH wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Herrmann, D., Flajoulot, S., Barre, P. et al. Comparison of morphological traits and molecular markers to analyse diversity and structure of alfalfa (Medicago sativa L.) cultivars. Genet Resour Crop Evol 65, 527–540 (2018). https://doi.org/10.1007/s10722-017-0551-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-017-0551-z