
Abstract
Higher phytase activity in food and feedstuffs is desirable in order to counter the antinutritional effects of phytate. The most promising platform where this might be achieved through plant breeding is wheat and its Triticeae relatives. They already accumulate notable amounts of phytase in the grains and higher expression of the responsible PAPhy_a gene can increase the activity further. Here we provide a survey of the genetic diversity of PAPhy_a in wild and extant relatives of wheat. Fifty sequences of the structural gene were obtained from 34 samples representing 21 species or subspecies. A phylogenetic tree is presented, demonstrating that the three gene copies in hexaploid wheat have been inherited from its diploid ancestors. This finding is not only relevant for applied research, it also provides further evidence regarding the origin of the genomes in polyploid wheat. T. urartu Tumanian ex Gandilyan and Ae. tauschii Coss. are confirmed as donors of the A and D genomes, respectively, and the hypothesis of a common origin (Ae. speltoides Tausch) of the B and G genomes is supported. The survey suggests that the allele variation available for breeding is increased by the inclusion of the closest relatives of wheat. This effort should benefit greatly from molecular methods because specimens of the same species may have either novel- or alleles-identical to those of wheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The PAPhy_a gene expresses a purple acid phosphatase phytase during grain filling and is the main source of mature grain phytase activity (MGPA) in cereals belonging to the Triticeae tribe (Brinch-Pedersen et al. 2013). Phytate represents the largest pool of phosphorus in cereal seeds and it chelates nutritionally important metals including calcium, iron and zinc. Monogastric animals excrete the majority of phytate bound phosphorus and metals unless the feed has sufficient phytase activity to degrade the phytate (Brinch-Pedersen et al. 2002). PAPhy enzyme activity therefore exerts a positive influence on the nutritional value of cereals as well as the environmental impact of farming. Accordingly, it is of major interest to find ways of increasing the activity in elite cultivars (Brinch-Pedersen et al. 2013). A transformation mediated gene duplication of PAPhy_a resulted in approximately doubled phytase activity in barley (Hordeum vulgare L.) (Holme et al. 2012). This demonstrated that there is no physiological barrier against a significantly higher expression of the PAPhy_a gene. Increased levels of activity will be within reach through conventional breeding, providing that more transcriptionally active PAPhy_a alleles are available. One potential strategy is to utilize the genetic diversity among wild and extant relatives of the major crop species wheat (Triticum aestivum L.) and barley. In cases where donor and recipient species are sexually compatible, favourable alleles can be introduced into elite cultivars by crossbreeding. Alleles from more distant relatives may be introduced by transformation or serve as templates for genome editing. However, there is very little background information available to guide the effort to increase MGPA. The PAPhy_a gene was only recently described and its sequence is only known for a handful of species (Madsen et al. 2013). Isolated PAPhy_a genes were mapped to chromosome 5H in H. vulgare and chromosomes 5A, 5B and 5D in T. aestivum using aneuploid lines. A single PAPhy_a gene was also identified in the diploid species einkorn (T. monococcum L. subsp. monococcum) and Aegilops tauschii Coss. whereas two copies were identified in rye (Secale cereale L.) (Madsen et al. 2013). This suggests that PAPhy_a existed as single gene on chromosome 5 in the diploid common ancestor of Hordeum L., Triticum L. and Aegilops L. and further that the PAPhy_a complement in modern wheat is directly inherited from its diploid ancestors. The search for more favourable PAPhy_a alleles and the study of wheat genome evolution may therefore be mutually informative.
The Triticeae cereals include diploids such as einkorn, barley and rye. However, the economically most important Triticeae cereals are the allopolyploid wheats. The allopolyploid wheats belong either to the ‘emmer’ group or the ‘timopheevii’ group. The ‘emmer group’ is derived from wild emmer (T. turgidum subsp. dicoccoides (Körn. ex Asch. et Graebn.) Thell.) and include among others, the domesticated, tetraploids emmer (T. turgidum subsp. dicoccon (Schrank) Thell.), durum (T. turgidum subsp. durum (Desf.) Husn.) and the hexaploid bread wheat (T. aestivum). Similarly, the ‘timopheevii’ group is derived from the wild T. timopheevii subsp. armeniacum (Jakubz.) van Slageren (syn. T. araraticum Jakubz.) and includes the domesticated forms sanduri (T. timopheevii Zhuk. (Zhuk.) subsp. timopheevii) and Zhukowskys wheat (T. zhukovskyi Menabde et Ericz.) (Salamini et al. 2002; Wagenaar 1966). The ‘emmer’ group became crops of global importance whereas the ‘timopheevii’ group remained restricted to the Transcaucasus region. Nevertheless, the ‘timopheevii’ group represent an important genepool for the improvement of wheat and has been the donor of e.g., disease resistance genes (Perugini et al. 2008). The genomes included in the ‘emmer’ group are denoted A, B and D and in the ‘timophevii’ group A and G (Kilian et al. 2007).
The evolution of the Triticeae cereals, in particular the identity of the donors of the T. aestivum genomes, has been studied for almost a century and the literature on the subject is immense [see reviews by Peng et al. (2011) and Haider (2013)]. Briefly, the D genome has been assigned to Aegilops tauschii (Kihara 1944; McFadden and Sears 1944, 1946). The A genome was initially assigned to wild einkorn (T. monococcum spp. aegilopoides (Link) Thell.), but the recognition of T. urartu Tumanian ex Gandilyan as a separate species prompted new investigations. Strong evidence now supports the later species as the A genome donor (Dvorak et al. 1993). The superscripts A u and A m are used to distinguish between the genomes from T. urartu and T. monococcum, respectively. The potential donor of the B genome remains controversial. Based on morphology, the search has focused on the Sitopsis section of Aegilops consisting of Ae. speltoides Tausch, Ae. sharonensis Eig, Ae. longissimi Schweinf. et Muschl., Ae. bicornis (Forssk.) Jaub. et Spach and Ae. searsii Feldman et Kislev ex Hammer (Sarkar and Stebbins 1956; van Slageren 1994). Each of these species has been suggested as donors of the B genome, but the most compelling evidence has been accumulated for Ae. speltoides (Haider 2013), though this is not universally accepted (Fernández-Calvín and Orellana 1994; Huang et al. 2002; Salse et al. 2008). The hybridization which created T. turgidum subsp. dicoccoides (AB) happened approximately 3–500,000 years before present (yBP), whereas the hybridization that created T. aestivum (ABD) took place 10,000 yBP (Haider 2013). Whole genome sequencing provided more ancient estimates of 580,000–820,000 yBP for AB and 230,000–430,000 yBP for ABD (Marcussen et al. 2014). It is however likely that these estimates partially reflect population divergence prior to the hybridization events as discussed by the authors. The more recent estimate for the ABD hybridization event is supported by the evidence which connect the hybridization event to the spread of agriculture (Salamini et al. 2002). The origin of bread wheat is so recent that its genomes have not diverged much form the donors and favourable alleles can relatively easily be introduced in T. aestivum from Ae. tauschii and T. turgidum via synthetic hexaploids (Dreisigacker et al. 2008). The phylogenetic relationships of diploid Triticeae has received comparatively less attention since molecular methods were introduced. Indeed, most studies have a priori limited themselves to species that are Triticum or Aegilops by name. This is unfortunate, since Petersen et al. (2006) placed a number of less studied species within the Triticum/Aegilops clade. Amblyopyrum (Boiss.) Eig and Thinopyrum (Saÿvul. et Rayss) Á. Löve was for example found to be much closer relatives of the A genome group than any of the Aegilops. Recently, the availability of whole genome sequence data has prompted renewed interest in the phylogenetic relationships of the diploid Triticum and Aegilops. Marcussen et al. (2014) proposed that Ae. tauschii and by extension the D genome resulted from an ancient hybridization between ancestors of the A and B genomes. A more complex hybridization history was subsequently suggested (Li et al. 2015a, b). El Baidouri et al. (2017) expanded on this, suggesting a model in which both the B and D genome ancestors had undergone ancient hybridizations. Further, the concept of subgenome genome dominance was used to explain different mutation rates following polyploidization. While genomics is certainly providing intriguing new insights it is unfortunately still limited to a few taxa. Approaches which can be applied to many taxa, such as single gene phylogeny, are therefore still indispensable.
Here we provide a survey of the diversity of PAPhy_a alleles in the Triticeae with emphasis on the Triticum/Aegilops complex. The survey includes seven samples of Ae. tauschii and T. turgidum since these species are of special interest to the improvement of bread wheat. Also rye has been used in the improvement of wheat and for the generation of synthetic polyploids, triticale (X Triticosecale Witt.) (Oettler 2005; Rabinovich 1998). We have therefore included four Secale L. accessions. A phylogenetic tree was constructed to visualize the evolution of the gene. The closest relatives of the wheat alleles were subjected to pairwise comparisons in order to assess the potential for finding useful genetic variation. To meet this end, we analysed approximately 2300 bp conserved gene sequence encompassing the core promoter, some 3′UTR and all introns and exons from the PAPhy_a gene. Fifty sequences from 34 samples of 21 species were included.
Materials and methods
The 34 samples used representing 21 species are listed in Table 1, which also include the authors of the taxa used. Note that two samples labelled as T. urartu by the IPK genebank (Tri 11494 and Tri 11495) have been re-identified as T. monococcum subsp. aegilopoides. This was done by morphology according to van Slageren (1994).
DNA was extracted for the current study as described by Møller et al. (2003). Some DNA samples were reused from Petersen et al. (2006), see Table 1. PCR was performed with Herculase II polymerase (Agilent Technologies) in 50 µL reaction mixture with 6% DMSO according to the manufacturer’s instructions. The primers used are listed in Table 2. Primers 1, 2 and 3 were reused from (Madsen et al. 2013). Primer 4 was designed to overcome the poor performance of primer 3. It is shifted 9 bp in the upstream direction. I.e. amplicons of this primer are 9 bp shorter when excluding the primer sequences themselves. Most samples were amplified using primers 1 + 3 and/or 1 + 4. Combination 1 + 4 produce stronger bands than combination 1 + 3 and requires no pre-annealing (see below). However combination 1 + 4 is not as universal as combination 1 + 3. Samples which failed to produce amplicons with these primer sets (i.e., Secale) were amplified using primers 2 + 3. Reactions with primer 3 required a pre-annealing step with this primer alone: 98 °C, 5 min; 30 °C, 20 s; heating to 72 °C at 2.0 °C/s; 72 °C, 5 min; 4 °C, ∞. The forward primer was subsequently added, whereafter the PCR proceeded using the same conditions for all primer combinations: 96 °C, 2 min; 38× (96 °C, 1 min; 59 °C, 20 s; 72 °C, 2 min 30 s; 72 °C, 3 min); 72 °C, 3 min; 4 °C ∞.
Amplicons were cloned in the pCR 4Blunt-TOPO vector (Invitrogen) and examined with EcoRI digests. Clones with 2000–2500 bp inserts were selected for sequencing. The restriction pattern from internal EcoRI sites supported the identification of homeologous clones in samples from the polyploid species. Sequencing was performed with two vector specific primers and two internal primers (Table 2 primers 5–8) by Eurofins Genomics, Ebersberg, Germany. The reads were assembled in CLC Main Workbench and trimmed to remove vector and incorporated primer sequences and to standardize the length of amplicons produced by different primer combinations. At least two clones from independent PCR reactions were sequenced per locus until a consensus was reached. The PAPhy_a sequence of Ae. sharonensis was downloaded from the published genome sequence after blasting with the TaPAPhy_a1 full length genomic sequence at http://wheat-urgi.versailles.inra.fr/Seq-Repo<3sitory/BLAST. All new sequences are deposited in ENA. See Table 1 for accession numbers.
Sequences were aligned using the CLC Main Workbench with default settings (gap open cost 10 and gap extension cost 1) and subsequently adjusted manually (supplementary file 1). CLC Main Workbench uses a proprietary algorithm based on progressive alignment (CLCbio 2007; Feng and Doolittle 1987). A phylogenetic analysis was performed using the RAxML maximum likelihood algorithm (Stamatakis 2006) implemented in Geneious version 8.1.2 (Kearse et al. 2012). The analysis was done with data in four partitions (exons, introns, upstream and downstream regions) and using the GTR + GAMMA + I nucleotide model and running 1000 bootstrap replicates. Pseudoroegneria (Nevski) Á. Löve was used to root the tree in accordance with Petersen et al. (2006). The sequences from the taxa in the individual genome groups A, D and B/G/ Ae. speltoides were realigned using above mentioned phylogenetic analysis (Fig. 3) as a guide tree. Again, the alignments were generated using CLC Main Workbench with the default settings, but without manual adjustment. The alignments were used for pairwise comparisons of the full length nucleotide sequences within the three genome groups. Subsequently, the alignments were shortened to include only the promotor and 5′UTR sequences (5′flanking region). Separate pairwise comparisons were generated from this alignment. Finally, the encoded protein sequences were aligned and compared [excluding the signal peptide as defined in Dionisio et al. (2011)].
Results
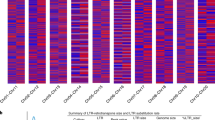
A total of 50 PAPhy_a sequences were obtained. The sequences varied in length from 2239 to 2598 bp and the alignment had a length of 3253 bp. The conservation along the alignment is compared to the structure of the gene in Fig. 1. The gene structure was determined using the TaPAPhy_a1 coding sequence as scaffold. The exons are more conserved than non-conding promoter, intron and UTR sequences. Lengthy insertions in a single sequence cause the three stretches of only 2% conservation at 178–397 bp (insertion in S. cereale cv. Picasso 2), 1882–1995 bp [intron gain in S. strictum NGB9846.1 (2)] and 2656–2933 bp (elongated intron in H. vulgare cv. Igri). A pairwise comparison of similarity was generated from the alignment (Fig. 2). The lowest similarity of 71% (851 nucleotide differences and indels) was found between H. vulgare cv. Igri and S. cereale cv. Picasso. Seventeen sequences were identical with at least one other sequence. With a single exception, all identical sequences were from the same species or from homologous genome groups (e.g., the A u genomes of tetraploid and hexaploid wheats). The maximum likelihood tree is shown in Fig. 3. Generally, there is no or only little bootstrap support (BS <50%) for the backbone of the tree whereas the various wheat genome groups and Secale are all strongly supported (BS = 100%). Accordingly, the data give only limited indication of relationships between the wheat genome groups. It does, however, allow us to infer the relationships within the groups including the relationships between polyploids and their diploid donor species. In the A genome group, sequences from T. monococcum and T. zhukovskyi are, with high support (BS = 100%), sister group to sequences from T. timopheevii, T. aestivum, T. turgidum and T. urartu. The highly supported D genome group (BS = 100%) consists only of sequences from Ae. tauschii and T. aestivum. The S′ genome group consists of sequences from three Aegilops species (Ae. bicornis, Ae. sharonensis and Ae. searsii) and is also highly supported (BS = 100%); within this group Ae. bicornis and Ae. sharonensis are most closely related (BS = 99%). Full length nucleotide sequences in the A genome group were between 94 and 100% similar (Fig. 4 top panel left). The sequences can be divided in three groups: sequences A–G represent the A u genome of the ‘emmer’ group and T. urartu. Sequences H–K represents the Au genome of the ‘timopheevii’ group and sequences L–O represent the A m genome. The full length nucleotide sequences within each group are >99% similar (Fig. 4, top panel left). The 5′flanking sequences were 67–100% similar (Fig. 4, top panel center). As for the full length sequences, the A genome 5′flanking region are highly similar within the subgroups. The pairwise comparison can therefore be simplified: A–G is 74–75% similar to H–K and 91–92% similar to L–O. The groups H–K and L–O are 67–68% similar. At the protein level, the T. aestivum cv. Bob White allele stands out because of a frame shift insertion. The remaining proteins in the A genome group has 0–10 amino acid differences but no indels.

Conservation along the PAPhy_a gene sequence. The top bar shows the gene structure. The 5′flanking region is blue, exons green, introns red and 3′UTR yellow. The hatch intron is only found in one sequence (S. cereale cv. Picasso 2). The conservation is shown as a percent similarity graph below the bar. (Color figure online)

Pairwise comparison based on the alignment of all isolated sequences. Below the diagonal, percent similarity; above the diagonal, number of differences

Maximum likelihood tree based on the four different partitions of the PAPhy_a gene. The numbers below or above the branches are bootstrap proportions

Pairwise comparisons of sequences from the taxa included in the A genome group (top), the B genome group (middle) and the D group (bottom). The comparisons include the full nucleotide sequences (left), the 5′flanking regions (middle) and the encoded proteins (right). The nucleotide sequences are compared according to % similarity below the diagonals and number of differences above the diagonals. The protein sequences were compared according to differences below and gaps above diagonals. Differences minus gaps equal amino acid substitutions
Three T. aestivum and four Ae. tauschii sequences belong to the D genome group (Fig. 4 bottom panel). The full length T. aestivum nucleotide sequences and one Ae. tauschii sequence (Ae231) are identical and all sequences are >99% similar (Fig. 4, bottom left panel). Also the 5′flanking region and proteins are highly conserved with maximum three nucleotide differences or amino acid substitutions respectively (Fig. 4, bottom panel centre and right).
The B/G/Ae. speltoides group include six sequences from the ‘emmer’ group (A–F), five sequences from the ‘timopheevii’ group (G–K) and three from Ae. speltoides (L–N) (Fig. 4 middle panel). The full length nucleotide sequences from the polyploid taxa are 89–100% similar, but none of the sequences from polyploid taxa are more than 94% similar to any of the Ae. speltoides sequences. One sequence of the ‘emmer’ group (T. aestivum cv. Bob White) is identical to sequences of the domesticated representatives of the ‘timopheevii’ group but only up to 90% similar to other members of the ‘emmer’ group. With this exception, sequences within the respective groups ‘emmer’ and ‘timopheevii’ are >98% similar. Excluding the Bob White allele, emmer group 5′flanking regions are >99% similar having just one nucleotide difference. The ‘timopheevii’ group display two distinct 5′flanking regions with 24 nucleotide differences. The proteins have 0-18 amino acid substitutions and no gaps (Fig. 4 middle panel right). The Bob White protein is identical to the proteins in the ‘timopheevii’ group and has 16 or 17 amino acid substitutions compared to other proteins in the ‘emmer’ group. Only one amino acid substitution is found among the remaining ‘emmer’ group proteins and there are no substitutions in the ‘timopheevii’ group.
Discussion
The present study includes samples of most, if not all, potential donors of all wheat genomes. For a more thorough representation of Triticeae diversity it also includes a number of wild, diploid species belonging to other genera. Further, it includes representatives of the frequently neglected ‘timopheevii’ group. Phylogenetic analysis and pairwise comparisons were based on a complete nuclear gene spanning more than 2.2 kb in all samples.
The three T. aestivum gene copies from cv. Chinese Spring have previously been mapped to chromosome 5A, 5B and 5D using aneuploid lines (Madsen et al. 2013). Their positions in the tree confirm that they have been inherited from the diploid genome donors and that these donors are T. urartu (A), Ae. speltoides (B) and Ae. tauschii (D) in line with the increasingly accepted view (Haider 2013; Peng et al. 2011). Including less or other taxa and accessions of both Triticum and Aegilops, largely the same patterns were found by Petersen et al. (2006). One interesting finding is that, Amblyopyrum which van Slageren (1994) excludes from the Aegilops based on morphology, here falls within Triticum and Aegilops though with no support. Petersen et al. (2006) found high support for a position within Triticum and Aegilops.
The A genome group
The sequences representing T. urartu and T. monococcum together with one sequence from each of the polyploid wheat samples form a monophyletic group with high support (BS = 100%). This A genome group has two distinct subgroups containing among others T. urartu and T. monococcum, respectively. These subgroups represent the A u and A m genome. The only polyploid taxon in the A m group is T. zhukovskyi. This is in agreement with Dvorak et al. (1993) who found evidence of A m as well as A u genomes in T. zhukovskyi and suggested it to originate from hybridization between T. timopheevii and T. monococcum. Accordingly an A u gene copy was also expected in T. zhukovskyi but we were unable to isolate it. It could be mutated in a way that prevents amplification with the current primes, but it is also possible that the A u gene copy has been replaced by the A m copy through heterogenous chromosome pairing. The apparent loss of A u DNA in T. zhukovskyi has also been observed by Baum and Bailey (2004) and Dvorak et al. (1993). Within the A u group, the sequences from the ‘timopheevii’ wheats form a well-supported clade (BS = 91%). A prominent characteristic of the ‘timopheevii’ group A u allele is a 69 bp deletion in the 5′flanking region, but also SNP’s contribute to differentiate this allele form other A u sequences. The deletion in the 5′flanking region includes cis acting regulatory elements previously identified (Madsen et al. 2013). It is therefore very likely that the gene regulation is affected. The A genome allele of the T. turgidum subsp. dicoccoides accession Tri18485 is identical to the allele of T. aestivum cv. Chinese Spring whereas those of Tri18508 and Tri18523 both have 8 differences resulting in 1 and 3 amino acid substitutions in the encoded proteins (Fig. 4). These results show that exotic breeding materials for the A genome may provide novel PAPhy_a alleles but they are not guaranteed to do so. It is therefore advisable to use molecular screening methods to support the breeding effort.
The D genome group
All sampled sequences from Ae. tauschii form a strongly supported group together with three identical sequences from T. aestivum, thus supporting that Ae. tauschii is the D genome donor of wheat. One sequence from Ae. tauschii (Ae 231) is identical to those of T. aestivum whereas the other Ae. tauschii sequences have 10 or 20 nucleotide differences, respectively (Fig. 4). One accession (AE229) has nucleotide differences in the 5′flanking region and the encoded protein (Fig. 4). Whereas the D genome PAPhy_a gene copy thus seems to be very conserved within T. aestivum, the variation among sequences in Ae. tauschii may potentially be useful for crop improvement. As for T. turgidum, it should be useful to apply molecular screening methods to ensure that Ae. tauschii specimens harbour novel alleles before crossing.
The B genome group
Aegilops speltoides has hitherto also been postulated to carry the S genome (Friebe et al. 1995). In the current tree it groups within a highly supported clade (BS = 100%) which also contains sequences from the B and G genome. This finding is in agreement with Kilian et al. (2007) and Golovnina et al. (2007) both of which found that the B and G genomes can be understood as two examples of Ae. speltoides haplotype diversity sequestered by independent hybridization events. Since the B and G genomes are both derived from Ae. speltoides, a more appropriate nomenclature for the group would be B b (emmer group), B g (timopheevii group) and B s (Ae. speltoides). Within the highly supported (BS = 100%) B genome group, sequences from the hexaploid wheats are placed in either of two strongly supported groups: one includes sequences from two T. aestivum cultivars and tetraploid T. turgidum, and the other includes sequences from one T. aestivum cultivar (Bob White), T. zhukovskyi and the four T. timopheevii accessions. A sequence from one of the accessions of Ae. speltoides is strongly supported (BS = 86%) as the sister group to the latter Bob White plus T. timopheevii and T. zhukovskyi clade and sequences from the remaining two accessions of Ae. speltoides are weakly supported (BS = 64%) as their sister group, thus rendering Ae. speltoides paraphyletic. It is reasonable to assume that the ‘timopheevii’ group sequences in this clade are representatives of the B g genome since the B b and B g genomes are known to be very similar (Kilian et al. 2007) and the other ‘timopheevii’ group sequences clearly belongs to the A genome group. The deepest bifurcation in the group splits a weakly supported (BS = 64%) clade with Ae. speltoides and the B g genome Triticum’s from the strongly supported (BS = 100%) B b genome Triticum’s. The position of the Bob White sequence appears to be an abnormality, which can be explained by the use of the T. timopheevii hybrid Timstein in breeding Bob White (GRIS database: http://wheatpedigree.net/sort/show/8192). Thus, it is an example of genetic variation of the PAPhy_a gene which has already been introduced in bread wheat from the ‘timopheevii’ group by chance. The five accessions belonging to the ‘timopheevii’ group produced only two different PAPhy_a sequences from the B g genome locus. The sequences distinguish domesticated and wild accessions and differ in 25 aligned positions due to one SNP and a 24 bp indel. Sequences assigned to the B b genome are more diverse than those of the B g genome. The least similar B b genome alleles are those of Tri18508 and Tri18523 which differ in 47 aligned positions because of 18 SNPs and 7 indels. On the other hand, identity was found between the sequences from Chinese Spring (a hexaploid) and Tri18485 (a tetraploid). The three T. turgidum sequences have identical 5′flanking sequences to Chinese Spring, whereas Villa Jurez has one SNP. Only the sequence Tri18508 encodes a different protein than the other B b alleles (one amino acid substitution).
Perspectives for the improvement of MGPA in bread wheat
It was demonstrated that direct progenitors of bread wheat, Ae. tauschii and T. turgidum, may carry PAPhy_a gene copies which are identical to those of bread wheat or copies with up to 44 nucleotide differences. Some of these differences were found in the 5′flanking region and others resulted in amino acid substitutions in the encoded proteins. Thus, it is very likely that alleles with different activity profiles are available. Further, it should be possible to evaluate them before introgression in bread wheat by direct comparison of the MGPA in candidate specimens with specimens of the same species carrying PAPhy_a gene(s) identical to those of bread wheat. The effect of amino acid substitutions on the enzyme properties can be evaluated after recombinant expression of the proteins as demonstrated by Dionisio et al. (2011).
Additional gene variation is found in T. timopheevii and T. zhukovskyi. As discussed above, the T. timopheevii B g genome allele has already been transferred to T. aestivum cv. Bob White (and presumably many other cultivars breed from Timstein). Bob White does not have a high MGPA (Madsen et al. 2013) so the B g genome allele does not appear to confer very expression levels or a much more active enzyme. It should be considered though, that Bob White also has an allele on the A genome which is most likely nonfunctional because of a frameshift insertion. It is therefore not possible to precisely evaluate the potential of the T. timopheevii allele based on Bob White MGPA. The T. timopheevii A u allele with its notable deletion of regulatory elements in the promotor is very likely to provide a different expression level but it cannot be predicted if it will be higher or lower. Also the A m allele of T. zhukovskyi and T. monococcum holds promise, since T. monococcum is known to have a high MGPA (Madsen et al. 2013).
However, the search for better expressed PAPhy_a alleles does not need to be restricted to very close relatives of wheat. Rye translocation lines have long been used in wheat breeding (Graybosch 2001) and it is clear from Fig. 3 that considerable genetic variation is available in the Secale. Rye has consistently shown higher MGPA compared to wheat and barley (Eeckhout and De Paepe 1994; Madsen et al. 2013; Steiner et al. 2007; Viveros et al. 2000), but it remains to be examined if the Secale genes will prove as active when transferred to wheat.
References
Baum BR, Bailey LG (2004) The origin of the A genome donor of wheats (Triticum: Poaceae)—a perspective based on the sequence variation of the 5S DNA gene units. Genet Resour Crop Evol 51:183–196
Brinch-Pedersen H, Sorensen LD, Holm PB (2002) Engineering crop plants: getting a handle on phosphate. Trends Plant Sci 7:118–125
Brinch-Pedersen H, Madsen CK, Holme IB, Dionisio G (2013) Increased understanding of the cereal phytase complement for better mineral bio-availability and resource management. J Cereal Sci 59:373–381
CLCbio (2007) White paper on alignment speed and quality. http://www.clcbio.com/files/whitepapers/wp_alignmentspeed_A4.pdf. Accessed 7 March 2016
Dionisio G et al (2011) Cloning and characterization of purple acid phosphatase phytases from wheat (Triticum aestivum L.), barley (Hordeum vulgare L.), maize (Zea maize L.) and rice (Oryza sativa L.). Plant Physiol 156:1087–1100
Dreisigacker S, Kishii M, Lage J, Warburton M (2008) Use of synthetic hexaploid wheat to increase diversity for CIMMYT bread wheat improvement. Aust J Agric Res 59:413–420
Dvorak J, Terlizzi P, Zhang HB, Resta P (1993) The evolution of polyploid wheats: identification of the A genome donor species. Genome 36:21–31
Eeckhout W, De Paepe M (1994) Total phosphorus, phytate-phosphorus and phytase activity in plant feedstuffs. Anim Feed Sci Technol 47:19–29
El Baidouri M et al (2017) Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol 213:1477–1486
Feng D-F, Doolittle RF (1987) Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol 25(4):351–360
Fernández-Calvín B, Orellana J (1994) Metaphase I-bound arms frequency and genome analysis in wheat-Aegilops hybrids. 3. Similar relationships between the B genome of wheat and S or S l genomes of Ae. speltoides, Ae. longissima and Ae. sharonensis. Theor Appl Genet 88:1043–1049
Friebe B, Tuleen NA, Gill BS (1995) Standard karyotype of Triticum searsii and its relationship with other S-genome species and common wheat. Theor Appl Genet 91:248–254
Golovnina K, Glushkov S, Blinov A, Mayorov V, Adkison L, Goncharov N (2007) Molecular phylogeny of the genus Triticum L. Plant Syst Evol 264:195–216
Graybosch RA (2001) Mini review: uneasy unions: quality effects of rye chromatin transfers to wheat. J Cereal Sci 33:3–16
Haider N (2013) The origin of the B-genome of bread wheat (Triticum aestivum L.). Russ J Genet 49:263–274
Holme IB, Dionisio G, Brinch-Pedersen H, Wendt T, Madsen CK, Vincze E, Holm PB (2012) Cisgenic barley with improved phytase activity. Plant Biotechnol J 10:237–247
Huang S, Sirikhachornkit A, Su X, Faris J, Gill B, Haselkorn R, Gornicki P (2002) Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc Natl Acad Sci USA 99:8133–8138
Kearse M et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kihara H (1944) Discovery of the DD-analyser, one of the ancestors of Triticum vulgare. Agric Hortic 19:13–14
Kilian B et al (2007) Independent wheat B and G genome origins in outcrossing Aegilops progenitor haplotypes. Mol Biol Evol 24:217–227
Li L-F, Liu B, Olsen KM, Wendel JF (2015a) Multiple rounds of ancient and recent hybridizations have occurred within the Aegilops–Triticum complex. New Phytol 208:11–12
Li L-F, Liu B, Olsen KM, Wendel JF (2015b) A re-evaluation of the homoploid hybrid origin of Aegilops tauschii, the donor of the wheat D-subgenome. New Phytol 208:4–8
Madsen CK, Dionisio G, Holme IB, Holm PB, Brinch-Pedersen H (2013) High mature grain phytase activity in the Triticeae has evolved by duplication followed by neofunctionalization of the purple acid phosphatase phytase (PAPhy) gene. J Exp Bot 64:3111–3123
Marcussen T et al (2014) Ancient hybridizations among the ancestral genomes of bread wheat. Science 345(6194):1250092. doi:10.1126/science.1250092
McFadden ES, Sears ER (1944) The artificial synthesis of Triticum spelta. Rec Genet Soc Am 13:26–27
McFadden ES, Sears ER (1946) The origin of Triticum spelta and its free-threshing hexaploid relatives. J Hered 37:81–107
Møller MG, Taylor C, Rasmussen SK, Holm PB (2003) Molecular cloning and characterisation of two genes encoding asparagine synthetase in barley (Hordeum vulgare L.). Biochim Biophys Acta 1628:123–132
Oettler G (2005) The fortune of a botanical curiosity—triticale: past, present and future. J Agric Sci 143:329–346
Peng J, Sun D, Nevo E (2011) Domestication evolution, genetics and genomics in wheat. Mol Breed 28:281–301
Perugini LD, Murphy JP, Marshall D, Brown-Guedira G (2008) Pm37, a new broadly effective powdery mildew resistance gene from Triticum timopheevii. Theor Appl Genet 116:417–425
Petersen G, Seberg O, Yde M, Berthelsen K (2006) Phylogenetic relationships of Triticum and Aegilops and evidence for the origin of the A, B, and D genomes of common wheat (Triticum aestivum). Mol Phylogenet Evol 39:70–82
Rabinovich SV (1998) Importance of wheat-rye translocations for breeding modern cultivar of Triticum aestivum L. Euphytica 100:323–340
Salamini F, Ozkan H, Brandolini A, Schäfer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441
Salse J et al (2008) New insights into the origin of the B genome of hexaploid wheat: evolutionary relationships at the SPA genomic region with the S genome of the diploid relative Aegilops speltoides. BMC Genom 9:1–12
Sarkar P, Stebbins GL (1956) Morphological evidence concerning the origin of the B genome in wheat. Am J Bot 43:297–304
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Steiner T, Mosenthin R, Zimmermann B, Greiner R, Roth S (2007) Distribution of phytase activity, total phosphorus and phytate phosphorus in legume seeds, cereals and cereal by-products as influenced by harvest year and cultivar. Anim Feed Sci Technol 133:320–334
van Slageren MW (1994) Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae): a revision of all taxa closely related to wheat, excluding wild Triticum species, with notes on other genera in the tribe Triticeae, especially Triticum. Wageningen Agricultural University papers; 94-7. ISBN-9067543772
Viveros A, Centeno C, Brenes A, Canales R, Lozano A (2000) Phytase and acid phosphatase activities in plant feedstuffs. J Agric Food Chem 48:4009–4013
Wagenaar EB (1966) Studies on the genome constitution of Triticum timopheevi Zhuk. II. The T. timopheevi complex and its origin. Evolution 20:150–164
Acknowledgements
This work was supported by the Danish Ministry of Food, Agriculture and Fisheries (Grant No. 3304-FVFP-08-M-07-01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Madsen, C.K., Petersen, G., Seberg, O. et al. Evolution and diversity of PAPhy_a phytase in the genepool of wheat (Triticum aestivum L., Poaceae). Genet Resour Crop Evol 64, 2115–2126 (2017). https://doi.org/10.1007/s10722-017-0501-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-017-0501-9